
Figure 1.
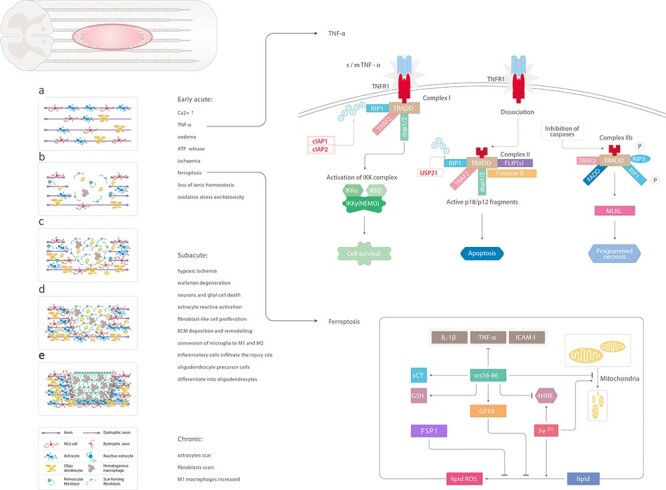
Schematic illustration of the main changes induced by spinal cord injury. (a) Uninjured spinal cord. (b) Early acute phase of spinal cord injury: increased level of Ca2+, TNF-α, edema, ATP release, ischemia, ferroptosis, loss of ionic homeostasis, oxidative stress excitotoxicity. Soluble TNF-α binds to TNFR1 on the cell membrane. RIP1 binds to other related proteins to form complex 1. cIAP1 and cIAP2 can ubiquitinate RIP1 and activate the NF-κB signaling pathway to promote cell survival. USP21 induces deubiquitination of RIP1 to form complex 2, leading to apoptosis. Fe2+ catalyzes the peroxidation of liposomes on the cell membrane and increases the production of reactive oxygen species. In addition, GPX4 is inactivated, thereby inducing iron-dependent death of cells. The iron-dependent death inhibitor, SRS 16–86, significantly inhibits the expression of inflammatory factors IL-1β, TNF-α and ICAM-1 and rescues the reduction of mitochondria and crest reduction. (c, d) Subacute phase: hypoxic ischemia, reactive astrocytosis, Wallarian degeneration, neurons and glial cell death, reactive activation of astrocytes, fibroblast-like cell proliferation, ECM deposition and remodeling, conversion of microglia to M1 and M2, infiltration of inflammatory cells at the injury site, differentiation of OPCs into oligodendrocytes. (e) Chronic phase: formation of astrocyte scars, fibroblast scars and increase in M1 macrophages. TNF- α tumor necrosis factor-alpha, ATP adenosine triphosphate, RIP1 receptor interacting protein 1, cIAP1 apoptosis inhibitory protein 1, GPX4 glutathione peroxidase 4, IL-1β interleukin-1β, ICAM-1 intercellular adhesion molecule-1, OPCs oligodendrocyte progenitor cells