

Abstract

The design and synthesis of butyl chain derivatives at the indane ring 3-position of our lead CD4-mimetic compound BNM-III-170 that inhibits human immunodeficiency virus (HIV-1) infection are reported. Optimization efforts were guided by crystallographic and computational analysis of the small-molecule ligands of the Phe43 cavity of the envelope glycoprotein gp120. Biological evaluation of 11–21 revealed that members of this series of CD4-mimetic compounds are able to inhibit HIV-1 viral entry into target cells more potently and with greater breadth compared to BNM-III-170. Crystallographic analysis of the binding pocket of 14, 16, and 17 revealed a novel hydrogen bonding interaction between His105 and a primary hydroxyl group on the butyl side chain. Further optimization of this interaction with the His105 residue holds the promise of more potent CD4-mimetic compounds.
Keywords: HIV, gp120, CD4, Entry inhibitor, Structure-based drug design, X-ray crystallography, Viral inhibition, Protein−protein interactions
In 2020 it was estimated that 38 million people were living with HIV-1 globally, with 1.5 million new infections reported that year.1 Although treatment options for HIV-infected individuals are available,2−4 the control of viral rebound after the cessation of antiretroviral therapy and the prevention of HIV-1 transmission remain important but still elusive goals.
CD4-mimetic compounds (CD4mc) can potentially contribute to the control of the HIV-1 pandemic. Work from our group5 and others6 demonstrated that compounds that mimic the T-cell receptor CD4 and its interaction with the HIV-1 viral envelope glycoprotein trimer (Env) are able to inhibit viral entry into host cells by competitive inhibition and by premature allosteric activation of the HIV Env. Such premature activation leads to a series of conformational changes in Env that result in deactivation of the virus.7 Furthermore, CD4mc can expose epitopes on the interior of the Env trimer that can be recognized by the host’s immune system.8 It is important to note that the HIV Env trimer is expressed on both the surface of viral particles and on HIV-1-infected cells.9 This occurrence permits dual pharmacological action of CD4mc. When the cell-surface Env is in an unliganded “closed” State 1 conformation, it cannot be recognized by most of the host antibodies generated during natural infection.10 However, when Env is stabilized by CD4mc in the open conformation, antibody epitopes become exposed that can be recognized by the host’s natural immune response. It has been demonstrated that CD4mc are able to stabilize open Env conformations, thus sensitizing HIV-1-infected cells to Antibody Dependent Cellular Cytotoxicity (ADCC).11 CD4mc that stabilize this “open” conformation are thus able both to neutralize the virus and stimulate ADCC of the infected cells to eradicate HIV-1.12 Moreover, evidence of viral eradication has recently been demonstrated in HIV-1-infected SRG-15 humanized mice. Upon treatment with CD4mc and CD4-induced antibodies, decreases in the viral reservoir and delay of viral rebound was observed in the infected mice after cessation of antiretroviral treatment.13 Given the potential applications of CD4mc, improvement of such small molecules would be of great utility.
In 2016, we reported the structure-based optimization of a CD4mc, based on a hit initially identified in the Debnath laboratory,14 to arrive at our current lead compound BNM-III-170 (Figure 1A).15 This compound displays low-micromolar (14.5 μM) inhibition of HIV-1JR-FL entry into human immune cells. X-ray crystal structures of this compound (Figure 1B) and congeners,5,15 as well as recent cryogenic electron microscopy structures,16 reveal that binding of CD4mc occurs in the Phe43 cavity on the surface of gp120 (the exterior, receptor-binding subunit of Env) as well as in the vestibule surrounding this cavity. Since our initial report, our group has explored substitution of the indane core at the 2-, 5-, 6-, and 7-position in efforts to optimize the structure further. To date, variations at the positions listed above have not yielded more active compounds than BNM-III-170. We therefore focused our efforts toward exploration of the 3-position of the indane core, given that this region is largely unexplored and harbors Env residues identified as important for engagement of the CD4 receptor.17
Figure 1.

(A) Chemical structure of BNM-III-170 with indane position numbering. (B) Co-crystal structure of BNM-III-170 in a gp120 core monomer from HIV Clade C1086.
