

Abstract
Leukemias are challenging diseases to treat due, in part, to interactions between leukemia cells and the bone marrow microenvironment (BMME) that contribute significantly to disease progression. Studies have shown that leukemic cells secrete C-chemokine (C-C motif) ligand 3 (CCL3), to disrupt the BMME resulting in loss of hematopoiesis and support of leukemic cell survival and proliferation. In this study, a murine model of blast crisis chronic myelogenous leukemia (bcCML) that expresses the translocation products BCR/ABL and Nup98/HoxA9 was used to determine the role of CCL3 in BMME regulation. Leukemic cells derived from CCL3−/− mice were shown to minimally engraft in a normal BMME, thereby demonstrating that CCL3 signaling was necessary to recapitulate bcCML disease. Further analysis showed disruption in hematopoiesis within the BMME in the bcCML model. To rescue the altered BMME, therapeutic inhibition of CCL3 signaling was investigated using bone-targeted nanoparticles (NP) to deliver Maraviroc, an inhibitor of C-C chemokine receptor type 5 (CCR5), a CCL3 receptor. NP-mediated Maraviroc delivery partially restored the BMME, significantly reduced leukemic burden, and improved survival. Overall, our results demonstrate that inhibiting CCL3 via CCR5 antagonism is a potential therapeutic approach to restore normal hematopoiesis as well as reduce leukemic burden within the BMME.
Keywords: nanoparticles, acute myeloid leukemia, peptide, drug delivery, small molecule drug, MIP1-alpha
1 |. INTRODUCTION
Leukemias are malignancies of the hematopoietic system associated with high risk for relapse and 5-year survival rates of 63%. The acutely progressing myelogenous leukemias, acute myeloid leukemia (AML), and blast crisis chronic myelogenous leukemia (bcCML), have a particularly poor prognosis and are associated with 5-year survival rates of ~28% and 33%, respectively.1,2 Improvements in survival for AML over the past several decades have been largely driven by advancements in supportive care and stem cell transplantation, while the discovery of novel treatment strategies has been limited.3 In chronic myeloid leukemia (CML), the beginning of tyrosine kinase inhibitor treatment in 2000 led to an improvement in the prognosis of CML patients. However, a sizable minority of patients will still progress to blast crisis, at which point the prognosis remains dismal.2,4 Therefore, the identification of additional therapeutic targets, including within the bone marrow microenvironment (BMME), are critical to improve leukemia survival rates.
The BMME provides a permissive niche for leukemia cell survival and plays a major role in patient relapse due to interplay between leukemic cells and the BMME.5,6 In particular, reduced or dysfunctional mesenchymal stem cells (MSCs) and osteoblasts (OBs) contribute to disease initiation and progression by creating a BMME that supports the leukemic cell survival.7–11 Furthermore, the disrupted BMME supports leukemic stem cells (LSCs), and provides a reservoir of therapy-resistant leukemic cells that can give rise to relapse.12
Recent literature has revealed that leukemic cells secrete C-chemokine (C-C motif) ligand 3 (CCL3) to disrupt the BMME.8,13–19 CCL3 is elevated in multiple murine leukemic models and patient samples of AML and bcCML, in myelodysplastic syndrome, B-cell lymphomas, and multiple myeloma, as well as multiple solid tumors.13–16 In murine models of myelogenous leukemias and multiple myeloma, elevated CCL3 mRNA and protein levels correlate with dysfunctional OBs in the BMME, which contributes to disease progression.8,17,18
To reduce the leukemic burden and restore the BMME, CCL3 signaling via the 7-transmembrane G protein-coupled receptors C-C chemokine receptor type 1, 4, and 5 (CCR1, CCR4, and CCR5) can be disrupted. To that end, small molecule inhibitors of CCR1 and CCR5, BX471 and Maraviroc (MVC), have been explored to disrupt CCL3 signaling in multiple myeloma, acute lymphoblastic leukemia, and AML.19–21 BX471, a CCR1 inhibitor, was originally developed for multiple sclerosis but failed in clinical trials, likely due to poor pharmacokinetics (eg, poor aqueous solubility and short circulation half-life).20–22 MVC is an FDA-approved CCR5 antagonist used in HIV therapy, however, MVC is also known to have subpar pharmacokinetics, with a half-life in mice of ~0.9 hours.20,23,24 The ligand/receptor interactions for C-C chemokines are known to be promiscuous. Thus, CCR1 and CCR5 both have multiple ligands, including CCL3, CCL5, CCL7, and CCL14 for CCR1 and CCL3, CCL4, and CCL5, and CCL14 for CCR5.25 This promiscuity makes targeting of the receptors appealing, as it has the potential to block signaling of multiple, possibly redundant ligands, many of which have previously been associated with the formation of a malignant microenvironment.26 While both drugs are promising inhibitors of CCL3 signaling, poor aqueous solubility and pharmacokinetics limit their therapeutic potential.
Drug delivery systems (DDS) increase drug solubility, stability, and circulation time.27 For example, polymer-based nanoparticles (NPs) self-assembled from amphiphilic poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS) diblock copolymers improve the overall bioavailability of poorly soluble drugs.28–30 NPs can enhance drug circulation half-lives and passively accumulate at sites of leaky vasculature, including the bone marrow, via the enhanced permeability and retention effect (EPR).31 Furthermore, drug solubility can be enhanced via loading into hydrophobic cores of polymer-based micelle NPs, protecting drugs from degradation, rapid clearance, and sequestration by off-target organs. Approaches to improve NP-based therapeutics for leukemia include introduction of targeting groups to enhance delivery to the BMME specifically. Here, the tartrate-resistant acid phosphatase (TRAP) binding peptide (TBP; sequence: TPLSYLKGLVTVG) was used to modify NPs to further enhance the drug delivery via marrow-targeting.32 TBP-NPs preferentially accumulate in bone due to osteoclast-specific TRAP deposition within endosteal bone resorption sites.30,33 In these studies, we hypothesized that NP-mediated delivery of CCL3 signaling antagonists will restore the disrupted BMME of the bcCML leukemia model. The bcCML system employed herein is used as a model of primary aggressive myelogenous leukemia.34 This model was chosen as it allows for the study of the BMME and normal hematopoiesis in mice without prior myeloablation or compromise of the immune system, providing an avenue to study the BMME in the presence of leukemia.
2 |. MATERIALS AND METHODS
2.1 |. Mice
All mouse studies were approved by the Institutional Animal Care and Use Committee at the University of Rochester School of Medicine and Dentistry. Male 6- to 8-week-old C57BL/6 (CD45.2), C57BL/002014 (CD45.1), and CCL3 knockout mice were purchased from the mouse breeding core facility at the Wilmot Cancer Institute and used for all studies.
