

Abstract
Precision oncology is a rapidly evolving interdisciplinary medical specialty. Comprehensive cancer panels are becoming increasingly available at pathology departments worldwide, creating the urgent need for scalable cancer variant annotation and molecularly informed treatment recommendations. A wealth of mainly academia-driven knowledge bases calls for software tools supporting the multi-step diagnostic process. We derive a comprehensive list of knowledge bases relevant for variant interpretation by a review of existing literature followed by a survey among medical experts from university hospitals in Germany. In addition, we review cancer variant interpretation tools, which integrate multiple knowledge bases. We categorize the knowledge bases along the diagnostic process in precision oncology and analyze programmatic access options as well as the integration of knowledge bases into software tools. The most commonly used knowledge bases provide good programmatic access options and have been integrated into a range of software tools. For the wider set of knowledge bases, access options vary across different parts of the diagnostic process. Programmatic access is limited for information regarding clinical classifications of variants and for therapy recommendations. The main issue for databases used for biological classification of pathogenic variants and pathway context information is the lack of standardized interfaces. There is no single cancer variant interpretation tool that integrates all identified knowledge bases. Specialized tools are available and need to be further developed for different steps in the diagnostic process.
Keywords: HiGHmed, personalized medicine, molecular tumor board, data integration, cancer therapy
Introduction
The availability of molecular diagnostics for routine healthcare is democratizing the field of precision oncology, enabling molecularly informed treatment and clinical trials for a steadily increasing number of patients worldwide [1–6]. In parallel, molecular tumor boards (MTBs) are established in a growing number of hospitals to interpret the therapeutic consequences of molecular alterations [7–11]. The MTB is highly interdisciplinary and its composition may vary by hospital. Participating disciplines include among others: oncologists, pathologists, geneticists, bioinformaticians as well as other scientists and physicians from other medical specialties involved in the treatment of the patient, e.g. surgeons, gynecologists and neurologists, depending on the tumor entity. This review focuses on the interpretation of variants based on tumor sequencing without a concurrent germline sequencing. Nevertheless, human geneticists have been added to the team, as they help to decide on the necessity for consecutive germline testing. While sequencing capacities are scalable, variant annotation and prioritization remain the bottleneck in the diagnostic process and are not harmonized across cancer centers [12]. The American Society of Clinical Oncology (ASCO) Omics and Precision Oncology Workshop identified the lack of a single comprehensive Knowledge Base (KB) and the need to search multiple, sometimes conflicting resources as major challenges for applying omics in healthcare [13]. High-quality sequencing data and reusable data processing pipelines for the annotation and interpretation of sequencing data are emerging as the pillars of MTBs.
A number of software tools have recently been proposed to support the diagnostic workflow in precision oncology by providing unified interfaces to selected KBs [14–41]. In the remainder of our work, we will refer to this class of software as cancer variant interpretation tools. These tools take as an input either a list of variants, e.g. provided by a VCF file, or expose a query-oriented interface and return aggregated information retrieved from individual KBs with varying levels of integration and harmonization. This information can then be used to filter and prioritize variants and ultimately derive treatment suggestions. In order to be integrated into any software tool, KBs need to provide programmatic access options, e.g. through a public application programming interface (API) or by providing a dump of the data for downloading.
Even if the aforementioned access options are available, integration of heterogeneous data sources into software solutions is tedious and error prone, e.g. due to varying programmatic interfaces, data models and formats. Therefore, interoperability of data sources, data annotation and algorithms are key success factors of collaborative research and collaborative therapy finding. The German Federal Ministry of Education and Research also identified these challenges. Therefore, it provides funding for four national consortia in the German Medical Informatics Initiative through 2022 to develop national exchange strategies for data beyond individual university hospitals [42]. The given work was developed in the context of the HiGHmed consortium, one of the four funded consortia in this funding program [43]. Similar activities are also taking place in other European countries [44, 45].
Within the consortium, medical experts share their real-world observations and provide valuable feedback from their daily routine. As a result, the requirements and feedback described in this work represent the opinion of clinical experts working in university hospitals across Germany. Beyond the data sources, the workflows from biomaterial sampling and sequencing (panel, whole genome, or exome) to variant annotation and interpretation of the results vary widely from institution to institution. This represents a major challenge for the harmonization of the analysis pipelines in the medical use cases of the HiGHmed consortium and comparable use cases in other projects [46]. On an international level, the Global Alliance for Genomics and Health (GA4GH) aims to set technical standards and has launched the Variant Interpretation for Cancer Consortium (VICC) as a driver project.
1.1 Contributions
In this database review, we provide a systematic overview and categorization of KBs relevant for variant interpretation in oncology, with a particular focus on the technical challenges of their integration into software tools. We propose a categorization of KBs along the diagnostic process of precision oncology and will therefore begin our survey with a brief description of this process and the respective need to access KBs during each of its steps. For all KBs within their assigned categories, we gather general technical and category-specific non-technical parameters as well as bibliographic data on literature citations. In order to prioritize KBs by their relevance in clinical practice, we additionally present results of a survey among molecular pathologists and translational oncologists at university hospitals in the HiGHmed consortium. Based on this categorization, relevancy ranking and KB parameters, we identify engineering challenges for developers of cancer variant interpretation tools and directions for future research.
2 Background: diagnostic process
The diagnostic process in precision oncology from genome sequencing to the discussion of potential therapeutic consequences of molecular alterations requires both a biological and a clinical classification of variants (Figure 1). The process is highly interdisciplinary and physicians are commonly supported by both life and data scientists. Since precision oncology is a new and evolving discipline, the composition and responsibilities vary between university centers.
Figure 1.
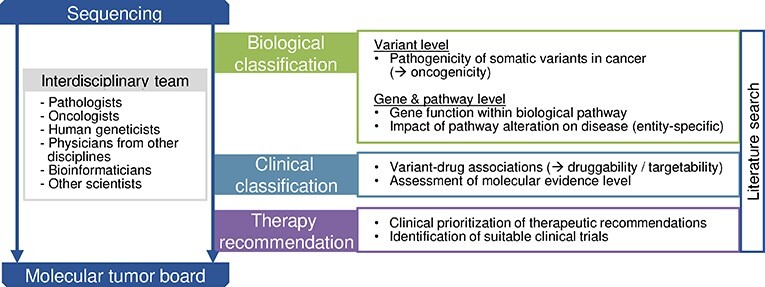
Overview of the diagnostic process in precision oncology. Within this process, knowledge bases can be attributed to the three main diagnostic steps biological classification of a variant, clinical classification and knowledge bases supporting therapy recommendations. Semi-automatic or manual literature search supports the entire process. The diagnostic process is performed by an interdisciplinary team of physicians and scientists. The exact composition and responsibilities within the team vary between university centers.
After obtaining the variant call format (VCF) file from the genetic data processing pipeline, biological classification of findings follows as the first diagnostic step. The term pathogenicity has been lend from the field of human genetics and its assessment includes population frequency, functional data, computational predictions, segregation and somatic frequency [47, 48]. The VICC suggests to use the term oncogenicity for the pathogenicity of somatic variants in cancer [49], but the nomenclature in the context of somatic analyses remains ambiguous.
Pathogenic variants are next interpreted in their pathway context and KBs are available that offer general information about the gene function within biological pathways and tissue- and tumor entity-specific expression patterns. However, most evidence about entity-specific pathway dependencies of variants has to be obtained by manual or semi-automated literature search, e.g. differences in oncogenic signaling between BRAF V600E-mutant melanomas and colorectal cancer.
The second step involves the clinical classification of variants, which is the identification of variant–drug or gene–drug associations, often referred to as druggability or targetability of a variant. In addition, this step includes the assessment of the molecular evidence level that is primarily defined by the clinical evidence of a study (prospective trial versus retrospective trial versus case study) as well as whether the study was performed within the same or a different tumor entity. In Germany, the National Center for Tumor Diseases (NCT) Heidelberg variant classification system is widely adapted. It provides evidence levels, which are of particular importance for the reimbursement of off-label molecularly informed drugs by German healthcare insurances. Leichsenring and colleagues present a comparison between the NCT and internationally recognized evidence levels [50].
The diagnostic process is, if available, concluded by therapy recommendations that are prioritized as the basis for discussion in the MTBs and for the search for suitable clinical trials.
3 Related work
In an early summary of the utility of NGS for cancer therapy, Gagan and Van Allen [51] recommend a set of 8 KBs to be used for the interpretation of somatic cancer variants. Tsang et al. [52] give an overview over catalogs of germline and somatic variants, functional annotation resources and resources linking cancer variants and clinical actionability. In addition, they describe software tools for manipulating variant datasets. Similarly, Prawira et al. [53] reviewed germline and somatic variant databases and in silico prediction tools. In contrast to the latter two reviews, we do not focus on effect predictions, unless they are part of a pre-computed databases such as dbNSFP [54]. Prediction of patient-individual effects describes another set of challenges beyond the scope of this study.
Zhang et al. [55] conducted an extensive review of computational resources (including databases, analysis tools and web platforms) for associations between diseases, genotypes, phenotypes and exposures. While the large set of KBs discussed in this work and ours overlap, we focus specifically on databases relevant for precision oncology with the goal to arrive at actionable treatment suggestions. Pallarz et al. [56] performed a qualitative and quantitative comparison of KBs for precision oncology and assessed their relative importance, concluding that each KB contains unique information relevant for MTB decisions.
In a recent, shorter review, Li and Warner [57] compiled a selection of publicly available and commercial KBs for determining therapeutic options for precision oncology, with detailed descriptions for each of the KBs. They also outline a high-level view of the sequencing process, whose steps ‘Interpretation’ and ‘Decision Making’ roughly correspond to the diagnostic process we described in Section 2. Also recently, Rao et al. [58] conducted a review of the landscape of tools and resources for the evaluation of cancer variants, including a survey of clinically relevant genomic data resources and KBs.
Our work aims to aggregate and extend the set of clinically relevant KBs for precision oncology discussed in the aforementioned articles, also including KBs of medical literature and registered clinical trials. In addition to existing comprehensive collections of KBs, we propose a categorization of KBs along the diagnostic process. Moreover, we attempt to guide implementers of cancer variant interpretation tools by an assessment of programmatic access options as well as a relevancy ranking based on feedback from clinical practitioners.
4 Methods
In the following, we share details about our involved methods to obtain the presented results.
4.1 Identification of relevant knowledge bases
First, we compiled a seed list of KBs based on prior reviews [52, 53, 57, 58] and guidelines for variant annotation by the medical societies ASCO [48] and ESMO [59]. This initial set contained 40 KBs (see the supplementary data). Second, we conducted a survey among a group of 10 selected medical professionals from hospital university centers in the HiGHmed consortium (university hospitals in Heidelberg, Göttingen, Cologne and the Hanover Medical School) and the German Cancer Research Center (DKFZ) to indicate which KBs are relevant for their daily work in the preparation of MTBs. This survey covered all involved parties in the recently implemented multi-site HiGHmed MTB. Participants were provided with the seed list and were asked to mark relevant and add missing KBs to the list. This way, 25 additional KBs were identified and added to the list, resulting in a total of 65 KBs. On the basis of our understanding of the critical difference between the biological and clinical variant interpretation introduced in Section 2, five participants of the survey were selected for their familiarity with biological variant interpretation (four molecular pathologists and one human geneticist), whereas the other five participants were selected from translational oncology departments (four oncologists and one bioinformatician from the German Cancer Research Center). The seed list of KBs and the survey responses were combined to assemble a complete list of KBs. The latter was sent to the participants once again, who had the opportunity to update their votes. The response options of the survey were yes, no and sometimes, e.g. when a KB was only used for specific tumor types. To simplify result presentation, we counted sometimes as yes and thus mapped the responses to a binary voting scheme. The detailed responses can be found in the supplementary data.