Computational analysis of de novo docking of CD4mc entry inhibitors was performed to target this unexplored region. From our docking studies, we found that addition of a butyl chain extending from the 3-position of the indane skeleton could serve as a template for installation of a diverse series of functional groups to enhance CD4mc binding. It was also determined that contact between the glycoprotein surface and the 3-position butyl chain was optimal when this chain was positioned “trans” to the methylguanidinium functionality at the 2-position. Additional docking studies revealed that incorporation of a secondary alcohol at C3 of the butyl chain has the potential to interact with His105 (Figure 2). The CD4mc’s pose in the CD4 binding site also suggested that installation of such an oxygenated butyl chain could form a hydrogen bond network between His105 and other nearby residues (Asp474, Arg476). In light of these potential interactions, we undertook synthetic efforts toward installation of a modular butyl side chain at the 3-position of BNM-III-170.
Figure 2.

Structure of BNM-III-170 with position numbering. Structure of a proposed indane 3-position analogue with numbering of the indane skeleton (blue) as well as the butyl side chain (red).
Early in this investigation, it was determined that a monosubstituted terminal alkene on the butyl side chain would be optimal for late-stage diversification. When all desired functional groups are installed on the indane skeleton, the molecule would be sensitive to strongly reducing conditions as well as strong bases or nucleophiles, whereas these groups show stability under a number of oxidative conditions. Therefore, we chose to pursue the installation of a terminal alkene as a common precursor to the desired oxygenated analogues due to the chemical versatility of a C=C double bond by virtue of oxidation.
With these design considerations in mind, we began the synthesis of common intermediate 10 (Scheme 1) bearing an alkene functional handle, exploiting our process synthesis of BNM-III-170 as a guide.18 Beginning with commercially available 5-bromoindanone, carboxylation of the 3-position was achieved by conversion of ketone 1 to the silyl-enol ether, which was then deprotonated to give an indene anion, which was trapped with CO2 to give the carboxylate. Upon acidic workup, the silyl-enol ether was hydrolyzed. After esterification of the carboxylic acid, the resulting racemic β-ketoester 2 was then subjected to a dynamic kinetic resolution utilizing the ruthenium catalyst RuCl[(R,R)-TsDPEN](p-cymene),19 which promoted transfer hydrogenation with excellent enantioselectivity (98% ee; see the