2.2 |. BCR/ABL Nup98/HoxA9 bcCML and MLL-AF9 AML models
BCR/ABL Nup98/HoxA9 bcCML mice were created using MSCV-BCR/ABL-IRES-green fluorescent protein (GFP) and MSCV-Nup98/HoxA9-yellow fluorescent protein (YFP) vectors.8,35 To obtain lineage-negative, Sca-1+, c-Kit+ cells (LSK), marrow hematopoietic stem, and progenitor cells of 6- to 8-week-old male C57BL/002014 (CD45.1) mice were enriched via FACS sorting. To create the leukemic cells, LSK cells were infected with MSCV-BCR/ABL-IRES-GFP and MSCV-Nup98/HoxA9-YFP vectors.8,35 Six- to 8-week-old male C57BL/6 (CD45.2) primary recipients sub-lethally irradiated with 6 Gy using a 137Cs radiation source (GAMMACELL-40) were tail-vein injected with 2 × 104 leukemic cells in 0.1 mL of FACS buffer (1x phosphate-buffered saline (PBS) containing 2% heat-inactivated fetal bovine serum (FBS)). The spleens of primary recipients were harvested after 15 days, crushed, strained, re-suspended in CryoStor CS10 (Biolife Solutions, WA) at a concentration of 2 × 107 cells/mL, and stored in liquid nitrogen.8 Cells were thawed and 2 × 105 cells in 0.1 mL of FACS buffer were injected via tail-vein into non irradiated 6- to 8-week-old male C57BL/6 (CD45.2) mice to create BCR/ABL and Nup98/HoxA9 (bcCML) mice. Mixed lineage leukemia-AF9 (MLL-AF9) mice were created using a retroviral construct encoding the MLL-AF9 fusion protein with a translocation at t(9;11)(p22;q23).36 Prior to injection of MLL-AF9 retroviral constructs, LSK cells from C57BL/002014 (CD45.1) 6- to 8-week-old male mice were enriched via FACS sorting and incubated overnight in Roswell Park Memorial Institute (RPMI) 1640 with 20% fetal calf serum, 20% Walter and Eliza Hall Institute of Medical Research (WEHI)-conditioned medium (R20/20), 20 ng/mL of stem cell factor (SCF), and 10 of ng/mL interleukin-6 (IL-6).36 MLL-AF9 leukemic cells were tail-vein injected with 2 × 105 leukemic cells in 0.1 mL of FACS buffer into 6- to 8-week-old male C57BL/6 primary recipients sub-lethally irradiated with 6 Gy using a 137Cs radiation source.
2.3 |. Flow cytometry
Analysis of spleen and marrow cell populations was performed as previously described8,13 using FlowJo version 9.6.5 (Tree Star). Further details are provided in the SI section.
2.4 |. Initiation of CCL3−/− murine AML models
CCL3 knockout (CCL3−/−) LSK cells were established using MSCV-BCR/ABL-IRES-GFP and MSCV-Nup98/HoxA9-YFP vectors or MLL-AF9 vectors transfected into marrow hematopoietic stem and progenitor cells of 6- to 8-week-old male CCL3−/− mice. CCL3 knockout LSK cells were isolated from CCL3−/− mice via FACS sorting and equal concentrations of CCL3−/− bcCML and MLL-AF9 LSKs or wild-type (WT) LSKs were injected via tail-vein into irradiated or non-irradiated WT mice. Survival of injected mice was monitored daily for up to 50 days for bcCML mice and up to 100 days for MLL-AF9 mice.
2.5 |. Colony forming assays
Following infection of marrow hematopoietic stem and progenitor cells with GFP/YFP vectors, sorted LSK cells were suspended in 100 μL of IMDM containing 10% FBS and added to 3 mL of M3534 methylcellulose containing media (Stemcell technologies). Cells were plated at a volume of 1.1 mL in 35 mm dishes. Following 11 days of culture at 37°C and 5% CO2 the number of colonies that were GFP+ and contained more than 35 cells were counted.
2.6 |. Engraftment of WT and CCL3−/− bcCML cells
The model of bcCML was initiated using LSK cells sorted from the marrow of WT or CCL3−/− mice. WT or CCL3−/− bcCML cells were generated and transplanted into non-irradiated WT recipient mice via tail-vein injection as described above. Bone marrow was analyzed by flow cytometry at 48 hours and 6 days post-transplantation.
2.7 |. Leukemia cell and osteoblast co-culture
Murine-derived bcCML leukemic cells were used for the co-culture system with MC3T3-E1 pre-osteoblastic cells. MC3T3-E1 cells were differentiated into mature OBs (28 days) using osteogenic media Minimum Essential Medium Eagle—alpha modification (α-MEM) with 10% fetal bovine serum (FBS), 100 U/mL of penicillin, 100 μg/mL of streptomycin (Invitrogen), 50 μg/mL of L-ascorbic acid (Sigma-Aldrich), and 10 mmol/L of glycerol 2-phosphate disodium salt hydrate (Sigma-Aldrich). On day 28, 1 × 104 bcCML leukemic cells were added to co-cultures, which were treated with 2 × 10−7 mol/L BX471 and 2 × 10−8 mol/L MVC and cultured for 6 days. On the day of analysis, non-adherent bcCML cells were removed by washing the plates four times with 1× PBS and MC3T3-E1, then OBs were analyzed for osteocalcin gene expression using qRT-PCR as previously described,8 with β-actin as the housekeeping/control gene. Total mRNA was extracted using the RNeasy kit (QIAGEN) according to the manufacturer’s instructions. The Quantitect Reverse Transcription kit (QIAGEN) was used to transcribe cDNA, which was then diluted 1:50 in dH20, combined with SYBR Green PCR master mix (Bio-Rad), and amplified using MyiQ Single-Color PCR detection systems and software under the following conditions: 95°C for 3 minutes followed by 40 cycles of 95°C for 15 seconds and 58°C (osteocalcin) or 60°C (CCL-3) for 30 seconds. Data were analyzed using a relative standard curve method, normalized to β-actin: murine β-actin 5′ primer: GCCACTGCCGCATCCTCTT; 3′ primer: GGAACCGCTCGTTGCCAATAG; murine osteocalcin 5′ primer: CCGCCTACAAACGCATCTACG; 3′ primer: GAGAGAGGACAGGGAGGATCAAG; and murine CCL3 5′ primer: AAGGATACAAGCAGCAGCGAGTA; 3′ primer: TGCAGAGTGTCATGGTACAGAGAA.
2.8 |. Treatment of bcCML model with CCL3 signaling antagonists
MVC (Selleck Chemicals) was prepared in 100% ethanol and diluted to a 5 mg/mL working solution in sterile PBS immediately prior to injection. Mice were treated with a 20 mg/kg dose of MVC by intraperitoneal injection twice daily with injections given 6 hours apart for 9 days, 1 day after bcCML initiation. BX471 (Sigma) was prepared in 40% w/v cyclodextrin and diluted to a 2.5 mg/mL working solution in sterile saline immediately prior to injection. Mice were treated with a 10 mg/kg dose of BX471 by intraperitoneal injection twice daily with injections given 6 hours apart for 9 days, 1 day after bcCML initiation. Mice were sacrificed on day 11 for flow cytometry analysis.