4.2 Categorization of knowledge bases along the diagnostic process
The KBs were categorized according to the diagnostic steps as introduced in Figure 1:
Biological classification
Clinical classification
Therapy recommendation
Within the biological classification category, KBs providing information on the variant level are differentiated from KBs that contain gene or pathway-level information. In addition, we have gathered sources for targeted literature research. The categorization of KBs along the diagnostic process was consented by the medical practitioners among the authors based on their experience in the preparation of MTB cases and presented to the survey participants. In case a KB offered content for more than one diagnostic step, it was assigned to the category most used by the survey participants.
4.3 Assessment of technical and non-technical KB parameters
For each of the KBs, an in-depth analysis regarding the license (academic, commercial), programmatic access options and update intervals was performed by the authors. For access options, we distinguish accessibility via API, i.e. the data reside with the database maintainer and a possibility to download a complete or partial dump of the dataset. We did not consider databases as accessible via APIs if the only possibility was scraping of web content, even if this was not discouraged (e.g. via a robot.txt) or inhibited (e.g. via rate limitation) by the database maintainer. Information regarding APIs and dump options could typically be found on the respective project websites. Complete details including links to APIs and file servers are included in the supplementary data. When there was a possibility to get a dump of the data, we also aimed to determine the update frequency of these dumps. When this was not stated on the project websites, we estimated the update intervals by checking the timestamps on the respective file servers (typically FTP).
In addition to these technical parameters, category-specific parameters were determined for each KB, e.g. the emphasis on somatic or germline variants for KBs used for biological classification on the variant level, or the availability of evidence tiers in clinical classification KBs.
For KBs with accompanying research articles, we determine the number of citations from Google Scholar and normalize the citations per year. Exact values can be found in the supplementary files. In case there are multiple articles, as commonly encountered for updated KB version, we report the value for the article that received most citations per year.
4.4 Identification of cancer variant interpretation tools
We identified cancer variant interpretation tools that access multiple KBs through a systematic review of literature indexed in PubMed. Details of the review process with the corresponding PRISMA statement can be found in the supplementary data. We require that tools included in this survey are accessible either through a public demo website or by enabling local installation. To determine which KBs are accessible through each tool, we checked the accompanying websites, research publications and, when available, the source code. A brief manual evaluation of tools providing an online demo was performed with a small set of known pathogenic variants to assess the core functionalities. As our focus is the coverage of accessible KBs, we did not perform an evaluation of the respective search results in terms of correctness, completeness or currency of the integrated KBs. Furthermore, we did only consider potential tools that provide an interface on the variant level, i.e. tools that work solely on the level of genes were excluded. In addition, tools were not considered that required a paid account (e.g. VarSome Clinical [60]), were no longer maintained (e.g. Oncotator [17]) or otherwise inaccessible for testing (e.g. the VMTB knowledge base described by Pishvaian et al. [61]). The assessment of covered KBs was performed during manuscript preparation and last checked on 14 December 2020.
5 Results
In this section, we share the results gathered by the literature- and survey-based collection of KBs and software tools for variant interpretation.
5.1 Cancer variant interpretation tools
As a result of the systematic literature search, 26 tools are included. Incorporating feedback from survey participants, three of these tools [16, 39, 40] were added manually to the result set. These citations were either not indexed by PubMed or the abstract was missing. Additionally, we include the file-based annotation tools ANNOVAR [14], SnpEff, or the professional version ClinEff respectively [15] and the Ensembl Variant Effect Predictor (VEP) [20]. These were used as common building blocks of many other identified tools, which then add additional oncology-specific functionality around them. Furthermore, we list GATK Funcotator [18], which has replaced the no-longer maintained tool Oncotator [17].
We present the identified tools in Table 1 and indicate whether they take as an input a user query (query based) or a file of variants, such as a VCF file (file based), as well as whether they create a local copy of the source data (materialized) or whether the source databases are queried on the fly (API based). In addition, we list the type of output that is produced, distinguishing file-based annotations, search result sets combined from different KBs or domain-specific reports, e.g. for use in MTB preparation. We also indicate if there is a documented update mechanism to keep the integrated KBs current, and if so, whether the update is provided as a bundled release or whether there are individual update routines per KB. If the update mechanism could not be determined from the publication or source code, we indicate this as unknown.
Table 1.
Overview of identified cancer variant interpretation tools, sorted by publication date. We indicate the type of interface (file or query based) for data input as well as the type of output, the accessibility of a online demo or the source code and the type of data integration and the corresponding update mechanisms. Details can be found in the supplementary data. PAS is available as an iOS app; therefore, online demo is marked in brackets.
| Tool | Cit. | Year | User interface | Output | Online demo | Source code | Data integration | Automatic KB update |
|---|---|---|---|---|---|---|---|---|
| ANNOVAR | [14] | 2010 | File based | Annotation | Materialized | Individual | ||
| SnpEff/ClinEff | [15] | 2012 | File based | Annotation | ✓ | Materialized | Bundled | |
| AnalyzeGenomes | [16] | 2014 | File based/query based | Result set | ✓ | Materialized | Individual | |
| GATK Funcotator | [17, 18] | 2015 | File based | Annotation | ✓ | Materialized | Bundled | |
| MyVariant.info | [19] | 2016 | Query based | Result set | ✓ | Materialized | Individual | |
| Ensemble Variant Effect Predictor (VEP) | [20] | 2016 | File based | Annotation | ✓ | ✓ | Materialized | Bundled |
| CanProVar | [21, 22] | 2017 | Query based | Result set | ✓ | Materialized | Unknown | |
| PathOS | [23] | 2017 | File based | Report | ✓ | ✓ | Materialized | Bundled |
| Houston Methodist Variant Viewer (HMVV) | [24] | 2017 | File based | Report | ✓ | Materialized | Bundled | |
| MTB-Report | [25] | 2018 | File based | Report | ✓ | Materialized | Unknown | |
| Smart Cancer Navigator | [26] | 2018 | Query based | Report | ✓ | ✓ | API based | API based |
| Clinical and Genomic Information System (CGIS) | [27] | 2018 | File based | Report | ✓ | Materialized | Unknown | |
| PanDrugs | [28] | 2018 | File based/ query based |
Report | ✓ | Materialized | Unknown | |
| PREDICT Variant Information System (VIS) | [29] | 2018 | Query based | Result set | ✓ | Materialized | Individual | |
| Sequence Variant Identification and Annotation Platform (SeqVItA) | [30] | 2018 | File based | Annotation | ✓ | Materialized | Bundled | |
| Precision Medicine Knowledgebase (PreMedKB) | [31] | 2019 | Query based | Report | ✓ | Materialized | Unknown | |
| Pathogenicity of Mutation Analyzer (PathoMAN) | [32] | 2019 | File based/ query based |
Result set | ✓ | Materialized | Unknown | |
| Variant Interpretation for Cancer (VIC) | [33] | 2019 | File based | Annotation | ✓ | Materialized | Bundled | |
| Translational Genomics expert (TGex) | [34] | 2019 | File based/ query based |
Report | ✓ | Materialized | Unknown | |
| PAS | [35] | 2020 | Query based | Result set | (✓) | Materialized | Individual | |
| AML Variant Analyzer (AMLVaran) | [36] | 2020 | File based | Report | ✓ | ✓ | Materialized | Bundled |
| Open Custom Ranked Analysis of Variants Toolkit (OpenCRAVAT) | [37] | 2020 | File based | Annotation | ✓ | Materialized | Individual | |
| VICC Meta-Knowledgebase (VICC MetaKB) | [38] | 2020 | Query based | Result set | ✓ | ✓ | Materialized | Individual |
| MIRACUM-Pipe | [39] | 2020 | File based | Report | ✓ | Materialized | Individual | |
| Molecular Tumor Board (MTB) Portal | [40] | 2020 | File based/ query based |
Report | ✓ | Materialized | Individual | |
| VarStack | [41] | 2020 | Query based | Result set | ✓ | Materialized | Unknown |
Especially in the case of tools with very active development, e.g. OpenCRAVAT, the coverage of KBs is likely to increase with future versions.
5.2 Knowledge bases and software support
In Tables 2–6, we list details on all identified KBs as well as their categorization along the diagnostic process. Figure 2 shows the ranking based on relevance in clinical practice indicated by subject-matter experts, as well as the coverage by the software tools introduced in the last section. We proceed by describing the main findings for each of the categories. For the first mention of a KB, we also report the percentage of participants responding in our survey to use the database in brackets.
Table 2.
Literature search KBs. For each KB in this and the following tables, we indicate the proportion of survey mentions, the free availability for academic use as well as programmatic access options through APIs and/or downloading options of a dump. If the API or dump of a KB is not freely accessible, usually because of a commercial business model, this is indicated by parentheses around the checkmark. We also list the stated update interval of downloadable KB dumps or the estimated update interval (*) if details were not given on the respective project websites. No update interval (--) is reported, when a download is not available. For details, including URIs, see the supplementary data. As additional specific parameters for the literature KBs in this table, we report the number of included citations ( on 19 February 2021) as well as the controlled vocabulary used for indexing.
on 19 February 2021) as well as the controlled vocabulary used for indexing.
Table 6.
Clinical trial registers and search engines. In addition to the technical parameters and survey responses as introduced in Table 2, we report the scope of trial locations, the number of registered trials ( on 19 February 2021) and the availability of an option to search by molecular markers in each KB.
on 19 February 2021) and the availability of an option to search by molecular markers in each KB.
| Database | Source | Survey | Acad. use | API | Dump | Update interval | Scope |
Number of trials
|
Molecular markers |
|---|---|---|---|---|---|---|---|---|---|
| ClinicalTrials.gov | [129] | 80% | ✓ | ✓ | ✓ | 1 d | International | 367 846 | |
| PCM | [130] | 70% | ✓ | – | – | – | International | 314 | ✓ |
| EU-CTR | [131] | 50% | ✓ | – | – | – | EU | 39 147 | |
| DKTK Trial Register | [132] | 20% | ✓ | – | (✓) | 1 m | Germany | 1056 | ✓ |
| DRKS | [133] | 20% | ✓ | – | – | – | Germany | 11 354 |
Figure 2.