Supporting Information for SFC analysis) and modest diastereoselectivity (4:1 d.r.). After setting the stereochemistry at the 3-position, we installed the butenyl side chain. To this end, the hydroxyl group at C1 of 3 was protected as the THP acetal, which resulted in a 1:1 mixture of diastereomers of subsequent intermediates that contained this functionality. In turn, lithium aluminum hydride reduction of the C3 ester, conversion of the resulting alcohol to the primary iodide, and substitution with allylmagnesium chloride furnished the desired butenyl side chain to give 4.
Scheme 1. Synthesis of 3,5-Substituted Common Intermediate 10.
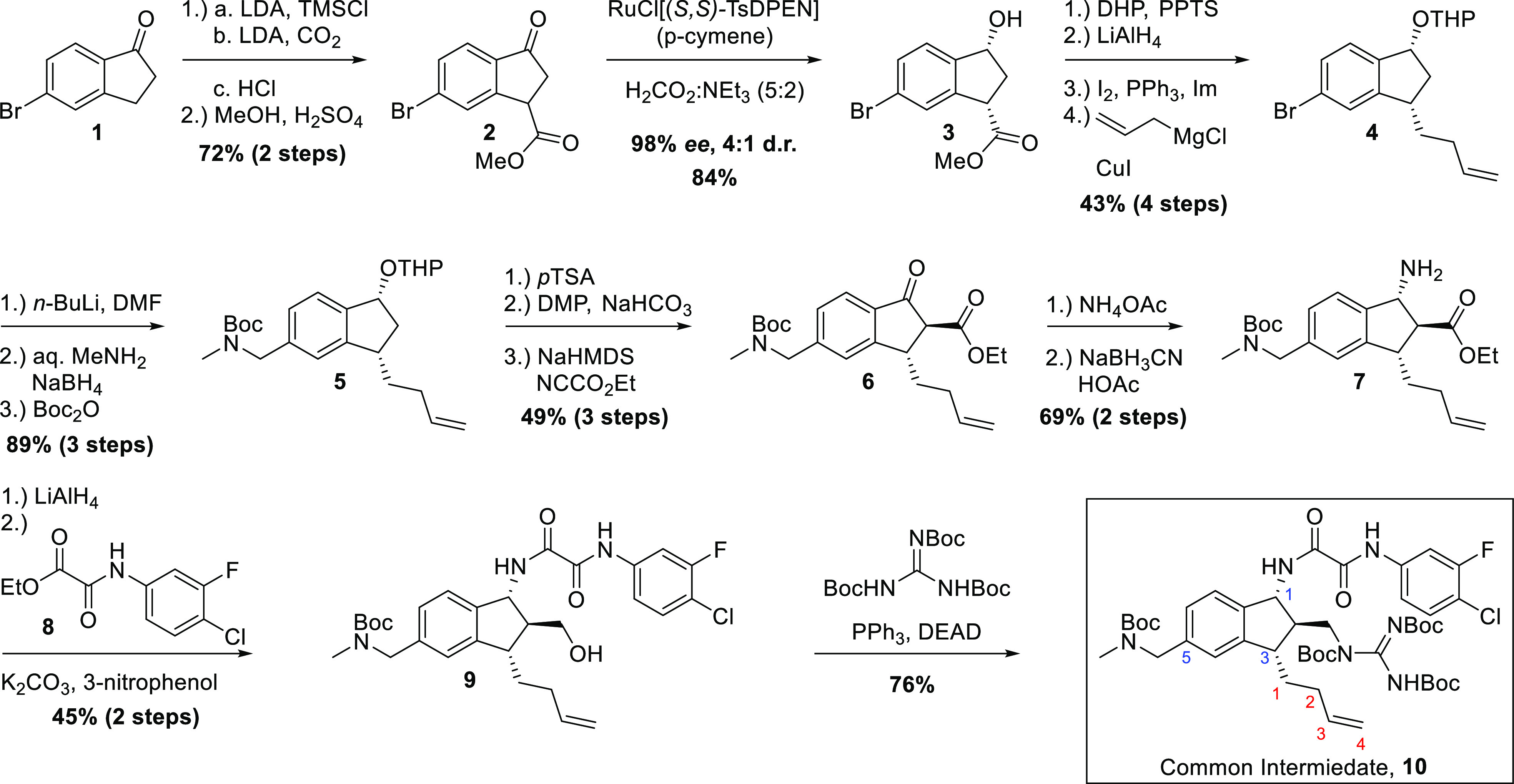
The synthesis of the 3,6-substituted common intermediate was the same as below starting from 6-bromoindanone. Further details can be found in the Supporting Information. Numbering in blue for common intermediate 10 indicates the position of substituents around the indane scaffold. The red numbering indicates carbons relating to the butyl side chain substitution.
Next, our aim was to install the C5 aryl methylamino methyl side chain. This was accomplished by lithium–halogen exchange of the bromide at C5 of 4, followed by formylation using DMF as the formyl source. With the aldehyde in hand, reductive amination with aqueous methylamine, followed by treatment with NaBH4 and subsequent Boc protection completed installation of the methylamino methyl side chain on 5. We then looked to append our previously optimized oxalamide and methyl guanidinium moieties at positions 1 and 2, respectively, on our indane core. To this end, the THP acetal of 5 was hydrolyzed, followed by oxidation of the alcohol to the ketone. Acylation was then achieved using ethyl cyanoformate to furnish β-ketoester 6. With the ketoester in hand, it was envisioned that the “trans–trans” stereoarray from C1 to C2 and C2 to C3 could be formed through directed reduction of an iminium, guided by the ester just installed.20 The β-ketoester 6 was thus converted to an enamine through condensation with ammonium acetate. Reduction using sodium cyanoborohydride with acetic acid then permitted the facial selectivity desired to give 7.
With the desired stereochemistry established, ester 7 was reduced to a primary alcohol followed by amide formation with ester 8 to install selectively the oxalamide at the primary amine of C1 to give 9. Finally, Mitsunobu coupling with tri-Boc-guanidine permitted installation of the methyl guanidinium moiety at C2 of the indane skeleton. Overall, the synthesis of common intermediate 10 was completed in 6.8% overall yield over 18 steps from commercially available 5-bromoindanone (1).