2.9 |. Complete blood counts
To determine complete blood counts, blood was collected in EDTA-coated tubes via the submandibular plexus and the hematology analyzer, scil Vet abc Plus+ was used as previously described.37
2.10 |. Diblock synthesis and NP self-assembly and characterization
One-step reversible addition-fragmentation chain transfer agent (RAFT) polymerization was used to synthesize amphiphilic diblock PSMA-b-PS copolymers whereby excess styrene was used to achieve a poly(styrene) chain upon exhaustion of maleic anhydride monomers.28,30 Briefly, distilled styrene (99%, ACS grade) and maleic anhydride recrystallized from chloroform ([STY]:[MA] = 5:1) were added to chain transfer agent, 4-cyano-4-dodecyl sulfanyltrithiocarbonyl sulfanyl pentanoic acid (DCT) ([STY]:[CTA] = 100:1) and the radical initiator, 2,2ʹ-Azo-bis(isobutylnitrile) (AIBN), recrystallized from methanol ([CTA]: [AIBN] = 5:1) in dioxane (50% w/w). After an initial sample was taken for proton nuclear magnetic resonance (1H NMR) analysis, the polymer solution was purged with nitrogen (N2) for 45 minutes and placed in a 60°C oil bath. After 72 hours, the reaction was terminated by exposing the polymer solution to air and the polymerized PSMA-b-PS diblock copolymers were diluted in acetone and precipitated using petroleum ether.28 Precipitated PSMA-b-PS diblock copolymers were dried in vacuum and stored at room temperature. Gel permeation chromatography (GPC) was used to determine the molecular weights by dissolving 2 mg of diblock copolymers into 1 mL of dimethylformide (DMF) + 0.05% lithium chloride (LiCl). GPC was performed using a Shimadzu LC-20AD equipped with a differential refractometer (Shimadzu RID-10A) and a light scattering detector (Wyatt Technology DAWN TREOS) with a 3 μm linear gel column (Tosoh TSK-Gel Super HM-N, 6.0 mm inner diameter × 15 cm) operating in a 60°C column oven. 1H-NMR was used to assess the complete conversion of maleic anhydride monomers by comparing pre- and post-polymerization samples dissolved in deuterated dimethyl sulfoxide (DMSO). To self-assemble diblock copolymers into NPs, diblock copolymers (200 mg) were dissolved in DMF (20 mL) and a syringe pump set at 24.4 μL/min was used to add ddH2O (31 mL). Self-assembled NPs were dialyzed against ddH2O for 72 hours using 6–8 kDa MW cutoff dialysis tubing (Spectrum Laboratories). NPs were filtered using 0.2 μm cellulose acetate filters (VWR international) prior to storage in 4°C. Gravimetric determination of NP concentrations was performed by weighing empty Eppendorf tubes, adding 0.5 mL of NP solution, lyophilizing, and weighing freeze dried product in tubes. NP physiochemical properties such as size and surface charge were analyzed using dynamic light scattering (DLS) and a Malvern Zetasizer at 0.2 mg NP/mL of 0.1 mol/L phosphate buffered solution (PBS), pH 7.4.
2.11 |. Peptide synthesis and conjugation
Microwave-assisted solid-phase peptide synthesis (CEM Corp, Liberty1 synthesizer) was used to generate tartrate-resistant acid phosphatase (TRAP) binding peptide (TBP) (sequence: TPLSYLKGLVTVG) using fluorenylmethyloxycarbonyl chloride (FMOC)-protected amino acids (AAPPTec and Peptides International).
Amino acid coupling was achieved with an activator mix of 0.5 mol/L O-(benzotriazole-1-yl)-N, N, N’, N’-tetramethyluronium hexafluorophosphate (HBTU) in DMF and activator base mix of 2 mol/L N, N-Diisopropylethylamine (DIEA) in 1-methyl-2-pyrrolidinone (NMP). Deprotection of individual amino acids during coupling was achieved with 5% piperazine in DMF. Synthesized TBP was cleaved from Fmoc-Gly-Wang resin (Millipore, MA) using 92.5% trifluoroacetic acid (TFA), 2.5% H2O, 2.5% 3, 6-dioxa-1,8-ocatanedithiol (DODT), and 2.5% triisopropylsilane (TIPS) for 2.5 hours and precipitated in ice-cold diethyl ether. TBP was dried in vacuum and validated using matrix-assisted laser desorption ionization time-of-light mass spectrometry (MALDI-TOF) (Brüker Autoflex III) whereby equal volumes of 10 mg TBP in 1 mL of 30:70 [v/v] acetonitrile: 0.1% TFA in H2O and 10 mg/mL α-cyano-4-hydroxycinnamic acid were spotted on a MALDI plate. Conjugation of TBP to NPs was performed via carbodiimide chemistry using 1-ethyl-3-(3-dimethylamino)propyl carbodiimide (EDC, Thermo Fisher) ([EDC]:[polymer] = 10:1), 5 mmol/L hydroxysulfosuccinimide (sulfo-NHS, Thermo Fisher), and TBP ([TBP]:[polymer] = 10:1 in 0.1 mol/L sodium phosphate buffer (pH 7.4)). The reaction was mixed overnight and dialyzed against ddH2O for 72 hours (MWCO 6–8 kDa) and conjugation efficiency was determined using fluoraldehyde o-phthaldialdehyde (OPA, Thermo Fisher, Ex/EM = 360 nm/455 nm) and nanoparticle tracking analysis (NTA, Nanosight NS300). For nanoparticle tracking analysis (NTA), five videos of 30 seconds recordings of the particle concentration within the 20–100 particles/frame range was collected. The measurement was performed at 25°C and a detection threshold of 3 was used to collect the sample data. Nanosight NTA software version 3.2 Dev Build 3.2.16 (Malvern Instruments Ltd.) was used for data collection and processing.
2.12 |. Drug loading and release characterization
To load PSMA-b-PS NPs, 23 mg of MVC was dissolved in a 1:5 vol/vol DMSO:chloroform mixture. TBP-NPs (90 mg/mL in ddH2O) were then added and the scintillation vial was covered with foil and stirred overnight. TBP-NPMVC were purified via two rounds of centrifugation to remove any aggregated-free drug (10 minutess at 4000 rpm) and two rounds of centrifugal filtrations using 100 000 kDa MW cutoff filters (Millipore, MA) to remove soluble-free drug (10 minutes at 2000 rpm). Release of MVC was performed using dialysis tubing with MWCO of 6–8 kDa (Spectrum Laboratories) and 0.1 mol/L PBS, pH 7.4 to emulate physiological conditions. For release, dialysis tubing filled with TBP-NPMVC or free MVC was submerged in PBS at 37°C and 100 μL samples were taken up to 72 hours. The PBS sink was replaced at t = 2, 8, 24, and 48 hours. Final solutions of purified drug-loaded peptide-conjugated NPs and release samples were analyzed via high-performance liquid chromatography (HPLC). Final solutions of purified TBP-NPMVC were dissolved in 9:1 methanol :ddH2O and analyzed via HPLC with a mobile phase consisting of A) HPLC grade water +0.05% formic acid and B) HPLC grade acetonitrile +0.05% formic acid. HPLC analysis was performed on a Kromasil C18 column (50 mm × 4.6 mm, 5 μm particle size, 100 Å pore size). Drug elution was monitored at 197 nm (Shimadzu). Flow conditions were set at 0.5 mL/min with a gradient elution (0–0.01 minutes 20%, 0.01–4 minutes 40% B, 4–7.50 minutes 70% B, 8.10–15 minutes 95% B, and 15.1–20 minutes 20% B). Loading capacity was calculated as mg of MVC/mg of NP. Release samples were analyzed via HPLC using the aforementioned procedure after dissolving sample in 9:1 methanol: ddH2O.
