
Fig. 1 |. Phenylalanine metabolism and PKU.
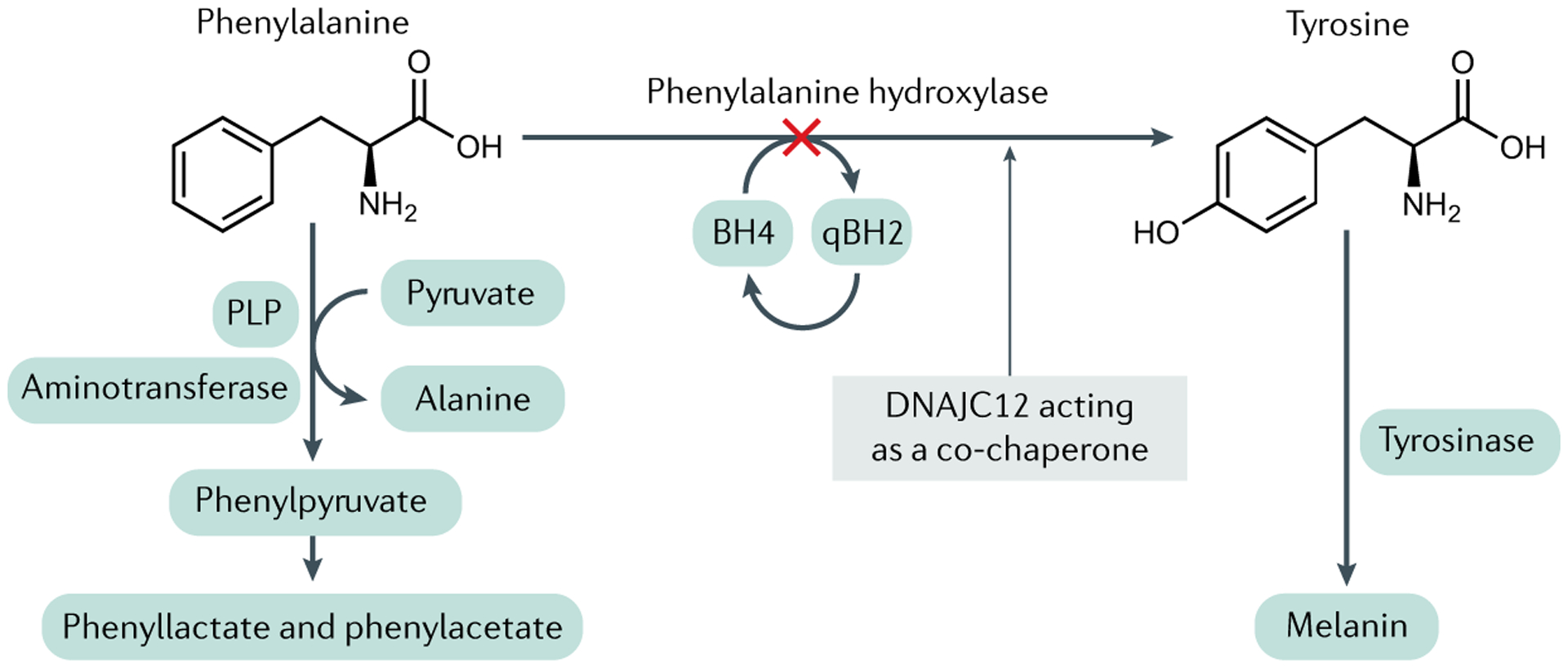
Phenylalanine hydroxylase (PAH) catalyses the hydroxylation of l-phenylalanine (Phe) to l-tyrosine (Tyr), a reaction that occurs pre-dominantly in the liver but also in the proximal renal tubules in the kidneys. The reaction requires the reduced pterin tetrahydrobiopterin (BH4), along with ferrous iron and molecular oxygen (not shown) as cofactors. BH4 is oxidized to quinonoid dihydrobiopterin (qBH2) in the course of the hydroxylation reaction; qBH2 is enzymatically recycled back to BH4 in order to support ongoing Phe hydroxylation. In individuals with recessively inherited pathogenetic variants in the PAH gene, PAH enzymatic activity is either entirely lacking or severely diminished. As approximately 90% of the daily dietary intake of Phe must be metabolized through this pathway, PAH deficiency causes the accumulation of Phe in the body, most readily measured as extreme elevations of Phe concentrations in the blood (hyperphenylalaninaemia). Blood Tyr concentration is also diminished relative to that in PAH-sufficient individuals, but hypotyrosinaemia is not typically severe, perhaps because of dietary Tyr intake. In the setting of hyperphenylalaninaemia, deamination of Phe forms phenylpyruvate and other phenylketones, which are readily excreted in the urine and are the source of the colloquial name phenylketonuria (PKU). PLP, pyridoxal 5′-phosphate.