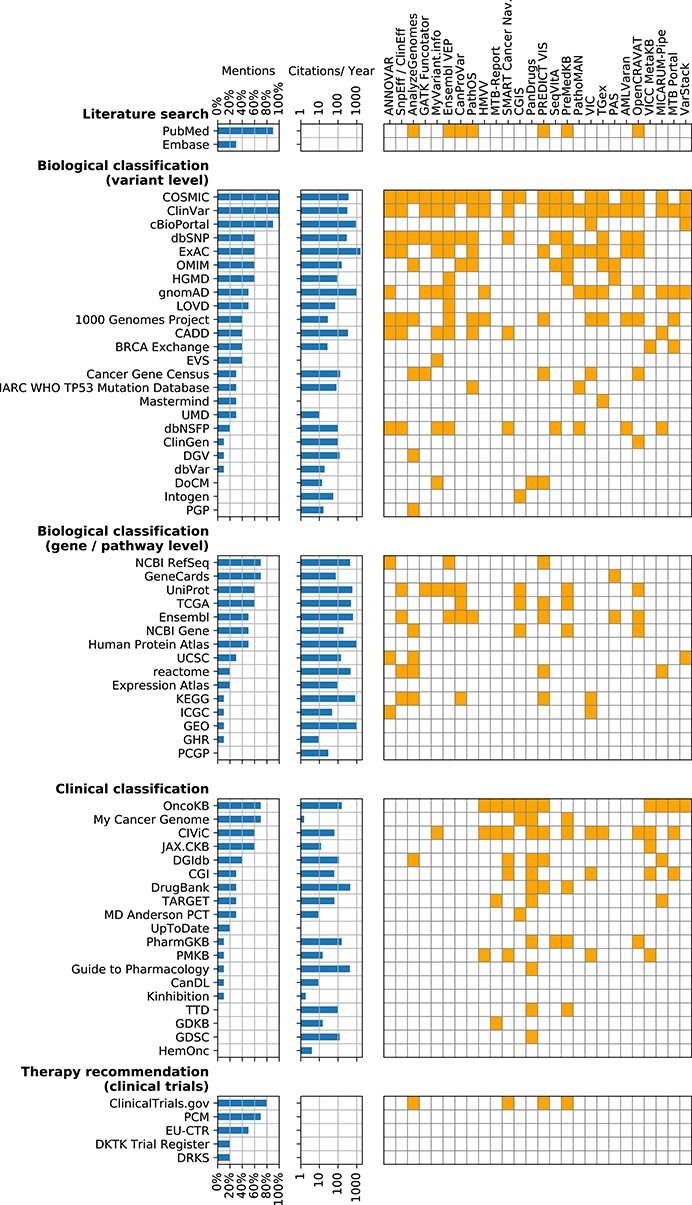
Survey results and tool integration for KBs. We report the overall fraction of survey participants who responded to use the KBs. The individual responses can be found in the supplementary data. We also report the number of citations per year according to Google Scholar. The right matrix indicates which KBs are accessible through each cancer variant interpretation tool.
Note that we do not report KBs, which are accessible through single tools but have not been identified through literature review or in the survey (see the Methods section). For instance, Molecular Match [62] is integrated into the VICC MetaKB but was not identified when compiling the list of KBs. Similarly, OpenCRAVAT integrates an ever increasing amount of KBs, many of which might not be of immediate relevance for clinical oncology and are therefore not discussed in this review.
We explicitly decide to report partially redundant KBs, if they have been named in the survey. For instance, prediction scores from the Combined Annotation Dependent Depletion (CADD) tool are available for downloading but also integrated into the Database of Human Nonsynonymous SNPs and Their Functional Predictions (dbNSFP). Similarly, The Exome Aggregation Consortium (ExAC) database has been completely migrated to the Genome Aggregation Database (gnomAD). In the case of partial or complete integration, this could reflect a preference of the survey participants to use the primary source of information. In the case of migration, it illustrates the challenge for tool developers to constantly update to the right version and location of each KB.
5.2.1 Literature search
The identified KBs of scientific literature are shown in Table 2.
PubMed (90%) is, by a large margin, the most relevant literature search engine in our survey. The data behind PubMed (the MEDLINE and PubMed Central databases) and other NCBI resources can be downloaded in their entirety or via incremental daily updates from public FTP servers. In addition, the Entrez Programming Utilities (eUtils) can be used for programmatic access to various NCBI resources. This way, PubMed search can be readily integrated into software tools, even though advanced indexing beyond Medical Subject Headings (MeSH) and search functionalities relevant for variant annotation would have to be implemented on top of the PubMed data [65–68].
Embase (30%) as a commercial search engine is used by three survey participants, due to the accessibility of the latest conference abstracts and richer search functionalities compared to PubMed. Embase has an API that can be accessed once a license has been obtained.
Advanced or more specific literature search services such as Trip [69] or Livivo [70], or databases of systematic literature reviews such as the Cochrane Database of Systematic Reviews [71] were not mentioned by survey participants.
5.2.2 Biological classification
In Tables 3 and 4, we show the KBs used for the biological classification on the level of variants, genes and pathways.
Table 3.
Biological classification (variant level) KBs. In addition to the technical parameters and survey responses as introduced in Table 2, we report whether a KB contains information on somatic variants and/or germline variants as well as pre-computed functional prediction scores. We do not apply the somatic/germline distinction when databases provide functional prediction results only. Note that germline variants in cBioPortal are not publicly available. Further, note that in the included population/healthy controls data sets (ExAC, 1000 Genomes Project, EVS, DGV and PGP), somatic variants may be found occasionally; therefore, the column is checked in parentheses.
| Database | Source | Survey | Acad. use | API | Dump | Update interval | Somatic variants | Germline variants | Prediction scores |
|---|---|---|---|---|---|---|---|---|---|
| COSMIC | [72] | 100% | ✓ | ✓ | ✓ | 3 m | ✓ | ||
| ClinVar | [73] | 100% | ✓ | ✓ | ✓ | 1 w | ✓ | ✓ | |
| cBioPortal | [74] | 90% | ✓ | ✓ | ✓ | 1 w–1 m* | ✓ | (✓) | |
| dbSNP | [75] | 60% | ✓ | ✓ | ✓ | Upon submission | ✓ | ✓ | |
| ExAC | [76] | 60% | ✓ | – | ✓ | – | (✓) | ✓ | |
| OMIM | [77] | 60% | ✓ | ✓ | ✓ | 1 d | ✓ | ✓ | |
| HGMD | [78] | 60% | ✓ | – | (✓) | 3 m | ✓ | ||
| gnomAD | [79] | 50% | ✓ | – | ✓ | Last: October 2020 | (✓) | ✓ | |
| LOVD | [80] | 50% | ✓ | ✓ | ✓ | 6 m | ✓ | ✓ | |
| 1000 Genomes Project | [81] | 40% | ✓ | ✓ | ✓ | Last: May 2013 | (✓) | ✓ | |
| CADD | [82] | 40% | ✓ | ✓ | ✓ | 1 y | – | – | ✓ |
| BRCA Exchange | [83] | 40% | ✓ | ✓ | ✓ | 2 m | ✓ | ||
| EVS | [84] | 40% | ✓ | – | ✓ | Unknown | (✓) | ✓ | ✓ |
| Cancer Gene Census | [85] | 30% | ✓ | – | ✓ | 3 m | ✓ | ✓ | |
| IARC WHO TP53 Mutation Database | [86] | 30% | ✓ | – | ✓ | 1 y | ✓ | ✓ | ✓ |
| Mastermind | [87] | 30% | (✓) | (✓) | – | – | ✓ | ✓ | |
| UMD | [88] | 30% | ✓ | – | ✓ | Last: January 2013 | ✓ | ||
| dbNSFP | [54] | 20% | ✓ | – | ✓ | 3–6 m | – | – | ✓ |
| ClinGen | [89] | 10% | ✓ | ✓ | – | – | ✓ | ||
| DGV | [90] | 10% | ✓ | – | ✓ | 1–4 y | (✓) | ✓ | |
| dbVar | [91] | 10% | ✓ | ✓ | ✓ | 1–2 m | ✓ | ✓ | |
| DoCM | [92] | 0% | ✓ | ✓ | – | – | ✓ | ||
| Intogen | [93] | 0% | ✓ | – | ✓ | Last: February 2020 | ✓ | ||
| PGP | [94] | 0% | ✓ | – | ✓ | Upon submission | (✓) | ✓ |
Table 4.
KBs for the biological classification on the gene/pathway level. In addition to the technical parameters and survey responses as introduced in Table 2, we indicate the available biological layers for each KB: genes (G), transcripts (T), proteins (P), gene expression (GE), protein expression (PE), pathways (PW) and other/multi-omics layers.
| Database | Source | Survey | Acad. use | API | Dump | Update interval | Biological layer | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G | T | P | GE | PE | PW | Other | |||||||
| NCBI RefSeq | [95] | 70% | ✓ | ✓ | ✓ | 1 d | ✓ | ✓ | ✓ | ||||
| GeneCards | [96] | 70% | (✓) | (✓) | – | – | ✓ | ✓ | ✓ | ✓ | |||
| UniProt | [97] | 60% | ✓ | ✓ | ✓ | 4 w | ✓ | ✓ | ✓ | ✓ | |||
| TCGA | [98] | 60% | ✓ | ✓ | ✓ | 1–3 m | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Ensembl | [99] | 50% | ✓ | ✓ | ✓ | 3 m | ✓ | ✓ | ✓ | ✓ | |||
| NCBI Gene | [100] | 50% | ✓ | ✓ | ✓ | 1 d | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Human Protein Atlas | [101] | 50% | ✓ | ✓ | ✓ | 6–12m | ✓ | ✓ | ✓ | ✓ | |||
| UCSC | [102] | 30% | ✓ | ✓ | ✓ | 2–8 w | ✓ | ✓ | ✓ | ||||
| reactome | [103] | 20% | ✓ | ✓ | ✓ | 3–4 m | ✓ | ✓ | |||||
| Expression Atlas | [104] | 20% | ✓ | ✓ | ✓ | 2–5 m | ✓ | ||||||
| KEGG | [105] | 10% | ✓ | ✓ | ✓ | 7 d | ✓ | ✓ | |||||
| ICGC | [106] | 10% | ✓ | ✓ | ✓ | Last: March 2019 | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| GEO | [107] | 10% | ✓ | ✓ | ✓ | Upon submission | ✓ | ||||||
| GHR | [108] | 10% | ✓ | ✓ | – | – | ✓ | ||||||
| PCGP | [109] | 0% | ✓ | – | (✓) | Unknown | ✓ | ✓ | ✓ | ||||
Variant level: databases in this category are mostly variant databases in structured tabular format. The most widely used KBs, ClinVar (100%) and the Catalogue Of Somatic Mutations In Cancer (COSMIC) (100%), are easily accessible through APIs and downloadable and have been integrated into a number of software tools. Opposed to that, cBioPortal (90%) is almost as widely used in practice and its data are accessible both via API and download, yet it has been integrated into only two of the considered tools.