2.13 |. Treatment of bcCML model with TBP-NPMVC
TBP-NPMVC were reconstituted in PBS prior to injections whereby 150 mg/kg NPs (30 mg/kg MVC) were injected intraperitoneally every other day for six doses in bcCML mice the day after injection of leukemic cells.
2.14 |. Flow cytometry
Spleen cells were isolated by crushing the spleen using a 40 μm pore size strainer and PBS + 2% FBS + 10 U/mL DNase. Bone marrow cells were liberated by flushing with PBS + 2% FBS (FACS buffer) using a 25 gauge needle. Liberated marrow cells were resuspended in 1 mg/mL collagenase type IV, 2 mg/mL dispase, and 10 U/mL DNase dissolved in Hank’s Balanced Salt Solution (HBSS). Red blood cell (RBC) lysis buffer (156 mmol/L NH4Cl, 127 μmol/L EDTA, and 12 mmol/L NaHCO3) was used to lyse RBCs for 5 minutes at room temperature. Samples were suspended in 100 μL of FACS buffer and analyzed on an LSRII flow cytometer: three lasers, 355, 488, and 633 nm (BD Biosciences) by collecting 1 × 106 events.
2.15 |. Treatment of ST2 cells with MVC
MSC surrogate, ST2 cells, were grown in alpha-minimum essential medium (α-MEM) supplemented with 10% of FBS, 100 U/mL of penicillin, and 100 μg/mL of streptomycin (Invitrogen). After reaching 80% confluency, cells were treated with MVC at— 0.1–100 μmol/L—for 24 hours. Cell number and viability were analyzed using the alamarBlue® assay (Thermo Fisher) as per the manufacturer’s protocol.
2.16 |. Statistical analysis
For all quantitative results, groups are reported as mean ± standard error mean (SEM) unless otherwise noted. Comparisons between two groups were performed using student’s unpaired two-tailed t-test for normally distributed data. Multiple group comparisons were performed using a one-way ANOVA test with Dunnett’s multiple comparison. For Kaplan-Meier survival curves, comparisons were performed using log-rank (Mantel-Cox) test. Statistical significance was set as P < .05.
3 |. RESULTS
3.1 |. Loss of CCL3 signaling improves survival in bcCML leukemia models
CCL3 signaling, which is elevated in myelogenous leukemias both in animal models and humans, has been shown to modify the BMME, and contribute to leukemia initiation and progression.8,17,38 We have previously shown that CCL3 signaling is elevated in the bcCML model, which contributes directly to a disrupted BMME.8 To further investigate the importance of CCL3 in bcCML mice, leukemic cells that were initiated in LSK cells isolated from CCL3−/− or WT control mice were transplanted via tail-vein injections into non-irradiated WT recipients (Figure 1A). At 48 hours and 6 days, post-transplant no difference was observed in the number of leukemic cells in the marrow of recipient mice that were transplanted with WT or CCL3−/− leukemic cells (Figure S1). Engraftment in the spleen was also analyzed indicating that CCL3−/− leukemic cells had increased homing at 48 hours and comparable levels of leukemic cells with WT at day 6 (Figure S1). This indicates that there is no defect in the homing or engraftment of CCL3−/− leukemic cells, and, combined with the colony forming assay results (Figure 1B), suggests that the defect in leukemogenic activity in CCL3−/− leukemic cells is perpetuated by interactions with the marrow microenvironment following engraftment. Though CCL3−/− leukemic cells form colonies to a similar extent as WT controls (Figure 1B, the percent of live GFP+ cells in spleen and marrow of CCL3−/− bcCML mice at day 10 were decreased by ~3.5-fold and ~20-fold (Figure 1C,D), showing that CCL3−/− bcCML cells did not cause disease in non-irradiated mice. Statistically significant and prolonged survival of >45 days was observed in CCL3−/− leukemic cells established in non-irradiated WT recipients compared to 23 days for WT bcCML initiated mice (Figure 1E). When CCL3−/− bcCML cells were implanted into irradiated recipients, engraftment in the bone marrow was achieved likely due to a radiation-induced disrupted BMME (Figure S2). To test the repopulation potential of CCL3−/− bcCML cells isolated from CCL3−/− bcCML mice, 3 × 104 FACS sorted GFP and YFP double positive cells (GFP+/YFP + bcCML cells) were serially transplanted from irradiated primary recipients of WT or CCL3−/− bcCML cells into non-irradiated recipient mice (Figure 1F). CCL3−/− bcCML cells did not engraft in non-irradiated recipient mice, resulting in no detectable GFP+ cells in the spleen and the marrow (Figure 1G,H). CCL3 expression levels in MLL-AF9 mice are similarly elevated, therefore, the contribution of CCL3 signaling to survival was also investigated.19 WT and CCL3−/− MLL-AF9 AML models were initiated using FACS-sorted WT and CCL3−/− LSK cells transplanted into irradiated mice (Figure 1I). Note that irradiated mice were used for MLL-AF9 studies because primary recipients of this model cannot reliably be established without irradiation.36 CCL3−/− MLL-AF9 mice exhibited statistically significant and prolonged survival of ~14 days compared to WT MLL-AF9 mice, although MLL-AF9 mice in both groups eventually succumbed to disease at days 82 and 96 (Figure 1J). Based on these findings, the bcCML model was the focus for additional experiments, as data suggest that CCL3 signaling is critical for leukemia progression in this model, similar to human disease.
FIGURE 1.