With common intermediate 10 in hand, we began late-stage diversification of the terminal alkene (Scheme 2). We first sought to simply evaluate the effect of adding a saturated butyl chain at the 3-position of the indane skeleton. To accomplish this, common intermediate 10 was subjected to Pd/C hydrogenation, followed by TFA deprotection of the Boc-protecting groups to give the TFA salt of 11. Biological evaluation of TFA salt 11 revealed that addition of the butyl chain did not increase potency against the JR-FL viral strain but, importantly, did not decrease the potency to a large degree (IC50 = 32.5 μM, Table 1). This result validated that a substituent at this position may well be tolerated. At this point, common intermediate 10 was deprotected to give TFA salt 12 (Scheme 2). With the alkene at the terminus of the butyl chain, a more active inhibitor of viral entry resulted (IC50 = 9.22 μM, Table 1) compared to the saturated congener 11.
Scheme 2. Late Stage Diversification of the 3,5-Substituted Common Intermediate 10 Resulting in the Corresponding TFA Salts 11–14.

Diversification of the corresponding 3,6-substituted common intermediate was the same as below. Further details can be found in the Supporting Information.
Table 1. SAR Exploration of the Indane 3-Positiona.


All IC50 values are reported as the mean ± standard deviation.
The IC50 was determined in Cf2Th-CD4/CCR5 cells infected with the HIV-1 virus of the strains indicated.
The IC50 in cells infected with HIV-1 pseudotyped with the envelope glycoproteins of amphotropic murine leukemia virus (A-MLV).
Next, analogues with varying levels of butyl chain oxidation were constructed. Starting from 10, oxidative cleavage, reduction of the resulting aldehyde, and TFA deprotection gave access to 13 (IC50 = 8.25 μM, Table 1), which bears a n-propanol substituent at the 3-position. Racemic dihydroxylation of the alkene followed by deprotection led to 14 (IC50 = 8.29 μM, Table 1) as a 1:1 diastereomeric mixture of alcohols at C3 of the butyl chain. Additionally, an overoxidation product of the dihydroxylation was isolated after TFA deprotection to give α-hydroxyketone 15 (IC50 = 4.97 μM, Table 1). As illustrated in Table 1, the TFA salts of 6-position aryl methylamino methyl side chain analogues were also investigated. To access these analogues, the same synthesis outlined in Scheme 1 was employed beginning from 6-bromoindanone. Late-stage diversification of the 3,6-substituted common intermediate was then carried out in the same fashion as outlined in Scheme 2 (see Supporting Information). Generally, the 3,6-substituted analogues proved less potent than their 3,5-substituted counterparts. Computational models suggest that transposition of the aryl methylamino methyl side chain from the 5-position to the 6-position for this series of analogues (see Table 1: 18–21 in comparison to 12–14 and 16) correlates with an increase in distance of the indane aromatic ring to Gly472 and thus decreased bioactivity.
Overall, many of the 3,5-substituted analogues were found to be similar in potency compared to BNM-III-170. However, upon examining these compounds against a wider panel of viral strains, it was determined that the diastereomeric mixture 14 was able to neutralize various Clade A viruses more effectively than BNM-III-170. As shown in Table 2, high concentrations of BNM-III-170 were required to achieve inhibition of virus strains A4 and BG505; high-micromolar inhibition was observed for strain 191084. Comparatively, CD4mc 14 was able to inhibit infection of these virus strains in the low-micromolar range. An exception was BG505, which required a high-micromolar concentration of 14 to achieve 50% inhibition of viral infection. Despite the observed high-micromolar inhibitory concentration, 14 remained more effective than BNM-III-170 against this viral strain.
Table 2. Comparison of the Bioactivity of Diastereomeric Mixture 14 and BNM-III-170 against Various HIV-1 Viral Strainsa.
| viral strain | amino acid 105 identity | IC50 (μM) 14b | IC50 (μM) BNM-III-170b |
|---|---|---|---|
| JR-FL | GIn | 8.29 ± 5.66 | 9.00 ± 5.14 |
| A4 | His | 98.9 ± 63.0c | 200 ± 42.7 |
| 191084 | His | 20.6 ± 3.10c | 95.9 ± 52.9 |
| BG505 | His | 130 ± 28.3c | 226 ± 47.4 |
All IC50 values are reported as the mean ± standard deviation.
The IC50 was determined in Cf2Th-CD4/CCR5 cells infected with the HIV-1 virus of the strains indicated.
Improvement in viral inhibiton compared to BNM- III-170 found to be statistically significant in a one-tailed unpaired t test (P < 0.05, N = 3).