Eight out of 17 databases most relevant for the survey participants (at least 30% mentions), namely cBioPortal, the Human Gene Mutation Database (HGMD) (60%), the Leiden Open Variation Database (LOVD) (50%), BRCA Exchange (40%), Exome Variant Server (EVS) (40%), the Universal Mutation Database (UMD) (30%), Mastermind (30%) and the International Agency for Research on Cancer (IARC) WHO TP53 Mutation Database (30%), have only been integrated into very few or no variant interpretation tools, even though programmatic access options exist. However, in the case of HGMD, the latest data are only available with a professional license and Mastermind provides a commercial API.
In contrast, the Database for Single Nucleotide Polymorphisms (dbSNP) (60%), Online Mendelian Inheritance in Man (OMIM) (60%), the 1000 Genomes Project (40%), the CADD (40%) database and Cancer Gene Census (30%) have been integrated into different tools. ExaC (60%) was also named as highly relevant, but has been migrated to gnomAD (50%) in the meantime, so the relative importance of gnomAD is expected to become larger in the future.
Less often used according to our survey, but publicly available and programmatically accessible resources are dbNSFP (20%), ClinGen (10%) and the Database of Genomic Variants (DGV) (10%), all which provide a download option or API access. The NCBI resource dbVar (10%) is mentioned in the ASCO guideline [48] and is regularly updated and programmatically accessible, yet it is not integrated into any considered tool.
While a large fraction of biological variant KBs are programmatically accessible and often integrated in a variety of tools, a more detailed investigation reveals that each one comes with their own individual data formats and interfaces. In effect, tools that access multiple of these databases have to implement separate ETL routines for each of the sources.
Three KBs (Personal Genomes Project (PGP) (0%), Database of Curated Mutations (DoCM) (0%) and Intogen (0%)) have been integrated into a few tools but are either unknown or irrelevant to our survey participants.
Gene and pathway level: the other category relevant for the biological classification of variants contains mainly KBs with information on the gene level. This includes general information about the gene function (NCBI RefSeq (70%), GeneCards (70%), Ensembl (50%), NCBI Gene (50%), the University of California Santa Cruz (UCSC) (30%) Genome Browser and Genetics Home Reference (GHR) (10%)), as well as insights about the tissue-specific (UniProt (60%), Human Protein Atlas (50%), Expression Atlas (20%), Gene Expression Omnibus (GEO) (10%)) or tumor entity-specific expression (The Cancer Genome Atlas (TCGA) (60%), International Cancer Genome Consortium (ICGC) (10%)).
A second type of KBs allow to query gene set level information in form of gene set enrichment analyses (reactome (20%), Kyoto Encyclopedia of Genes and Genomes (KEGG) (10%)).
An additional KB mentioned in the ASCO guideline on cancer variant interpretation [48], but not chosen in the survey, is the Pediatric Cancer Genome Project (PCGP) (0%).
Nearly all KBs in this category are programmatically accessible, yet none is integrated by more than a few tools, whereas in other categories, a set of a few important KBs are typically integrated by most tool providers.
5.2.3 Clinical classification
In Table 5, we show the KBs used for the clinical classification step of the diagnostic process.
Table 5.
Clinical classification KBs. In addition to the technical parameters and survey responses as introduced in Table 2, we report the availability of a system of evidence tiers, whether the curation process is based on manual curation by experts and/or a community, as well as the self-reported integration of text mining of scientific articles in the curation process.
| Database | Source | Survey | Acad. use | API | Dump | Update interval | Evidence tiers | Manual curation | Community | Text mining |
|---|---|---|---|---|---|---|---|---|---|---|
| OncoKB | [110] | 70% | ✓ | ✓ | ✓ | 1–4 m* | ✓ | ✓ | ||
| My Cancer Genome | [111] | 70% | ✓ | (✓) | – | – | ✓ | |||
| CIViC | [112] | 60% | ✓ | ✓ | ✓ | 1 m | ✓ | ✓ | ✓ | |
| JAX.CKB | [113] | 60% | (✓) | – | (✓) | 1 d | ✓ | ✓ | ||
| DGIdb | [114] | 40% | ✓ | ✓ | ✓ | 1 m | ✓ | ✓ | ||
| CGI | [115] | 30% | ✓ | ✓ | ✓ | Last: January 2018 | ✓ | ✓ | ||
| DrugBank | [116] | 30% | ✓ | ✓ | ✓ | 1–6 m* | ✓ | |||
| TARGET | [117] | 30% | ✓ | – | ✓ | Last: February 2015 | ✓ | |||
| MD Anderson PCT | [118] | 30% | ✓ | – | – | – | ✓ | ✓ | ✓ | |
| UpToDate | [119] | 20% | – | (✓) | – | – | ✓ | |||
| PharmGKB | [120] | 10% | ✓ | ✓ | ✓ | Upon submission | ✓ | |||
| PMKB | [121] | 10% | ✓ | ✓ | ✓ | Last: August 2019 | ✓ | ✓ | ||
| Guide to Pharmacology | [122] | 10% | ✓ | ✓ | ✓ | 1-3 m | ✓ | |||
| CanDL | [123] | 10% | ✓ | – | ✓ | Last: July 2015 | ✓ | ✓ | ||
| Kinhibition | [124] | 10% | ✓ | – | – | – | ||||
| TTD | [125] | 0% | ✓ | – | ✓ | Last: June 2020 | ✓ | ✓ | ||
| GDKB | [126] | 0% | ✓ | – | ✓ | Last: July 2017 | ✓ | ✓ | ||
| GDSC | [127] | 0% | ✓ | – | ✓ | 3 m–1 y* | ||||
| HemOnc | [128] | 0% | ✓ | – | – | – | ✓ | ✓ | ✓ |
There are a number of widely used, programmatically accessible KBs with information on the clinical actionability of cancer variants. OncoKB (70%) and the community KB Clinical Interpretation of Variants in Cancer (CIViC) (60%) are accessible via API and dump options and are integrated into many recent annotation tools. Likewise, the The Drug Gene Interaction Database (DGIdb) (40%) provides similar programmatic access options and receives monthly updates since the major 4.x release. Cancer Genome Interpreter (CGI) (30%) provides a REST API and can be downloaded but has not been updated since 2018. Similarly, the data of Tumor Alterations Relevant for Genomics-Driven Therapy (TARGET) (30%) and Cancer Driver Log (CanDL) (10%) can still be downloaded but have not been updated since 2015. Somewhat less important resources, albeit still regularly updated, are DrugBank (30%), the Pharmacogenomics Knowledgebase (PharmGKB) (10%), the Precision Medicine Knowledge Base (PMKB) (10%) and Guide To Pharmacology (10%) with either API access and download options.
Commercial or otherwise programmatically inaccessible KBs play a comparatively important role when it comes to clinical variant classification. MyCancerGenome (70%) provides an API only on demand through its licensee GenomOncology. JAX.CKB (60%) has not been integrated into any tool besides the VICC MetaKB, as downloading of JAX.CKB requires a paid account. The web pages MD Anderson Personalized Cancer Therapy (PCT) (30%) and Kinhibition (10%) also do not provide any documented programmatic access options.
Similarly to variant-level biological classification, KBs with information on targeted therapies are usually available in a structured tabular format. However, for clinical classification, there have been harmonization efforts for a range of KBs through the VICC Consortium [38]. From this category, the VICC MetaKB incorporates OncoKB, CIViC, JAX.CKB, Cancer Genome Interpreter, PMKB and Molecular Match. Indeed, this seems to cover most of the identified clinical classification KBs that provide their data for downloading.
Synthesized evidence appears to play only a minor role in precision oncology. Commercial resources such as UpToDate (20%) have not been integrated into any tools under consideration. Other forms of evidence synthesis, such as clinical practice guidelines, also seem to be of little relevance in variant interpretation, as targeted therapies are mostly beyond the scope of guideline recommendations. Also, expectedly, resources on clinical classifications tend to be cancer specific (see the supplementary data).
The Gene Drug Knowledge Base (GDKB) (0%), Therapeutic Target Database (TTD) (0%) and Genomics of Drug Sensitivity in Cancer (GDSC) (0%) are accessible through single tools but were either unknown or irrelevant to our survey participants. The Wiki-based HemOnc (0%) has been named by prior reviews on the subject (e.g. Li and Warner [57]) but is not integrated into any of the tools nor was it mentioned by survey participants.
Even though evidence levels for gene–drug or variant–drug associations are not standardized across KBs, it is helpful if they include a form of evidence grading to guide identification of molecularly informed cancer treatments. KBs that provide some form of evidence tiers include OncoKB, CIViC, MD Anderson PCT, JAX.CKB, Cancer Genome Interpreter, Gene Drug Knowledge Base, PMKB, HemOnc, TTD and CanDL.
While most of the KBs in this category ultimately consist of curated evidence from the primary literature, only very few maintainers report to make use of text mining in the curation process.
5.2.4 Therapy recommendation (clinical trials)
Finally, the KBs of clinical trials are shown in Table 6.
ClinicalTrials.gov (80%) is the most relevant database to search for clinical trials. It can be easily accessed via a REST API and downloaded in an XML format with daily updates. In effect, ClinicalTrials.gov has been integrated into a few different software tools and can be augmented with indices for variants and genes [134]. Precision Cancer Medicine (PCM) (70%) provides a specialized trials search engine to find trials focusing on targeted therapies. Among survey participants, it is perceived as almost as important as ClinicalTrials.gov but in contrast does not provide means for programmatic access and has therefore not been integrated into any of the software tools we consider. Even though the curation process of PCM is unclear, its widespread use emphasizes the demand for specialized search engines for biomarker driven clinical trials.
In addition to well-known international trial registers, finding matching local trials is highly relevant for actual treatment suggestions, as location will be an important factor for inclusion in ongoing trials. In effect, study registers have been mentioned to be relevant which are specific to Europe, such as the EU Clinical Trials Register (EU-CTR) (50%), or to Germany, such as the German Clinical Trials Register (DRKS) (20%) or the German Cancer Consortium (DKTK) Trial Register (20%), which integrates local trial registers from individual sites of the German Cancer Consortium. We expect additional trial registers to be relevant in other countries.
Except for ClinicalTrials.gov, programmatic accessibility to clinical trial data is very restricted. For instance, the EU-CTR does not provide an official API or download options, even though the plain text content of the EU-CTR website could potentially be scraped fairly easily [135].
5.3 Analysis of citations
The number of citations per year for each KB is displayed in Figure 2. As literature search engines and clinical trial registers are typically not explicitly referenced, the number of citations for these categories are 0. Spearman’s rank correlation coefficients between survey mentions, citations per year and number of tools that integrate a certain KB for each of the other three categories are given in Table 7.
Table 7.
Correlation of citations and survey mentions of KBs with their availability through software tools per category. The left columns show the Spearman rank correlation between the number of citations per year and the number of tools integrating a certain KB. The right columns show the Spearman rank correlation between the number of survey mentions and the number of tools integrating a certain KB. Significant values (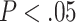 ) are underlined.
) are underlined.
| Citations/no. tools | Survey/no. tools | |||
|---|---|---|---|---|
|
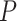
|
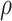
|
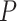
|
|
| Biological classification (variant level) | .65 | <.001 | .66 | <.001 |
| Biological classification (gene/pathway level) | .38 | .167 | .40 | .138 |
| Clinical classification | .46 | .057 | .58 | .011 |
There is a significant correlation between both the number of citations and mentions in the survey and the number of integrating tools for variant level biological classification KBs. For gene/pathway level KBs, there is no such correlation. For clinical classification KBs, the correlation with tool support is only significant for our survey results, not for citations in the literature. Again, this can most likely be attributed to the fact that some KBs in this category are less driven by academia.