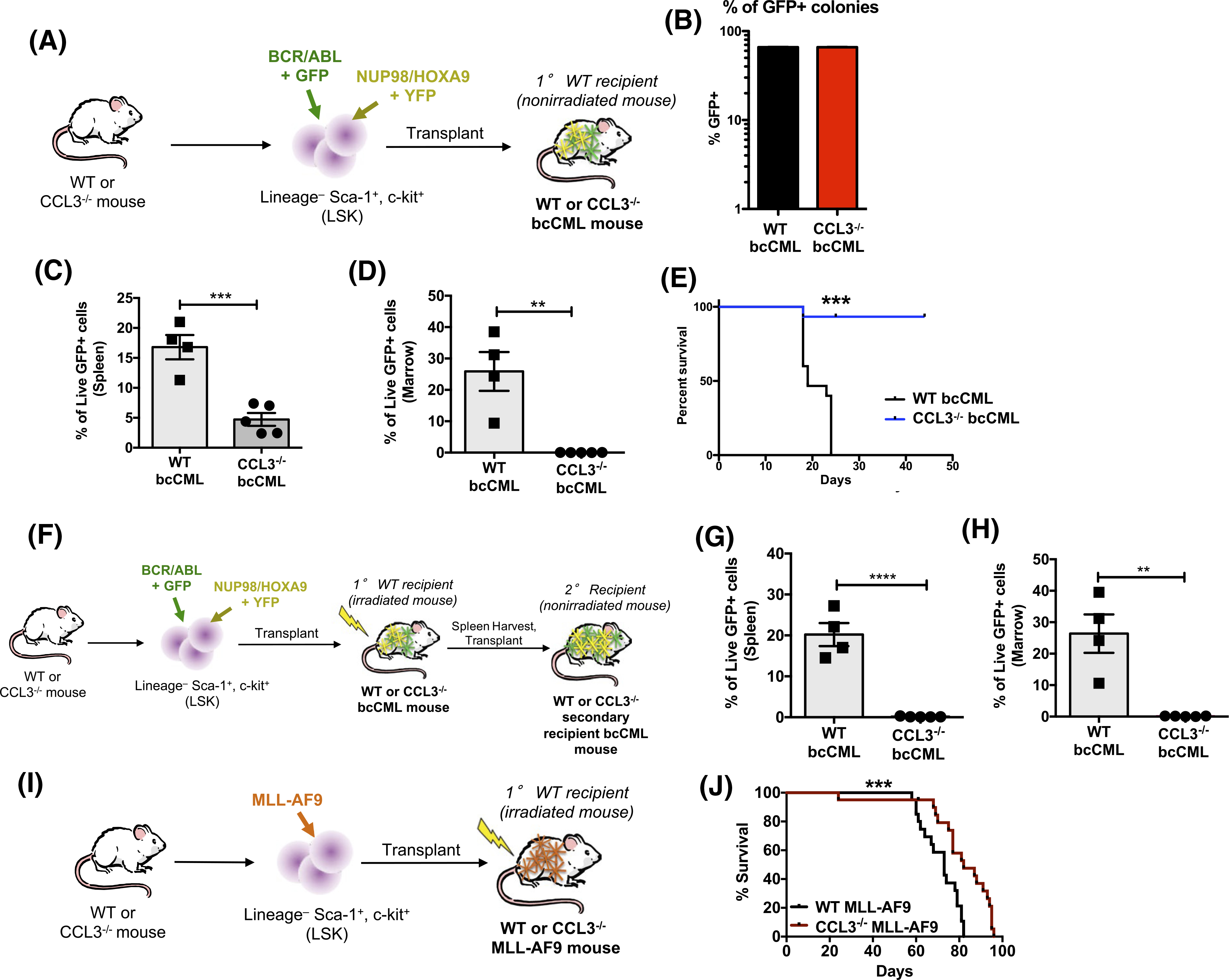
Loss of CCL3 signaling improves survival of bcCML mice. A, Schematic showing initiation of CCL3−/− bcCML model in non-irradiated mice. B, Colony forming assays. C and D, Percent live GFP+ cells in spleen and marrow from CCL3−/− bcCML mice at day 10. Data represents mean ± SEM (n = 4–5). Significance tested via unpaired two-tailed t-tests. **P < .01 and ***P < .001. E, Percent survival of mice injected with CCL3−/− leukemic cells and wild-type leukemic cells (n = 25). F, Schematic illustrating serial transplantation of CCL3−/− bcCML in non-irradiated mice. G and H, Percent of live GFP+ cells in spleen and marrow from serially transplanted CCL3−/− bcCML mice. I, Schematic showing initiation of CCL3−/− MLL-AF9 model. J, Percent survival of MLL-AF9 murine model injected with CCL3−/− leukemic cells and wild-type leukemic cells (n = 15). Log-rank (Mantel-Cox) test indicates significant differences with **P < .01 and ***P < .001 vs wild-type leukemic cell injections
3.2 |. The marrow hematopoietic system and microenvironment is disrupted in bcCML mice
To investigate the phenotype of bcCML mice for subsequent studies, splenic cells from primary recipients of GFP+/YFP + transfected LSKs were harvested and transplanted into non-irradiated recipient mice and phenotypic populations were analyzed in the bone marrow, spleen, and peripheral blood 10 days after transplantation of bcCML cells (Figure 2A,B). In the peripheral blood, white blood cells were increased >2-fold, platelets were decreased by 2-fold, and mature RBCs were unchanged, suggesting a dysfunctional hematopoietic system (Figure S3). In the marrow, GFP+ cells were significantly increased, indicating successful leukemia progression (Figure 2C). Flow cytometric analysis of HSPC and MPP populations were completed according to previously established markers on GFP− cells (Figure 2B). Data are presented as the percent of live GFP− cells, normalized to GFP− cells isolated from nonleukemic WT mice. The frequency of long-term HSCs (GFP−/LSK/Flt3−/CD48−/CD150+), short-term HSCs (GFP−/LSK/Flt3−/CD48−/CD150−), myeloid-biased MPP subsets, MPP2 (GFP−/LSK/Flt3−/CD48+/CD150+), and MPP3 (GFP−/LSK/Flt3−/CD48+/CD150−), and lymphoid-primed MPP4 cells (GFP−/LSK/Flt3+/CD48+/CD150−) were significantly increased in bcCML mice compared to WT controls, again demonstrating a disruption in normal hematopoiesis (Figure 2D–H). Specifically, LT-HSCs were increased by 20-fold, ST-HSCs were increased by ~10-fold, MPP2s were increased by >20-fold, MPP3s were increased by >5000-fold, and MPP4s were increased by 4-fold in bcCML mice. However, no difference in HSPC populations were observed between mice transplanted with CCL3−/− bcCML cells and non-leukemic mice (Figure S4). Marrow microenvironmental cells including MSCs were defined according to literature.9,37,39,40 MSCs, defined as CD45−/Lin−/CD31−/CD51+/ Sca1+ (MSC)9 (Figure S5), CD45−/Lin−/CD140a+/Sca1+ (PαS)39 (Figure S5), or CD45−/Lin−/CD31−/CD51+/140a+ (PααV)40 (Figure 2J), were significantly increased in bcCML mice compared to WT controls (Figure 2J–L). Altogether, the HSC, MPP, and MSC increases and platelet cell decreases suggest that the BMME is disrupted in bcCML mice.
FIGURE 2.