To understand the structural basis of the increased bioactivity, TFA salt 14 (Figure 3A) was soaked into preformed crystals of gp120 from HIV Clade C1086, and diffraction data were obtained at 2.6 Å Bragg spacings. The parent compound, BNM-III-170, was readily fitted to difference density, which also included density for the 3-substituted butanediol; however, the evident admixture of enantiomeric diols was not readily deconvolved. Therefore, the (S) diastereomer was placed into the 2Fo-Fc density map, as this conformation fit the density best. While missing good density about the secondary alcohol, this structure revealed a novel interaction between the primary hydroxyl group of the butanediol substituent and the gp120 residue His105. Ultimately, after the structures of 16 and 17 were determined (see below), the structure of 14 was refined as a mixture of diastereomers. The interaction with His105 was found to involve a hydrogen bond, of which the distance was measured to be an average of 2.98 Å from the primary hydroxyl groups of 16 and 17 (Figure 3B). Based on this structure, viruses that have the His105 residue (191084, A4, or BG505; Table 2) are expected to display increases in susceptibility to 14, which is consistent with our observations. It is important to note that the His105 residue is conserved only in about 80% of available HIV-1 Env sequences in the NIH Los Alamos HIV database.21 The H105Q polymorphism is found in the JR-FL Env (Table 2),22 which may explain the comparable levels of viral entry inhibition between 14 and BNM-III-170.
Figure 3.

(A) Chemical structure of 14. (B) Crystal structure of 14 in a gp120 core monomer from Clade C1086. 14 is shown as a mixture of its diastereomers 16 (cyan) and 17 (pink). The occupancy of 16:17 was refined at 0.7:0.3. A novel hydrogen bond was observed with His105 at distance of 2.94 Å (16) and 3.01 Å (17).
Given the promising activity of 14, and recognizing that 14 was a diastereomeric mixture, we were interested to know if one of the two diastereomers would be more active than the other. To further investigate the potential difference of the two diastereomers, docking calculations were performed for 16 and 17 using a core monomeric gp120 from HIV Clade C1086. In contrast to 17, only 16 displayed hydrogen bonding between the primary hydroxyl group of the butanediol substituent and His105 with a distance of 2.75 Å. Additionally, a hydrogen bond between the secondary hydroxyl group of 16 and Asp474 was observed with a distance of 2.95 Å. Altogether, structural and computational data of mixture 14 indicated that the (S) diastereomer at the secondary hydroxyl group would be superior to the (R) diastereomer.
With the promising crystallographic and computational data of 16 and 17 in mind, we pursued the corresponding pure diastereomers to validate the models. To do so, common intermediate 10 was subjected to Sharpless asymmetric dihydroxylation23 followed by deprotection with TFA. AD-mix α gave the (S) alcohol 16 in a 4:1 diastereomeric ratio. Unfortunately, obtaining the pure (R) alcohol 17 with AD-mix β proved to be more challenging (1:1 d.r.). After many unsuccessful attempts at diastereoselective installation of the alcohol at the C3 carbon using synthetic methods, physical methods of separation were investigated. Fortunately, after protecting the diastereomeric mixture of 14 as a 1,1-dimethyl acetonide (see Supporting Information), the diastereomers could be separated using chiral HPLC, and subsequently deprotected with TFA to give the pure diastereomers 16 and 17. Based on the JR-FL IC50, 16 and 17 were found to be similar in terms of viral entry inhibition (JR-FL IC50 = 17.7 and 22.0 μM, respectively; not found to be statistically significant in a two-tailed unpaired t test, P > 0.05), and neither diastereomer displayed a statistically significant difference in viral inhibition compared to the diastereomeric mixture 14 (two-tailed unpaired t test was utilized for 14/16 and 14/17; P > 0.05 in both cases).
To better understand the comparable potency of these analogues, TFA salts 16 and 17 were individually soaked into preformed crystals of gp120 from HIV Clade C1086, and diffraction data were obtained at 2.6 Å Bragg spacings (see Supporting Information). The ligands and associated gp120 interactions from the respective crystal structures are shown in Figure 4C,D. Due to flexibility in the butanediol moiety, despite inversion at the asymmetric carbon, the primary hydroxyl group remains in the same locus in both crystal structures and within hydrogen bonding distances to His105 at 3.00 and 3.13 Å for the (S) and (R) isomers, respectively. Additionally, the secondary hydroxyl groups are also at hydrogen bonding distances of 2.98 and 2.85 Å to a carboxyl oxygen atom of Asp474 for both 16 and 17, respectively. The structural information on the two diastereomers suggests that the flexibility of the butanediol moiety allows the ligand to adopt similar binding poses on the protein surface, which may explain the comparable levels of viral entry inhibition among 14, 16, and 17.