5.4 Update intervals
The identified update intervals of downloadable resources range from immediate updates upon submission, regular updates in larger intervals (weekly, monthly, yearly) to irregular major releases in the range of months to years. Many databases integrated into some of the tools have not been updated in multiple years.
With few exceptions (e.g. some of the NCBI resources), most KB providers do not provide straightforward options to perform incremental updates. This poses a substantial challenge for the implementation of automated updating mechanisms in tools based on materialized integrated KBs, as the different update intervals of every source database need to be considered.
Our data show a correlation between documented dump options, update intervals and integration of KBs by software tools. As almost all tools are based on materialized representations, KBs without a dump option have been hardly integrated into any tools (see Tables 2–6 and Figure 2). For the subset of tools with verifiable update intervals (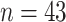 ), the length of the update interval (or the time since the last update) is significantly negatively correlated with both the proportion of mentions in the survey (Spearman rank correlation test
), the length of the update interval (or the time since the last update) is significantly negatively correlated with both the proportion of mentions in the survey (Spearman rank correlation test 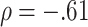 ,
, 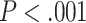 ) and the number of integrating tools (
) and the number of integrating tools (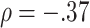 ,
, 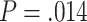 ), i.e. KBs with frequent updates tend to be integrated by more tool developers.
), i.e. KBs with frequent updates tend to be integrated by more tool developers.
6 Discussion
6.1 Tool support for variant interpretation
Molecular diagnostics in precision oncology is an inter-disciplinary process comprising several steps. The review of cancer variant interpretation tools revealed that not a single tool covers all KBs required for all steps equally. Depending on the diagnostic steps, the survey results suggest the usage of specific tools. In addition, the present work allows to determine the possibility to interrogate which tools and which KBs might be integrated in an individualized automated workflow.
We found that for earlier diagnostic steps, especially the biological classification on the variant level, there was a stronger consensus among tool developers about relevant KB to integrate. This consensus is reflected by the strong correlation between the number of integrating tools, citations and mentions in the survey. KBs containing information on the gene or pathway level were identified to be underrepresented in cancer variant interpretation tools and were mentioned by relatively fewer survey participants. While some of these KBs are highly cited in the scientific literature, the relevance according to these citations and also the mentions in our survey are not reflected by the number of integrating tools. However, this step might gain more importance in the future when clinical-grade exome sequencing and RNA-seq become available.
6.2 Standards and interoperability
Our investigation regarding programmatic accessibility reveals notable disparities among categories of KBs used along the diagnostic process. Ongoing standardization efforts within the VICC consortium of the GA4GH initiative address mostly KBs with information on targeted therapies. Defining such harmonized interfaces, based on syntactic and semantic standards, will also facilitate the integration of in-house databases within and across hospitals. However, the relative importance of commercial or otherwise programmatically inaccessible KBs will remain an obstacle for creating a comprehensive integrated KB.
Surprisingly, programmatic access to ongoing clinical trial information, even though in essence publicly available, is very restricted, with ClinicalTrials.gov being an exception. Opposed to that, while most important KBs used for biological classification provide programmatic access options, there is little harmonization and standardization across database providers, imposing a significant burden for the implementation and maintenance of cancer variant interpretation tools integrating these KBs.
6.3 Update intervals and mechanisms
Aligning software tool release cycles with KB releases is a major challenge to be addressed by tool providers. Vastly different update intervals and a lack of incremental updates for many KBs makes updating materialized representations on a per-KB basis challenging. Tools that still enable such updates have to implement KB specific extract-transform-load routines. In practice, users will also need to know if the currently used KB version is the latest version available. Managing these updates will be an additional burden to users.
Tools that provide bundled releases might be more convenient in this regard but add an additional layer of indirection that might incur additional time lags in translation of new results into clinical practice. Moreover, the exact update mechanism has been not clearly documented for a number of identified annotation tools, although this information will be essential for the integration into clinical workflows.
6.4 Usability
Integration of a large set of relevant KBs is a deciding factor, but not the only design aspect motivating the choice of a particular cancer variant interpretation tool. Software usability will be an important feature for adoption by clinical practitioners. For instance, a query-based interface requiring exact input of variant coordinates can impede an explorative use of a tool, in particular in later stages of the diagnostic process. Potentially large results sets from different KBs need to be presented in a manageable fashion, e.g. by providing filtering and sorting mechanisms with sensible defaults. In addition, a usable tool should provide adequate export functionalities to integrate well with applications downstream in the treatment process, e.g. tumor board reporting tools.
6.5 Implementation of an audit trail
A crucial consideration regarding the technical mechanisms used to integrate different KBs is the implementation of an audit trail. When data in public KBs is used to give treatment recommendations endorsed by an MTB, there needs to be a possibility to refer and keep track of the version of the data the decision was based on. Apart from data privacy and performance considerations, this is a key aspect motivating the use of materialized representations of the source data as opposed to an API-based integration. The application of automated versioning of these data in a database management system would be one natural way to implement such an audit trail. In addition to these technical parameters, metadata about the quality of the data sources, the curation process and the data integration pipelines are needed. Usually, these data are not provided in the standard interfaces.
This audit trail would include the information about the queried KBs, their version, the query result and the influence of the result on the MTB recommendation [136, 137]. Furthermore, findability, accessibility, interoperability and reusability of the provided data and pipelines are mandatory to allow implementation of the FAIR guiding principles for scientific data [138]. Most of the aspects are already considered in this review (see Tables 2–6), yet the undetermined update cycles of some data sources are problematic in terms of findability and reusability.
6.6 Annotation quality and redundancy
In previous studies, it has been established that subsets of the identified KBs contain partially overlapping information [26, 38, 56, 139]. Performing a similar analysis on the large set of KBs covered in this survey is likely to reveal similar partial or complete subsumption among KBs, as well as disagreement in annotations. Reliable identification of such conflicts is a prerequisite for developing strategies for their resolution [29]. While this information would be extremely valuable to implementers of cancer variant interpretation tools, such an analysis is hardly feasible without prior restructuring of all KBs to a canonical data format, as it was done in the aforementioned studies, and therefore remains an open problem.
6.7 Determination of molecular evidence levels
The principles of evidence-based medicine also apply to precision oncology. Different variant classification systems have been proposed assigning evidence to variant-drug associations in the context of the individual tumor entity [50]. Main principles of molecular evidence levels include a higher evidence for studies in the same tumor entity versus a different entity and for prospective studies versus retrospective studies or case reports. While several KBs (see Table 5) list evidence levels, they rely on manual curation and are far from being complete. As of today, clinical assertions in these KBs also cannot be systematically and automatically derived through, e.g. text mining of publications, even though this is an active area of research [140, 141]. In Germany, the molecular evidence level determines whether an off-label therapy qualifies for reimbursement by health insurances and is hence an essential part of the MTB report but not a single KB offers an annotation according the NCT evidence levels. Hence, the determination of molecular evidence levels remains a mainly manual and time-consuming step in precision oncology.
6.8 Limitations
6.8.1 Selection of knowledge bases
While we based the selection of KBs on a variety of existing reviews, guidelines and a survey across multiple hospital sites in a large national consortium, a different choice of survey participants could have resulted in a deviating set of KBs. This would likely be the case in particular for KBs, which were included because they were named by a single survey participant. However, as there was substantial agreement regarding the most important KBs, we expect the overall ranking to be informative. In addition, we expect further national databases to be important in other countries, for instance when it comes to finding matching local clinical trials. As the field of precision oncology is evolving at a rapid pace, the list of KBs, their relative importance and coverage by tools are bound to be a temporal snapshot of the current state-of-the-art in variant interpretation.
6.8.2 Variability across institutions
A number of issues regarding the integration of KBs into the diagnostic process have not been considered in this survey. In particular, different sequencing technologies and variant calling pipelines are used across university centers that may or may not already include variant filters. The impact of this variability on the relevance of used KBs as indicated in the survey needs to be considered when interpreting our results. A better understanding of the differences between variant calling pipelines will be crucial to harmonize variant annotation and prioritization workflows in the future.
6.9 Relevance of commercial solutions
In this work, we deliberately chose to only include non-commercial cancer variant interpretation tools to support open science and to enable a unified software support within multiple academic medical centers in Germany independent of individual licenses. Alongside these ongoing efforts at university hospitals, several commercial software solutions are widely used at hospitals and private practices in Germany including the NAVIFY Mutation Profiler (Roche), QIAGEN Clinical Insight (QCI) Interpreter, CureMatch Bionov, Molecular Health Guide and Sophia Genetics. Common advantages include user friendly front ends, end-to-end workflows from sequencing files to reports and audit trails. However, a direct comparison of the KB integration of academic and commercial is not feasible since the exact use and weighting of data sources is in most cases protected and the closed source architecture impedes an integration with other KBs deemed important for the individual tumor of study.
7 Conclusion and outlook
The landscape of KBs for variant annotation and interpretation in precision oncology is constantly evolving. In this database review, our goal was to give an up-to-date overview of the most important precision oncology KBs relevant for molecular pathologists and translational oncologists. In addition, we discussed programmatic access options of KBs and their integration into cancer variant interpretation tools. While it only shows a point-in-time record and may not reflect a European or international opinion, it still provides major building blocks for them.
When adopting any of the KBs in diagnostic workflows, attention must be given to the curation process and the resulting data quality. This is an urgent matter as comprehensive gene panel analysis is becoming a routine diagnostics method at an increasing number of academic cancer centers worldwide. Therefore, completeness and currency of the results derived from interpretation tools does not only depend on automated and errorless data integration mechanisms but also on the reliability of the underlying data sources. A quantitative evaluation of different cancer variant interpretation tools in this regard will be an important direction for future research.
A major future challenge for software tools supporting the diagnostic process in precision oncology is the integration of additional biological layers, particularly the transcriptome and methylome. RNA sequencing has been pivotal to reliably detect cancer gene fusions [142] and help guide cancer of unknown primary diagnostics [143–146]. In addition, an RNA overexpression of candidate driver genes could be identified as predictive biomarkers in the absence of genetic alterations in the same gene [147, 148]. The methylome is gaining increasing significance for diagnostic purposes, e.g. in the classification of central nervous tissue tumors [149] and cancers of unknown primary [150, 151]. Another emerging layer that could aid to increase the performance of drug response predictions is proteomics [152–154].
Lastly, as more and more patients receive targeted treatment based on molecular diagnostics, a key question for future projects will be how outcome data of these cases can be obtained, shared and used to inform evidence-based decision making. Community KBs are a step in this direction, but maintenance and sharing of in-house databases within and across hospitals will become pressing issues in the near future.
Key Points
 Variant interpretation in precision oncology requires access to a variety of knowledge bases in different parts of the diagnostic process.