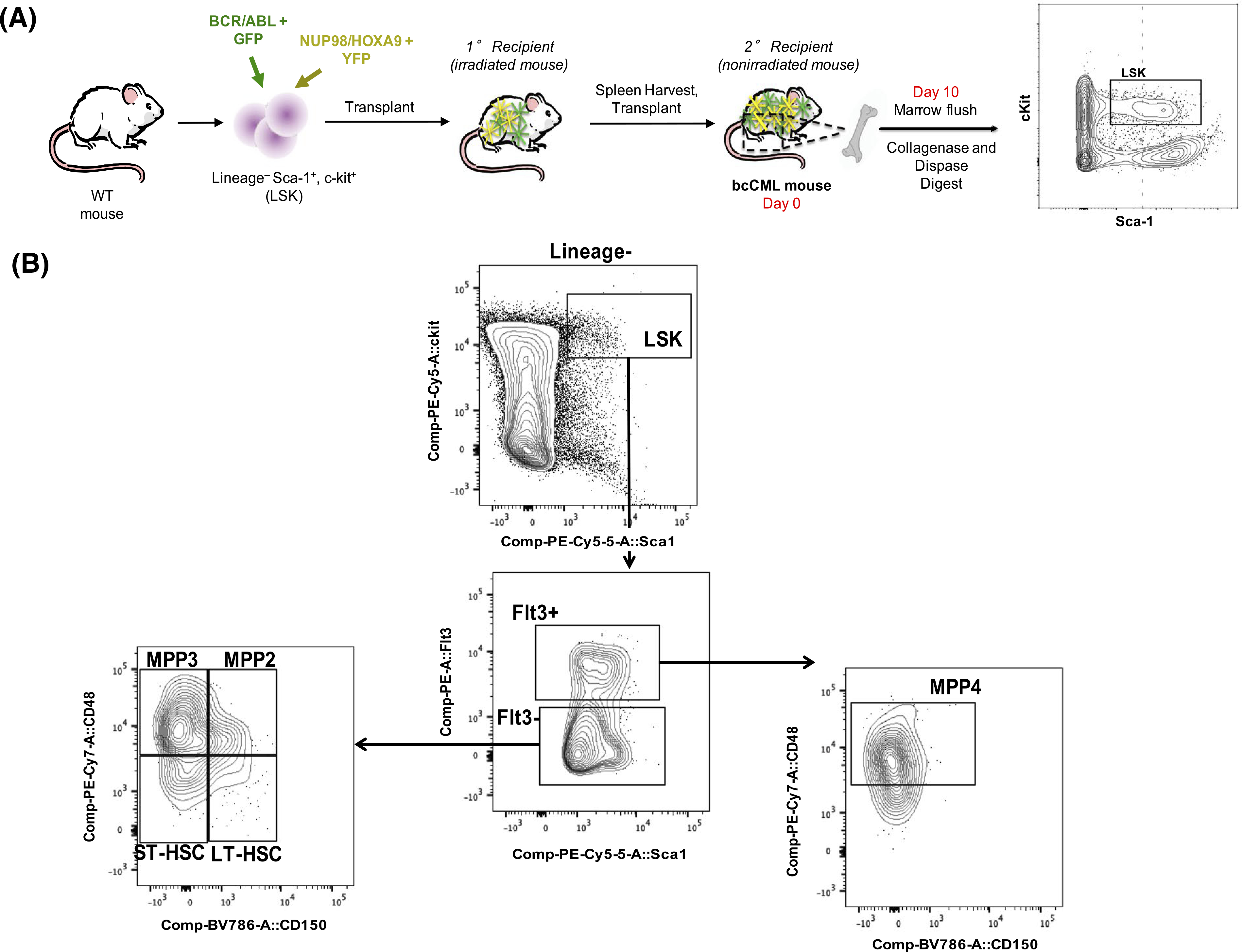
The hematopoietic system and BMME in bcCML mice are disrupted. A, Schematic of bcCML initiation and sample processing. B, Flow cytometry plots defining HSC and MPP populations. Flow cytometry analysis of marrow derived HSPCs demonstrates increases in LT-HSCs, ST-HSCs, MPP2, MPP3, and MPP4 populations at Day 10 post-engraftment. C, GFP+ marrow cells (D) GFP-LT-HSCs (E) GFP-ST-HSCs (F) GFP-MPP2 (G) GFP-MPP3. H, GFP-MPP4. I, Flow cytometry plots defining MSC populations. J, MSC population defined as CD45−/Lin−/CD31−/CD51+/140a+. Data represents mean ± SEM (n = 5–9). Unpaired two-tailed t-tests were performed. *P < .05 and ****P < .0001
3.3 |. MVC decreases bcCML tumor burden in the spleen but not in the marrow
Elevated CCL3 signaling inhibits osteogenic differentiation in multiple malignancies, which may directly or indirectly exacerbate disease progression.13–16 As a measure of osteoblast function, in vitro osteocalcin gene expression was evaluated for 28-day differentiated osteoblast cells alone, osteoblast cells cultured in the presence of bcCML cells, and osteoblast cells cultured in the presence of bcCML cells and the CCL3 signaling inhibitors BX471 and MVC, which antagonize CCR1 and CCR5, respectively (Figure 3A). Osteoblasts cultured with bcCML cells exhibited significantly decreased osteocalcin gene expression (<20% vs no leukemic cells) (Figure 3B). Treatment with inhibitors rescued osteocalcin gene expression of OBs in the presence of bcCML cell to 50% of osteoblast cells alone. To test the therapeutic efficacy of blocking CCR1 and CCR5 receptors in vivo, bcCML models were treated with BX471 or MVC (Figure 3C). MVC only showed significantly decreased splenic leukemic cells from 14% to 7% GFP+ cells (Figure 3D). In the bone marrow, however, there was no decrease in leukemic cells following treatment with either BX471 or MVC (Figure 3E), potentially owing to poor pharmacokinetics of both drugs in rodents. Based on the observed reduction of splenic leukemic burden in the MVC-treated groups and the success of MVC as an FDA approved drug for other indications, MVC was selected for further studies. However, due to the lack of an effect on the marrow, a targeted nanoparticle approach for the delivery of MVC was used to increase its marrow accumulation.30
FIGURE 3.