Figure 4.

(A) Chemical structure of 16. (B) Chemical structure of 17. (C) Crystal structure of 16 (cyan, S) in a gp120 core monomer from Clade C1086. The hydrogen bond distance from the primary hydroxyl group to the His105 side chain is measured to be 3.00 Å. The hydrogen bond distance from the secondary (S) hydroxyl group to the Asp474 side chain is measured to be 2.98 Å. (D) 17 (pink, R) in a gp120 core monomer from Clade C1086. The hydrogen bond distance from the primary hydroxyl group to the His105 side chain is measured to be 3.13 Å. The hydrogen bond distance from the secondary (R) hydroxyl group to the Asp474 side chain is measured to be 2.85 Å.
With these structures in hand, the further refinement of the 14-gp120 complex was possible. We were then able to determine the relative occupancies of the individual diastereomers in the 14-gp120 complex as being 0.7 and 0.3 for the (S) and (R) diastereomers, respectively (see Supporting Information). The slight differences between occupancies for the (S) and (R) diastereomers in the crystal structure of 14 are compatible with the statistical equivalence of IC50 value for inhibition of viral entry.
In summary, by exploring substitutions at the 3-position of the CD4mc indane scaffold, we have identified a new target residue for engagement on the HIV-1 Env glycoprotein surface, His105. With this starting point, further exploration and optimization will be necessary to better engage this target residue and create more effective CD4mc compounds. Importantly, this series of 3-substituted indane analogues has greater breadth of viral neutralization than any other series of CD4mc synthesized to date. Further exploration of the 3-position analogues is anticipated to lead to more potent congeners with increased activity against more viral strains.
Acknowledgments
We thank Irwin Chaiken and all the members of the P01 Consortium Structure-Based Antagonism of HIV-1 Envelope Function in Cell Entry. Dr. Charles Ross and Dr. Jun Gu (University of Pennsylvania) are also acknowledged for their assistance obtaining mass and NMR spectra, respectively. Instrumentation was supported by the NSF Major Research Instrumentation Program (award NSF CHE-1827457) and the Vagelos Institute for Energy Science and Technology used in this study.
Glossary
ABBREVIATIONS
- HIV-1
human immunodeficiency virus type 1
- A-MLV
amphotropic murine leukemia virus
- SFC
supercritical fluid chromatography
- THP
tetrahydropyran
- LDA
lithium diisopropylamide
- DHP
3,4-dihydropyran
- Im
imidazole
- DMF
N,N-dimethylformamide
- pTSA
p-toluenesulfonic acid
- THF
tetrahydrofuran
- Boc
tert-butoxycarbonyl
- HMDS
hexamethyldisilazide
- DPEN
diphenylethylenediamine
- DEAD
diethylazodicarboxylate
- TFA
trifluoroacetic acid
- PPTS
pyridinium para-toluenesulfonic acid
- DMP
Dess–Martin periodinane
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00437.
Synthesis, experimental methods, characterization and crystal data, structures and chromatograms, and bond distances (PDF)
Author Present Address
▼ Schrödinger, Inc., 120 West 45th Street, New York, NY 10036, United States
Author Present Address
△ J-STAR Research, Inc., 3001 Hadley Road, South Plainfeild, NJ 07080, United States
Author Contributions
¶ C.J.F. and S.L. contributed equally to this work.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding was provided by a Program Project (NIH AI1 50471) to W.A.H., A.B.S., and J.G.S.
The authors declare no competing financial interest.
Notes
Accession Codes, atomic coordinates, and structure factors have been deposited in the Protein Data Bank with the following accession numbers: 7RSX (16), 7RSY (17) and 7RSZ (14).
Supplementary Material
References
- UNAIDS Fact Sheet; United Nations, 2021; https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf. 2021.