Variant interpretation in precision oncology requires access to a variety of knowledge bases in different parts of the diagnostic process. Through a review of literature and guidelines for variant interpretation as well as a survey among clinical practitioners, we derive a comprehensive list of knowledge bases with a categorization along the diagnostic process.
Through a review of literature and guidelines for variant interpretation as well as a survey among clinical practitioners, we derive a comprehensive list of knowledge bases with a categorization along the diagnostic process. We assess programmatic access options for all identified knowledge bases and existing integrations into cancer variant interpretation tools .
We assess programmatic access options for all identified knowledge bases and existing integrations into cancer variant interpretation tools .
Supplementary Material
Acknowledgments
We thank the HiGHmed Use Case Oncology leaders (Benedikt Brors, Volker Ellenrieder), the Use Case Oncology executive board members (Andreas Beyer, Alexander König, Thomas Wirth, Christoph Springfeldt) and the members of the HiGHmed working group Analytics for their constant support and valuable input contributing to our research.
In addition, we thank all colleagues from the HiGHmed consortium and participants of the survey: Arndt Vogel, Sebastian Uhrig, Thomas Zander, Olaf Neumann, Bernd Auber, Ulrich Lehmann, Kirsten Reuter-Jessen, Li Beißbarth, Sabine Merkelbach-Bruse and Janna Siemanowski.
We thank Susanne Ibing for fruitful discussions regarding biological layers and multi-omics.
Florian Borchert is a computer scientist and PhD student at the Digital Health Center of the Hasso Plattner Institute (HPI) for Digital Engineering in Potsdam.
Andreas Mock is a physician–scientist at the National Center for Tumor Diseases (NCT) Heidelberg, German Cancer Research Center (DKFZ) and Heidelberg University Hospital.
Aurelie Tomczak is a bioinformatician at the Institute of Pathology Heidelberg, Heidelberg University Hospital and manages the patient register of the Liver Cancer Center Heidelberg.
Jonas Hügel is a computer scientist and PhD student in the Translational Research Informatics research group at the Department of Medical Informatics at the University Medical Center Göttingen (UMG).
Mohammad Samer Alkarkoukly is a physician and currently working as a medical data steward at the University Hospital Cologne for the HiGHmed project.
Alexander Knurr is a computer scientist working for more than 10 years at the NCT Heidelberg and the DKFZ.
Anna-Lena Volckmar is a biologist and performs routine diagnostic genetic testing at the Center for Molecular Pathology at the Institute of Pathology, Heidelberg University Hospital.
Albrecht Stenzinger heads the Center for Molecular Pathology at the Institute of Pathology, Heidelberg University Hospital.
Peter Schirmacher is the medical director of the Department of General Pathology and Pathological Anatomy and the managing director of the Institute of Pathology at Heidelberg University Hospital.
Jürgen Debus is the medical director of the Department of Radiation Oncology and a managing director of the NCT Heidelberg at Heidelberg University Hospital.
Dirk Jäger is the medical director of the Department of Medical Oncology at Heidelberg University Hospital, managing director of the NCT Heidelberg and head of the Clinical Cooperation Unit Applied Tumor-Immunity.
Thomas Longerich is the Deputy Medical Director of the Department of General Pathology and Pathological Anatomy at the Institute of Pathology, Heidelberg University Hospital.
Stefan Fröhling is a managing director of the NCT Heidelberg and head of the Department of Translational Medical Oncology at NCT Heidelberg and the DKFZ.
Roland Eils is the founding director of the Center for Digital Health at Berlin Institute of Health (Charité, Berlin) and the Health Data Science unit at the Medical Faculty of Heidelberg University.
Nina Bougatf is a computer scientist with high expertise in oncology and heads the NCT Cancer Registry and a data science research group at the Department of Radiation Oncology.
Ulrich Sax is a medical informaticist heading the Translational Research Informatics research group in the department of Medical Informatics at UMG.
Matthieu-P. Schapranow is Scientific Manager Digital Health Innovations at the HPI and head of the working group ‘In-Memory Computing for Digital Health’ at the Digital Health Center.
Contributor Information
Florian Borchert, Digital Health Center, Hasso Plattner Institute (HPI), University of Potsdam, Prof.-Dr.-Helmert-Str. 2-3, 14482 Potsdam, Germany.
Andreas Mock, Department of Translational Medical Oncology (TMO), National Center for Tumor Diseases (NCT) Heidelberg, German Cancer Research Center (DKFZ) Heidelberg, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany; Department of Medical Oncology, National Center for Tumor Diseases (NCT) Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany.
Aurelie Tomczak, Institute of Pathology Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 224, 69120 Heidelberg, Germany; Liver Cancer Center Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany.
Jonas Hügel, Department of Medical Informatics, University Medical Center Göttingen, Von-Siebold-Str. 3, 37099 Göttingen, Germany; Campus Institute Data Science, Göttingen, Germany.
Samer Alkarkoukly, CECAD, Faculty of Medicine and University Hospital Cologne, University of Cologne, Joseph-Stelzmann-Straße 26, 50931 Cologne.
Alexander Knurr, Division of Medical Informatics for Translational Oncology, German Cancer Research Center (DKFZ) Heidelberg, Im Neuenheimer Feld 280, 69120 Heidelberg, Germany.
Anna-Lena Volckmar, Institute of Pathology Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 224, 69120 Heidelberg, Germany.
Albrecht Stenzinger, Institute of Pathology Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 224, 69120 Heidelberg, Germany.
Peter Schirmacher, Institute of Pathology Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 224, 69120 Heidelberg, Germany; Liver Cancer Center Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany.
Jürgen Debus, Department of Radiation Oncology, Heidelberg University Hospital, Im Neuenheimer Feld 400, 69120 Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg University Hospital, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany; Clinical Cooperation Unit Radiation Oncology, German Cancer Research Center (DKFZ) Heidelberg, Im Neuenheimer Feld 280, 69120 Heidelberg, Germany; Heidelberg Ion-Beam Therapy Center (HIT), Department of Radiation Oncology, Heidelberg University Hospital, Im Neuenheimer Feld 450, 69120 Heidelberg, Germany; Heidelberg Institute of Radiation Oncology (HIRO), Heidelberg University Hospital, Im Neuenheimer Feld 400, 69120 Heidelberg, Germany.
Dirk Jäger, Department of Medical Oncology, National Center for Tumor Diseases (NCT) Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany; Clinical Coorporation Unit Applied Tumor-Immunity, German Cancer Research Center (DKFZ) Heidelberg, Im Neuenheimer Feld 280, 69120 Heidelberg, Germany.
Thomas Longerich, Institute of Pathology Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 224, 69120 Heidelberg, Germany; Liver Cancer Center Heidelberg, Heidelberg University Hospital, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany.
Stefan Fröhling, Department of Translational Medical Oncology (TMO), National Center for Tumor Diseases (NCT) Heidelberg, German Cancer Research Center (DKFZ) Heidelberg, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany; German Cancer Consortium (DKTK), 69120 Heidelberg, Germany.
Roland Eils, Health Data Science Unit, Heidelberg University Hospital, Im Neuenheimer Feld 267, 69120 Heidelberg, Germany; Center for Digital Health, Berlin Institute of Health and Charité Universitötsmedizin Berlin, Kapelle-Ufer 2, 10117 Berlin, Germany.
Nina Bougatf, Department of Radiation Oncology, Heidelberg University Hospital, Im Neuenheimer Feld 400, 69120 Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg University Hospital, Im Neuenheimer Feld 460, 69120 Heidelberg, Germany; Clinical Cooperation Unit Radiation Oncology, German Cancer Research Center (DKFZ) Heidelberg, Im Neuenheimer Feld 280, 69120 Heidelberg, Germany; Heidelberg Ion-Beam Therapy Center (HIT), Department of Radiation Oncology, Heidelberg University Hospital, Im Neuenheimer Feld 450, 69120 Heidelberg, Germany; Heidelberg Institute of Radiation Oncology (HIRO), Heidelberg University Hospital, Im Neuenheimer Feld 400, 69120 Heidelberg, Germany.
Ulrich Sax, Department of Medical Informatics, University Medical Center Göttingen, Von-Siebold-Str. 3, 37099 Göttingen, Germany; Campus Institute Data Science, Göttingen, Germany.
Matthieu-P Schapranow, Digital Health Center, Hasso Plattner Institute (HPI), University of Potsdam, Prof.-Dr.-Helmert-Str. 2-3, 14482 Potsdam, Germany.
Funding
German Federal Ministry of Research and Education (01ZZ1802); Physician-Scientist Program of the University of Heidelberg, Faculty of Medicine, DKTK (German Cancer Consortium) School of Oncology and the Cancer Core Europe TRYTRAC program (to A.M.); MTB-Report project (VolkswagenStiftung ZN3424) (to J.H.).