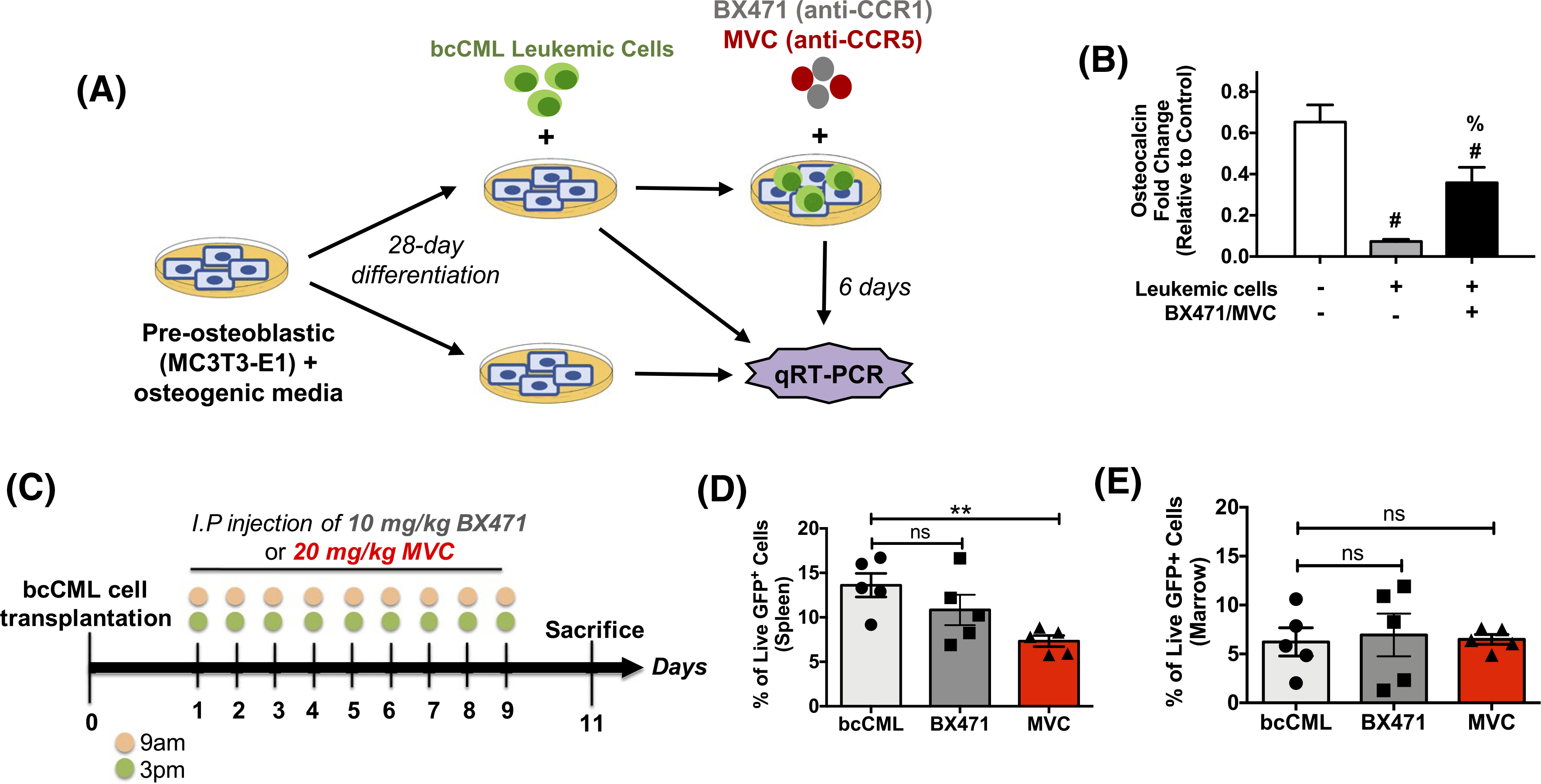
BX471/MVC treatment restores osteocalcin in vitro but is unable to reduce leukemic burden in vivo. A, Schematic showing co-culture system used to measure osteoblast function in the presence of leukemic cells. B, Osteocalcin gene expression levels using qRT-PCR after treatment with combination BX471 and MVC. Data represents mean ± standard deviation (std) (n = 3–5). Significant difference noted between groups as determined via one-way ANOVA followed by Tukey’s multiple comparisons test. #P < .01 compared to control of no treatment and no leukemic cells, % P < .01 compared to no treatment in the presence of leukemic cells. C, Treatment paradigm used in the in vivo assessment of BX471 and MVC. D, Flow cytometry analysis of leukemic cells in the spleen and (E) bone marrow of bcCML mice via flow cytometry. Data represents mean ± std (n = 5). Significant difference noted between treatment groups and saline control as determined via one-way ANOVA followed by Dunnett’s multiple comparisons. *P < .05 and ns = not significant
3.4 |. Bone-targeted NPs load and release MVC
Polymer diblocks were synthesized via one-step RAFT polymerizations using maleic anhydride (MA) and styrene (STY) monomers to produce diblock copolymers, PSMA83-b-PS310 (Figure 4A).28–30 In the presence of water, PSMA83-b-PS310 diblocks self-assembled into micelle NPs with 51 ± 1 nm overall sizes and −43 ± 2 mV surface charge.28 We have previously shown that PSMA-b-PS NPs functionalized with a targeting ligand specific to TRAP deposited by osteoclasts dramatically increase bone biodistribution of drugs.30 Therefore, PSMA-b-PS NPs were conjugated with ~4000 TRAP binding peptide (TBP) per NP (Figure 4B). Loading of MVC indicated 0.24 ± 1 mg of MVC per mg of NP and release of MVC from TBP-NPs indicated slower release rate of 0.1 h−1 compared to 0.3 h−1 of free drug (Figure 4C).
FIGURE 4.

MVC loading and release in synthesized PSMA-b-PS NPs. A, One-step RAFT polymerizations were used to synthesize amphiphilic diblock copolymers using maleic anhydride (MA) and styrene (STY) monomers. In the presence of water, the diblock copolymers self-assemble into micelle NPs with a hydrophilic corona and a hydrophobic core. B, PSMA-b-PS NPs are functionalized with TRAP-binding peptide (TBP) via carbodiimide chemistry and loaded with MVC to produce TBP-NPMVC. C, Release of free MVC and MVC from TBP-NPs in 0.1 mol/L PBS pH 7.4, 37°C show sustained release from TBP-NPs. Data represents mean ± std. (n = 3). Statistical differences were tested via a paired two-way ANOVA followed by Sidak’s multiple comparisons test for free MVC and TBP-NPMVC at each time point. *P < .05, ****P < .0001, ns = not significant
3.5 |. Nanoparticle-mediated delivery of MVC reduces leukemic burden and partially restores HSCs and BMME
To determine whether CCL3 inhibition would have an effect on leukemic burden and the BMME, MVC was delivered via TBP-NPs. Based on the release profile of MVC from TBP-NPs, namely that >75% of loaded drug releases by 48 minutes, bcCML mice were treated every other day for six doses (Figure 5A). Based on previously established understanding in the field that HSCs home to the BMME and establish at the endosteal surface within 12 hours of injections, treatments were initiated 24 hours after bcCML cell injections.41 The percentage of GFP+ cells in the bone marrow were reduced via NP-mediated delivery of MVC as analyzed by flow cytometry whereby MVC alone did not reduce GFP+ cells without a carrier (Figure 5B). Trends toward normalization of HSPC subsets were observed in both free MVC and TBP-NP-mediated delivery of MVC with no statistical differences noted between the treatment groups (Figure 5C–G). Specifically, LT-HSCs were unaffected by free MVC and TBP-NPMVC treatment, however, there was rescue of ST-HSCs (>5-fold), MPP2 (~10-fold), and MPP3 (>50-fold) populations in the marrow upon treatment and no differences in MPP4 populations (Figure 5C–G). Interestingly, there was a significant reduction in the HSC supportive, PααV mesenchymal stem cell population40 via TBP-NP-mediated delivery of MVC, which was not observed in groups treated with free MVC (Figure 5H, Figure S6), and which was not attributable to cytotoxic effects of MVC on MSCs, as indicated by in vitro data (Figure S6). Most importantly, there was prolonged survival of bcCML mice treated with TBP-NPMVC compared to free drug and vehicle-treated mice, thereby demonstrating the effectiveness of nanoparticle delivery of MVC to the bone marrow (Figure 5I).
FIGURE 5.