- Celum C.; Baeten J. M. Tenofovir-based pre-exposure prophylaxis for HIV prevention: evolving evidence. Curr. Opin. Infect. Dis. 2012, 25 (1), 51–57. 10.1097/QCO.0b013e32834ef5ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdool Karim Q.; Abdool Karim S. S.; Frohlich J. A.; Grobler A. C.; Baxter C.; Mansoor L. E.; Kharsany A. B. M.; Sibeko S.; Mlisana K. P.; Omar Z.; Gengiah T. N.; Maarschalk S.; Arulappan N.; Mlotshwa M.; Morris L.; Taylor D. Effectiveness and Safety of Tenofovir Gel, an Antiretroviral Microbicide, for the Prevention of HIV Infection in Women. Science 2010, 329 (5996), 1168. 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clercq E. D. The design of drugs for HIV and HCV. Nat. Rev. Drug Discovery 2007, 6 (12), 1001–1018. 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- Courter J. R.; Madani N.; Sodroski J.; Schön A.; Freire E.; Kwong P. D.; Hendrickson W. A.; Chaiken I. M.; LaLonde J. M.; Smith A. B. Structure-Based Design, Synthesis and Validation of CD4-Mimetic Small Molecule Inhibitors of HIV-1 Entry: Conversion of a Viral Entry Agonist to an Antagonist. Acc. Chem. Res. 2014, 47 (4), 1228–1237. 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttisintong K.; Kaewchangwat N.; Thanayupong E.; Nerungsi C.; Srikun O.; Pungpo P. Recent Progress in the Development of HIV-1 Entry Inhibitors: From Small Molecules to Potent Anti-HIV Agents. Curr. Top. Med. Chem. 2019, 19 (18), 1599–1620. 10.2174/1568026619666190712204050. [DOI] [PubMed] [Google Scholar]
- Haim H.; Si Z.; Madani N.; Wang L.; Courter J. R.; Princiotto A.; Kassa A.; DeGrace M.; McGee-Estrada K.; Mefford M.; Gabuzda D.; Smith A. B. 3rd; Sodroski J. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog. 2009, 5 (4), e1000360 10.1371/journal.ppat.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsahafi N.; Bakouche N.; Kazemi M.; Richard J.; Ding S.; Bhattacharyya S.; Das D.; Anand S. P.; Prévost J.; Tolbert W. D.; Lu H.; Medjahed H.; Gendron-Lepage G.; Ortega Delgado G. G.; Kirk S.; Melillo B.; Mothes W.; Sodroski J.; Smith A. B. 3rd; Kaufmann D. E.; Wu X.; Pazgier M.; Rouiller I.; Finzi A.; Munro J. B. An Asymmetric Opening of HIV-1 Envelope Mediates Antibody-Dependent Cellular Cytotoxicity. Cell Host Microbe 2019, 25 (4), 578–587. 10.1016/j.chom.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Prévost J.; Alsahafi N.; Ding S.; Finzi A. Impact of HIV-1 Envelope Conformation on ADCC Responses. Trends Microbiol. 2018, 26 (4), 253–265. 10.1016/j.tim.2017.10.007. [DOI] [PubMed] [Google Scholar]
- Munro J. B.; Gorman J.; Ma X.; Zhou Z.; Arthos J.; Burton D. R.; Koff W. C.; Courter J. R.; Smith A. B.; Kwong P. D.; Blanchard S. C.; Mothes W. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346 (6210), 759. 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillette M.; Coutu M.; Richard J.; Batraville L.-A.; Dagher O.; Bernard N.; Tremblay C.; Kaufmann Daniel E.; Roger M.; Finzi A.; Silvestri G. The HIV-1 gp120 CD4-Bound Conformation Is Preferentially Targeted by Antibody-Dependent Cellular Cytotoxicity-Mediating Antibodies in Sera from HIV-1-Infected Individuals. J. Virol. 2015, 89 (1), 545–551. 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Pacheco B.; Gohain N.; Veillette M.; Ding S.; Alsahafi N.; Tolbert W. D.; Prévost J.; Chapleau J.-P.; Coutu M.; Jia M.; Brassard N.; Park J.; Courter J. R.; Melillo B.; Martin L.; Tremblay C.; Hahn B. H.; Kaufmann D. E.; Wu X.; Smith A. B.; Sodroski J.; Pazgier M.; Finzi A. Co-receptor Binding Site Antibodies Enable CD4-Mimetics to Expose Conserved Anti-cluster A ADCC Epitopes on HIV-1 Envelope Glycoproteins. EBioMedicine 2016, 12, 208–218. 10.1016/j.ebiom.