References
- 1. Ortiz MV, Kobos R, Walsh M, et al. . Integrating genomics into clinical pediatric oncology using the molecular tumor board at the Memorial Sloan Kettering Cancer Center. Pediatr Blood Cancer 2016; 63(8): 1368–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horak P, Klink B, Heining C, et al. . Precision oncology based on omics data: the NCT Heidelberg experience. Int J Cancer 2017. [DOI] [PubMed] [Google Scholar]
- 3. Freedman AN, Klabunde CN, Wiant K, et al. . Use of next-generation sequencing tests to guide cancer treatment: results from a nationally representative survey of oncologists in the United States. JCO Precis Oncol 2018; 2:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knepper TC, Bell GC, Hicks JK, et al. . Key lessons learned from Moffitt’s molecular tumor board: the clinical genomics action committee experience. Oncologist 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singer F, Irmisch A, Toussaint NC, et al. . SwissMTB: establishing comprehensive molecular cancer diagnostics in Swiss clinics. BMC Med Inform Decis Mak 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van der Velden DL, Hoes LR, van der Wijngaart JM, et al. . The drug rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature 2019. [DOI] [PubMed] [Google Scholar]
- 7. Luchini C, Lawlor RT, Milella M, et al. . Molecular tumor boards in clinical practice. Trends Cancer 2020. [DOI] [PubMed] [Google Scholar]
- 8. Horak P, Kreutzfeldt S, Mock A, et al. . Comprehensive genomic and transcriptomic profiling in advanced-stage cancers and rare malignancies: clinical results from the MASTER trial of the German Cancer Consortium. Ann Oncol 2019. [Google Scholar]
- 9. Schwaederle M, Parker BA, Schwab RB, et al. . Molecular tumor board: the University of California San Diego Moores Cancer Center Experience. Oncologist 2014; 19(6): 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brian Dalton W, Forde PM, Kang H, et al. . Personalized medicine in the oncology clinic: implementation and outcomes of the Johns Hopkins molecular tumor board. JCO Precis Oncol 2017; 1:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoefflin R, Geißler A-L, Fritsch R, et al. . Personalized clinical decision making through implementation of a molecular tumor board: a German single-center experience. JCO Precis Oncol 2018; 2:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singer J, Irmisch A, Ruscheweyh H-J, et al. . Bioinformatics for precision oncology. Brief Bioinform 2017; 20(3): 778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hughes KS, Ambinder EP, Hess GP, et al. . Identifying health information technology needs of oncologists to facilitate the adoption of genomic medicine: recommendations from the 2016 American Society of Clinical Oncology Omics and Precision Oncology Workshop. J Clin Oncol 2017; 35(27): 3153–9. [DOI] [PubMed] [Google Scholar]
- 14. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38(16): e164–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cingolani P, Platts A, Wang LL, et al. . A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012; 6(2): 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Plattner H, Schapranow MP. High-Performance In-Memory Genome Data Analysis: How In-Memory Database Technology Accelerates Personalized Medicine. Cham, Switzerland: Springer Science & Business Media, 2014. [Google Scholar]
- 17. Ramos AH, Lichtenstein L, Gupta M, et al. . Oncotator: cancer variant annotation tool. Hum Mutat 2015; 36(4): E2423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DePristo MA, Banks E, Poplin R, et al. . A framework for variation discovery and genotyping using next-generation dna sequencing data. Nat Genet 2011; 43(5): 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xin J, Mark A, Afrasiabi C, et al. . High-performance web services for querying gene and variant annotation. Genome Biol 2016; 17(1): 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McLaren W, Gil L, Hunt SE, et al. . The ensembl variant effect predictor. Genome Biol 2016; 17(1): 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li J, Duncan DT, Zhang B. Canprovar: a human cancer proteome variation database. Hum Mutat 2010; 31(3): 219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang M, Wang B, Xu J, et al. . Canprovar 2.0: an updated database of human cancer proteome variation. J Proteome Res 2017; 16(2): 421–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doig KD, Fellowes A, Bell AH, et al. . PathOS: a decision support system for reporting high throughput sequencing of cancers in clinical diagnostic laboratories. Genome Med 2017; 9(1): 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Christensen PA, Ni Y, Bao F, et al. . Houston methodist variant viewer: an application to support clinical laboratory interpretation of next-generation sequencing data for cancer. J Pathol Inform 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perera-Bel J, Hutter B, Heining C, et al. . From somatic variants towards precision oncology: evidence-driven reporting of treatment options in molecular tumor boards. Genome Med 2018; 10(1): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Warner JL, Prasad I, Bennett M, et al. . Smart cancer navigator: a framework for implementing asco workshop recommendations to enable precision cancer medicine. JCO Precis Oncol 2018; 2:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jang Y, Choi T, Kim J, et al. . An integrated clinical and genomic information system for cancer precision medicine. BMC Med Genomics 2018; 11(2): 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piñeiro-Yáñez E, Reboiro-Jato M, Gómez-López G, et al. . Pandrugs: a novel method to prioritize anticancer drug treatments according to individual genomic data. Genome Med 2018; 10(1): 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Starlinger J, Pallarz S, Ševa J, et al. . Variant information systems for precision oncology. BMC Med Inform Decis Mak 2018; 18(1): 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dharanipragada P, Seelam SR, Seqvita NP. Sequence variant identification and annotation platform for next generation sequencing data. Front Genet 2018; 9:537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu Y, Wang Y, Xia Z, et al. . Premedkb: an integrated precision medicine knowledgebase for interpreting relationships between diseases, genes, variants and drugs. Nucleic Acids Res 2019; 47(D1): D1090–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ravichandran V, Shameer Z, Kemel Y, et al. . Toward automation of germline variant curation in clinical cancer genetics. Genet Med 2019; 21(9): 2116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. He MM, Li Q, Yan M, et al. . Variant interpretation for cancer (VIC): a computational tool for assessing clinical impacts of somatic variants. Genome Med 2019; 11(1): 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dahary D, Golan Y, Mazor Y, et al. . Genome analysis and knowledge-driven variant interpretation with TGex. BMC Med Genomics 2019; 12(1): 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ahmed Z, Zeeshan S, Mendhe D, et al. . Human gene and disease associations for clinical-genomics and precision medicine research. Clin Transl Med 2020; 10(1): 297–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wünsch C, Banck H, Müller-Tidow C, et al. . Amlvaran: a software approach to implement variant analysis of targeted ngs sequencing data in an oncological care setting. BMC Med Genomics 2020; 13(1): 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pagel KA, Kim R, Moad K, et al. . Integrated informatics analysis of cancer-related variants. JCO Clin Cancer Inform 2020; 4:310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wagner AH, Walsh B, Mayfield G, et al. . A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat Genet 2020; 52(4): 448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Metzger P, Scheible R, Hess M, et al. . Miracum-pipe. https://github.com/AG-Boerries/MIRACUM-Pipe (17 August 2020, date last accessed). [DOI] [PMC free article] [PubMed]
- 40. Tamborero D, Dienstmann R, Rachid MH, et al. . Support systems to guide clinical decision-making in precision oncology: the Cancer Core Europe Molecular Tumor Board Portal. Nat Med 2020; 26(7): 992–4. [DOI] [PubMed] [Google Scholar]
- 41. Howard M, Kane B, Lepry M, et al. . VarStack: a web tool for data retrieval to interpret somatic variants in cancer. Database 2020; 2020(11): baaa092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamdje-Wabo G, Gradinger T, Löbe M, et al. . Towards structured data quality assessment in the german medical informatics initiative: initial approach in the mii demonstrator study. Stud Health Technol Inform 2019; 264:1508–9. [DOI] [PubMed] [Google Scholar]
- 43. Haarbrandt B, Schreiweis B, Rey S, et al. . Highmed—an open platform approach to enhance care and research across institutional boundaries. Methods Inf Med 2018; 57(Suppl 1): e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Louis RJ, Ramòn T-PJ, Sylvain P, et al. . SPHN/PHRT—MedCo in action: empowering the swiss molecular tumor board with privacy-preserving and real-time patient discovery. Stud Health Technol Inform 2020; 270:1161–2. [DOI] [PubMed] [Google Scholar]
- 45. Cuggia M, Combes S. The French Health Data Hub and the German Medical Informatics Initiatives: two national projects to promote data sharing in healthcare. Yearb Med Inform 2019; 28(01): 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Buechner P, Hinderer M, Unberath P, et al. . Requirements analysis and specification for a molecular tumor board platform based on cbioportal. Diagnostics 2020; 10(2): 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Richards S, Aziz N, Bale S, et al. . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li MM, Datto M, Duncavage EJ, et al. . Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017; 19(1): 4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. VICC Knowledge Curation and Interpretation Standards . Standard Operating Procedure for the Interpretation of Oncogenicity of Somatic Variants (Draft Version 1.9.1). https://cancervariants.org/research/standards/onc_path_sop/ (25 January 2021, date last accessed).
- 50. Leichsenring J, Horak P, Kreutzfeldt S, et al. . Variant classification in precision oncology. Int J Cancer 2019; 145(11): 2996–3010. [DOI] [PubMed] [Google Scholar]
- 51. Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Med 2015; 7(1): 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsang H, Addepalli KD, Davis SR. Resources for interpreting variants in precision genomic oncology applications. Front Oncol 2017; 7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prawira A, Pugh TJ, Stockley TL, et al. . Data resources for the identification and interpretation of actionable mutations by clinicians. Ann Oncol 2017; 28(5): 946–57. [DOI] [PubMed] [Google Scholar]
- 54. Liu X, Wu C, Li C, et al. . dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 2016; 37(3): 235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang W, Zhang H, Yang H, et al. . Computational resources associating diseases with genotypes, phenotypes and exposures. Brief Bioinform 2018. bby071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pallarz S, Benary M, Lamping M, et al. . Comparative analysis of public knowledge bases for precision oncology. JCO Precis Oncol 2019; 3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li X, Warner JL. A review of precision oncology knowledgebases for determining the clinical actionability of genetic variants. Front Cell Dev Biol 2020; 8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rao S, Pitel B, Wagner AH, et al. . Collaborative, multidisciplinary evaluation of cancer variants through virtual molecular tumor boards informs local clinical practices. JCO Clin Cancer Inform 2020; 4:602–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mateo J, Chakravarty D, Dienstmann R, et al. . A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO scale for clinical Actionability of molecular targets (ESCAT). Ann Oncol 2018; 29(9): 1895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kopanos C, Tsiolkas V, Kouris A, et al. . Varsome: the human genomic variant search engine. Bioinformatics 2019; 35(11): 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pishvaian MJ, Blais EM, Bender RJ, et al. . A virtual molecular tumor board to improve efficiency and scalability of delivering precision oncology to physicians and their patients. JAMIA Open 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. MolecularMatch, Inc. . MolecularMatch. https://www.molecularmatch.com/ (17 August 2020, date last accessed).
- 63. McEntyre J, Lipman D. PubMed: bridging the information gap. CMAJ 2001; 164(9): 1317–9. [PMC free article] [PubMed] [Google Scholar]
- 64. Elsevier . Embase. https://www.elsevier.com/solutions/embase-biomedical-research (17 August 2020, date last accessed).
- 65. Poon H, Quirk C, DeZiel C, et al. . Literome: Pubmed-scale genomic knowledge base in the cloud. Bioinformatics 2014; 30(19): 2840–2. [DOI] [PubMed] [Google Scholar]
- 66. Schapranow M-P, Kraus M, Perscheid C, et al. . The medical knowledge cockpit: real-time analysis of big medical data enabling precision medicine. In 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Washington, DC, USA: IEEE, 2015, 770–5. [Google Scholar]
- 67. Allot A, Peng Y, Wei C-H, et al. . Litvar: a semantic search engine for linking genomic variant data in PubMed and PMC. Nucleic Acids Res 2018; 46(W1): W530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wei C-H, Allot A, Leaman R, et al. . PubTator Central: automated concept annotation for biomedical full text articles. Nucleic Acids Res 2019; 47(W1): W587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Trip Database Limited . Trip. http://www.tripdatabase.com/ (17 August 2020, date last accessed).
- 70. Pössel J. LIVIVO: das neue ZB MED-Suchportal Lebenswissenschaften. GMS Medizin Bibliothek Information 2015; 15(3). [Google Scholar]
- 71. Starr M, Chalmers I, Clarke M, et al. . The origins, evolution, and future of the Cochrane database of systematic reviews. Int J Technol Assess Health Care 2009; 25(S1): 182–95. [DOI] [PubMed] [Google Scholar]
- 72. Forbes SA, Bindal N, Bamford S, et al. . Cosmic: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 2010; 39(suppl_1): D945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Landrum MJ, Lee JM, Benson M, et al. . Clinvar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016; 44(D1): D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cerami E, Gao J, Dogrusoz U, et al. . The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2(5): 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sherry ST, Ward M-H, Kholodov M, et al. . dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29(1): 308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lek M, Karczewski KJ, Minikel EV, et al. . Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536(7616): 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hamosh A, Scott AF, Amberger JS, et al. . Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005; 33(suppl_1): D514–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cooper DN, Ball EV, Krawczak M. The Human Gene Mutation Database. Nucleic Acids Res 1998; 26(1): 285–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Karczewski KJ, Francioli LC, Tiao G, et al. . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581(7809): 434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fokkema IFAC, Taschner PEM, Schaafsma GCP, et al. . LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 2011; 32(5): 557–63. [DOI] [PubMed] [Google Scholar]
- 81. Clarke L, Zheng-Bradley X, Smith R, et al. . The 1000 Genomes Project: data management and community access. Nat Methods 2012; 9(5): 459–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rentzsch P, Witten D, Cooper GM, et al. . CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019; 47(D1): D886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cline MS, Liao RG, Parsons MT, et al. . BRCA exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet 2018; 14(12): e1007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. NHLBI GO Exome Sequencing Project . Exome Variant Server. http://evs.gs.washington.edu/EVS/ (17 August 2020, date last accessed).