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajashekar J. K.; Richard J.; Beloor J.; Prévost J.; Anand S. P.; Beaudoin-Bussières G.; Shan L.; Herndler-Brandstetter D.; Gendron-Lepage G.; Medjahed H.; Bourassa C.; Gaudette F.; Ullah I.; Symmes K.; Peric A.; Lindemuth E.; Bibollet-Ruche F.; Park J.; Chen H. C.; Kaufmann D. E.; Hahn B. H.; Sodroski J.; Pazgier M.; Flavell R. A.; Smith A. B. 3rd; Finzi A.; Kumar P. Modulating HIV-1 envelope glycoprotein conformation to decrease the HIV-1 reservoir. Cell Host Microbe 2021, 29 (6), 904–916. 10.1016/j.chom.2021.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Ma L.; Jiang S.; Lu H.; Liu S.; He Y.; Strick N.; Neamati N.; Debnath A. K. Identification of N-phenyl-N•-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339 (2), 213–225. 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Melillo B.; Liang S.; Park J.; Schön A.; Courter J. R.; LaLonde J. M.; Wendler D. J.; Princiotto A. M.; Seaman M. S.; Freire E.; Sodroski J.; Madani N.; Hendrickson W. A.; Smith A. B. Small-Molecule CD4-Mimics: Structure-Based Optimization of HIV-1 Entry Inhibition. ACS Med. Chem. Lett. 2016, 7 (3), 330–334. 10.1021/acsmedchemlett.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jette C. A.; Barnes C. O.; Kirk S. M.; Melillo B.; Smith A. B.; Bjorkman P. J. Cryo-EM structures of HIV-1 trimer bound to CD4-mimetics BNM-III-170 and M48U1 adopt a CD4-bound open conformation. Nat. Commun. 2021, 12 (1), 1950. 10.1038/s41467-021-21816-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Schön A.; Freire E. Optimization of CD4/gp120 Inhibitors by Thermodynamic-Guided Alanine-Scanning Mutagenesis. Chem. Biol. Drug Des. 2013, 81 (1), 72–78. 10.1111/cbdd.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Park J.; Kirk S. M.; Chen H.-C.; Li X.; Lippincott D. J.; Melillo B.; Smith A. B. Development of an Effective Scalable Enantioselective Synthesis of the HIV-1 Entry Inhibitor BNM-III-170 as the Bis-trifluoroacetate Salt. Org. Process Res. Dev. 2019, 23 (11), 2464–2469. 10.1021/acs.oprd.9b00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rast S.; Modec B.; Stephan M.; Mohar B. γ-Sultam-cored N, N-ligands in the ruthenium(ii)-catalyzed asymmetric transfer hydrogenation of aryl ketones. Org. Biomol. Chem. 2016, 14 (6), 2112–2120. 10.1039/C5OB02352A. [DOI] [PubMed] [Google Scholar]
- Guillon J.; Dallemagne P.; Stiebing S.; Bovy P. R.; Rault S. First Synthesis of 1-Phenyl-3-pyrrol-1-ylindan-2-carboxylic Acid, a New Scaffold of Potential Non-peptide Endothelin Receptor Antagonists. Synlett 1999, 1999 (08), 1263–1264. 10.1055/s-1999-2793. [DOI] [Google Scholar]
- Prévost J.; Tolbert William D.; Medjahed H.; Sherburn Rebekah T.; Madani N.; Zoubchenok D.; Gendron-Lepage G.; Gaffney Althea E.; Grenier Melissa C.; Kirk S.; Vergara N.; Han C.; Mann Brendan T.; Chénine Agnès L.; Ahmed A.; Chaiken I.; Kirchhoff F.; Hahn Beatrice H.; Haim H.; Abrams Cameron F.; Smith Amos B.; Sodroski J.; Pazgier M.; Finzi A.; Zhou T.; Smithgall Thomas E. The HIV-1 Env gp120 Inner Domain Shapes the Phe43 Cavity and the CD4 Binding Site. mBio 2020, 11 (3), e00280-20 10.1128/mBio.00280-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-c.; Tang M.; Zhang M.-Y.; Majeed S.; Montabana E.; Stanfield R. L.; Dimitrov D. S.; Korber B.; Sodroski J.; Wilson I. A.; Wyatt R.; Kwong P. D. Structure of a V3-Containing HIV-1 gp120 Core. Science 2005, 310 (5750), 1025. 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. C.; VanNieuwenhze M. S.; Sharpless K. B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994, 94 (8), 2483–2547. 10.1021/cr00032a009. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.