- 85. Sondka Z, Bamford S, Cole CG, et al. . The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 2018; 18(11): 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bouaoun L, Sonkin D, Ardin M, et al. . TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat 2016; 37(9): 865–76. [DOI] [PubMed] [Google Scholar]
- 87. Chunn LM, Nefcy DC, Scouten RW, et al. . Mastermind: a comprehensive genomic association search engine for empirical evidence curation and genetic variant interpretation. Front Genet 2020; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Béroud C, Collod-Béroud G, Boileau C, et al. . UMD universal mutation database: a generic software to build and analyze locus-specific databases. Hum Mutat 2000; 15(1): 86–94. [DOI] [PubMed] [Google Scholar]
- 89. Rehm HL, Berg JS, Brooks LD, et al. . N Engl J Med 2015; 372(23): 2235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. MacDonald JR, Ziman R, Yuen RKC, et al. . The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014; 42(D1): D986–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lappalainen I, Lopez J, Skipper L, et al. . DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res 2013; 41(Database issue): D936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ainscough BJ, Griffith M, Coffman AC, et al. . DoCM: a database of curated mutations in cancer. Nat Methods 2016; 13(10): 806–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Martínez-Jiménez F, Muiños F, Sentís I, et al. . A compendium of mutational cancer driver genes. Nat Rev Cancer 2020. [DOI] [PubMed] [Google Scholar]
- 94. Church GM. The Personal Genome Project. Mol Syst Biol 2005; 1(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pruitt KD, Maglott DR. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res 2001; 29(1): 137–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rebhan M, Chalifa-Caspi V, Prilusky J, et al. . GeneCards: integrating information about genes, proteins and diseases. Trends Genet 1997; 13(4): 163. [DOI] [PubMed] [Google Scholar]
- 97. The UniProt Consortium . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019; 47(D1): D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Weinstein JN, Collisson EA, Mills GB, et al. . The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013; 45(10): 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Aken BL, Ayling S, Barrell D, et al. . The Ensembl gene annotation system. Database 2016; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Maglott D, Ostell J, Pruitt KD, et al. . Entrez gene: gene-centered information at ncbi. Nucleic Acids Res 2005; 33(suppl_1): D54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Uhlén M, Fagerberg L, Hallström BM, et al. . Tissue-based map of the human proteome. Science 2015; 347(6220): 1260419. [DOI] [PubMed] [Google Scholar]
- 102. Kent WJ, Sugnet CW, Furey TS, et al. . The human genome browser at UCSC. Genome Res 2002; 12(6): 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Croft D, Mundo AF, Haw R, et al. . The reactome pathway knowledgebase. Nucleic Acids Res 2014; 42(D1): D472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Papatheodorou I, Moreno P, Manning J, et al. . Expression atlas update: from tissues to single cells. Nucleic Acids Res 2020; 48(D1): D77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000; 28(1): 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhang J, Bajari R, Andric D, et al. . The International Cancer Genome Consortium Data Portal. Nat Biotechnol 2019; 37(4): 367–9. [DOI] [PubMed] [Google Scholar]
- 107. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30(1): 207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mitchell JA, Fun J, McCray AT. Design of genetics home reference: a new NLM consumer health resource. J Am Med Inform Assoc 2004; 11(6): 439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Downing JR, Wilson RK, Zhang J, et al. . The pediatric cancer genome project. Nat Genet 2012; 44(6): 619–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chakravarty D, Gao J, Phillips S, et al. . OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017; 1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Micheel CM, Lovly CM, Levy MA. My Cancer Genome. Cancer Genet 2014; 207(6): 289. [Google Scholar]
- 112. Griffith M, Spies NC, Krysiak K, et al. . CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet 2017; 49(2): 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Patterson SE, Liu R, Statz CM, et al. . The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Hum Genomics 2016; 10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Freshour SL, Kiwala S, Cotto KC, et al. . Integration of the drug–gene interaction database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res 2020; gkaa1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tamborero D, Rubio-Perez C, Deu-Pons J, et al. . Cancer genome interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med 2018; 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wishart DS, Feunang YD, Guo AC, et al. . DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018; 46(D1): D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. van Allen EM, Wagle N, Stojanov P, et al. . Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med 2014; 20(6): 682–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Dumbrava EI, Meric-Bernstam F. Personalized cancer therapy-leveraging a knowledge base for clinical decision-making. Cold Spring Harb Mol Case Stud 2018; 4(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. UpToDate Inc . UpToDate. https://www.uptodate.com (17 August 2020, date last accessed).
- 120. Hewett M, Oliver DE, Rubin DL, et al. . PharmGKB: the Pharmacogenomics Knowledge Base. Nucleic Acids Res 2002; 30(1): 163–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Huang L, Fernandes H, Zia H, et al. . The cancer precision medicine knowledge base for structured clinical-grade mutations and interpretations. J Am Med Inform Assoc 2017; 24(3): 513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Armstrong JF, Faccenda E, Harding SD, et al. . The IUPHAR/BPS guide to pharmacology in 2020: extending immunopharmacology content and introducing the IUPHAR/MMV guide to malaria pharmacology. Nucleic Acids Res 2020; 48(D1): D1006–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Damodaran S, Miya J, Kautto E, et al. . Cancer driver log (CanDL): catalog of potentially actionable cancer mutations. J Mol Diagn 2015; 17(5): 554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bello T, Gujral TS. KInhibition: A Kinase Inhibitor Selection Portal. Iscience 2018; 8:49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wang Y, Zhang S, Li F, et al. . Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res 2020; 48(D1): D1031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Dienstmann R, Jang S, Bot B, et al. . Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov 2015; 5(2): 118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yang W, Soares J, Greninger P, et al. . Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2012; 41(D1): D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Warner JL, Cowan AJ, Hall AC, et al. . Hemonc.org: a collaborative online knowledge platform for oncology professionals. J Oncol Pract 2015; 11(3): e336–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zarin DA, Tse T, Williams RJ, et al. . Trial reporting in ClinicalTrials.gov—the final rule. N Engl J Med 2016; 375(20): 1998–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dana-Farber/Brigham and Women’s Cancer Center . Precision Cancer Medicine. http://www.precisioncancermedicine.org/ (17 August 2020, date last accessed).
- 131. European Medicine Agency . EU Clinical Trials Register. https://www.clinicaltrialsregister.eu/ (17 August 2020, date last accessed).
- 132. Ihrig K, Battke P, Batz D, et al. . Das DKTK-Studienregister: die zentrale Informationsplattform für Therapiestudien onkologischer Spitzenzentren der Krebsmedizin. ONKOLOGIE Heute 2018:38–41. [Google Scholar]
- 133. Dreier G, Hasselblatt H, Antes G, et al. . Das deutsche register Klinischer Studien: Begründung, technische und inhaltliche Aspekte, internationale Einbindung. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 2009; 52(4): 463–8. [DOI] [PubMed] [Google Scholar]
- 134. Xu J, Lee H-J, Zeng J, et al. . Extracting genetic alteration information for personalized cancer therapy from ClinicalTrials.gov. J Am Med Inform Assoc 2016; 23(4): 750–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Herold R. Ctrdata: R package to aggregate and analyse information on clinical trials from public registers, 2018.
- 136. Sinaci AA, Núñez-Benjumea FJ, Gencturk M, et al. . From raw data to FAIR data: the FAIRification workflow for health research. Methods Inf Med 2020; 59(S 01): e21–32. [DOI] [PubMed] [Google Scholar]
- 137. Parciak M, Bender T, Sax U, et al. . Applying FAIRness: redesigning a biomedical informatics research data management pipeline. Methods Inf Med 2019; 58(06): 229–34. [DOI] [PubMed] [Google Scholar]
- 138. Wilkinson MD, Dumontier M, Aalbersberg IJJ, et al. . The FAIR guiding principles for scientific data management and stewardship. Sci Data 2016; 3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sakai K, Takeda M, Shimizu S, et al. . A comparative study of curated contents by knowledge-based curation system in cancer clinical sequencing. Sci Rep 2019; 9(1): 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lever J, Jones MR, Danos AM, et al. . Text-mining clinically relevant cancer biomarkers for curation into the civic database. Genome Med 2019; 11(1): 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lever J, Barbarino JM, Gong L, et al. . PGxMine: text mining for curation of PharmGKB. In Pac Symp Biocomput, Vol. 25. Kohala Coast, Hawaii: World Scientific, 2020, 611–22. [PMC free article] [PubMed] [Google Scholar]
- 142. Heyer EE, Deveson IW, Wooi D, et al. . Diagnosis of fusion genes using targeted RNA sequencing. Nat Commun 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Monzon FA, Lyons-Weiler M, Buturovic LJ, et al. . Multicenter validation of a 1,550-gene expression profile for identification of tumor tissue of origin. J Clin Oncol 2009. [DOI] [PubMed] [Google Scholar]
- 144. Tothill RW, Shi F, Paiman L, et al. . Development and validation of a gene expression tumour classifier for cancer of unknown primary. Pathology 2015. [DOI] [PubMed] [Google Scholar]
- 145. Horlings HM, van Laar RK, Kerst JM, et al. . Gene expression profiling to identify the histogenetic origin of metastatic adenocarcinomas of unknown primary. J Clin Oncol 2008. [DOI] [PubMed] [Google Scholar]
- 146. Hainsworth JD, Rubin MS, Spigel DR, et al. . Molecular gene expression profiling to predict the tissue of origin and direct site-specific therapy in patients with carcinoma of unknown primary site: a prospective trial of the Sarah cannon research institute. J Clin Oncol 2013. [DOI] [PubMed] [Google Scholar]
- 147. Schuler M, Cho BC, Sayehli CM, et al. . Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol 2019. [DOI] [PubMed] [Google Scholar]
- 148. Rodon J, Soria JC, Berger R, et al. . Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Capper D, Jones DTW, Sill M, et al. . DNA methylation-based classification of central nervous system tumours. Nature 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Moran S, Martínez-Cardús A, Sayols S, et al. . Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 2016. [DOI] [PubMed] [Google Scholar]
- 151. Moran S, Martinez-Cardús A, Boussios S, et al. . Precision medicine based on epigenomics: the paradigm of carcinoma of unknown primary, 2017. [DOI] [PubMed]
- 152. Klaeger S, Heinzlmeir S, Wilhelm M, et al. . The target landscape of clinical kinase drugs. Science 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wahjudi LW, Bernhardt S, Abnaof K, et al. . Integrating proteomics into precision oncology. Int J Cancer 2020. [DOI] [PubMed] [Google Scholar]
- 154. Frejno M, Meng C, Ruprecht B, et al. . Proteome activity landscapes of tumor cell lines determine drug responses. Nat Commun 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.