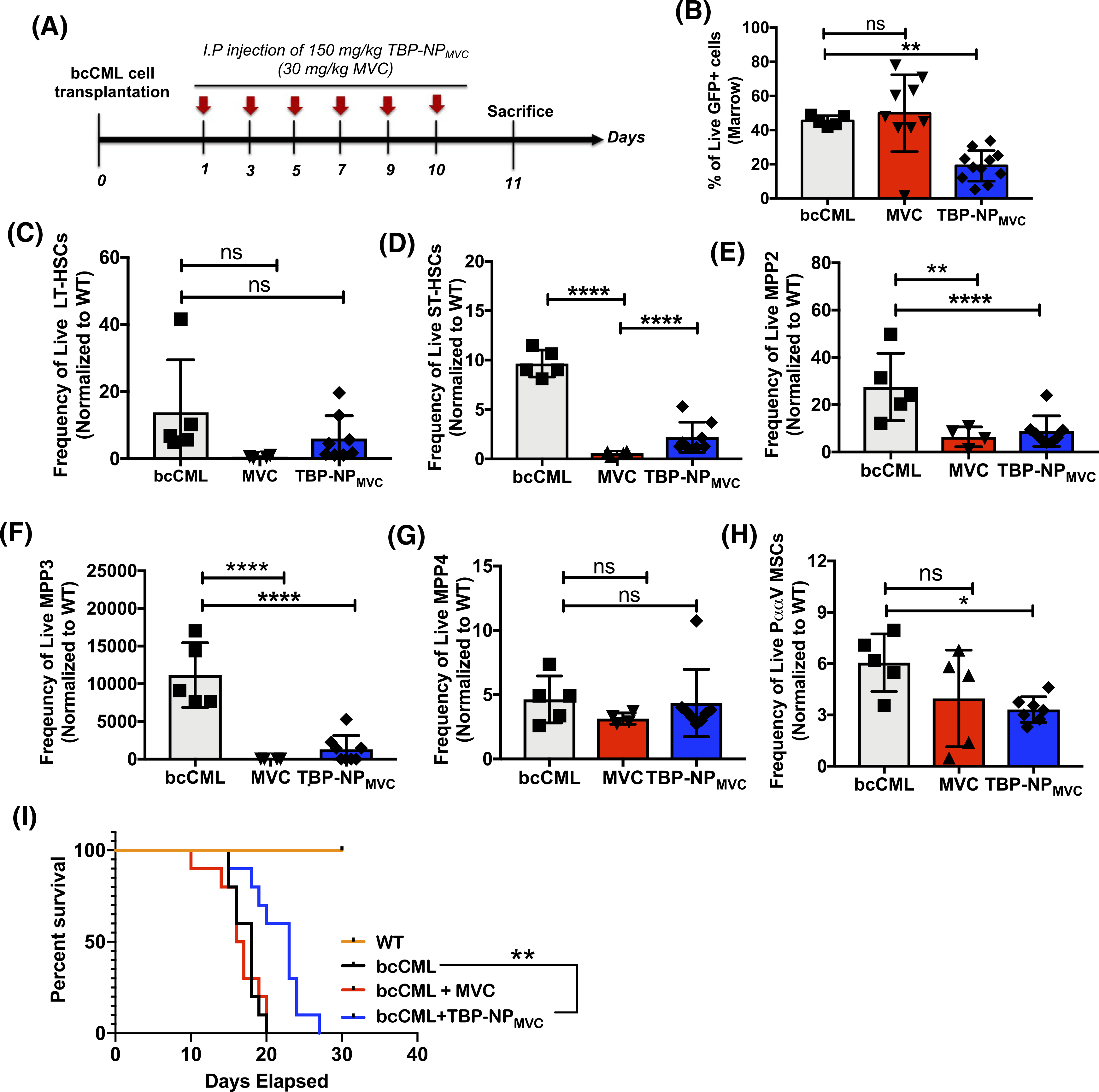
NP delivery of MVC decreases leukemic burden in the marrow and partially restores HSC populations in the marrow. A, Treatment paradigm used in experiments. B, GFP+ cells in the bone marrow. C, GFP-LT-HSC. D, GFP-ST-HSCs. E, GFP-MPP2. F, GFP-MPP3. G, GFP-MPP4. H, PααV cells as defined as CD45−Lin−CD31−CD51+140a+. Data were pooled from two independent experiments, n = 3–4 per group (mean ± SEM). One-way ANOVA was used to assess the differences between groups followed by Dunnett’s multiple comparisons to bcCML control without treatment. *P < .05, **P < .01, ****P < .0001 ns = not significant. I, Percent survival of WT, bcCML injected with saline, and bcCML mice after treatment with free MVC and TBP-NPMVC (n = 10). Log-rank (Mantel-Cox) test indicates significant differences with **P < .01 vs bcCML injected with saline
4 |. DISCUSSION
Leukemias represent a diverse group of marrow malignancies. Interaction with the bone marrow is postulated to confer resistance to therapy and enhance leukemia cell self-renewal. Therefore, to identify novel therapeutic targets that are associated with leukemic-BMME interactions, mechanisms that lead to BMME dysfunction were investigated. Furthermore, the combination of a mechanistic approach and unique drug delivery platform that enhances drug accumulation in the BMME allowed for greater specificity of delivery and sustained the release of small molecules that specifically target the BMME.
We have previously reported that mature osteoblastic cells are lost in a murine model of bcCML and that the chemokine CCL3, a known mediator of microenvironmental dysfunction in several malignancies, was elevated both in the murine model and in ~75% of primary AML samples. In the current studies, the bcCML model was used to characterize a dysfunctional BMME and loss of normal hematopoiesis. In this model, hematopoietic stem cells (HSCs) and MSCs, that under normal conditions support hematopoiesis, are elevated while terminally differentiated platelets are lost. This shows that though ample HSCs are present, differentiation is lost, demonstrating hematologic dysfunction. These findings are significant as they support previous studies that show leukemia disrupts normal hematopoiesis and damages the BMME.6 Leukemia cells have been shown to produce CCL3 to decouple osteoblast/osteoclast processes leading to overall loss of bone volume.8 Additionally, elevated CCL3 disrupts normal erythropoiesis,19 while CCL3−/− mice retain functional HSCs and hematopoiesis, though myeloid differentiation is partially blocked in the absence of CCL3.42 CCL3 is also elevated in the majority of AML patient RNA and protein samples.8,17 Therefore, inhibiting CCL3 signaling was hypothesized to be an effective therapeutic strategy for reducing leukemic burden without compromising remaining functional hematopoiesis.18,19,38 To this end, CCL3 signaling was shown to be necessary for leukemia progression in the bcCML model. Though loss of CCL3 prolonged survival in the murine MLL-AF9 model of AML, all of the mice ultimately succumbed to disease, indicating that individual leukemias may have different levels of dependence on CCL3 signaling. These findings are similar to observations in chronic myeloid leukemia (CML) and juvenile myelomonocytic leukemia (JMML) models18,38 whereby CCL3 signaling was implicated as a requirement for disease initiation, rather than progression, further supporting the hypothesis that inhibiting CCL3 pharmacologically is a viable therapeutic strategy.
To block CCL3 signaling and recapitulate the effects observed in CCL3 knockout studies, the CCR1 antagonist, BX471, and CCR5 antagonist, MVC, were investigated. BX471 is a safe, selective CCR1 inhibitor that was previously developed to treat chronic inflammatory diseases by blocking CCL3-CCR1 binding.22 MVC is a well-tolerated FDA approved CCR5 inhibitor for HIV patients, that can bind to CCR5 with nanomolar affinity.24 Combination BX471 and MVC treatment restores expression of the osteoblast specific marker osteocalcin in in vitro co-cultures of OBs and leukemic cells, suggesting antagonism of both CCR1 and CCR5 can effectively reverse leukemic cell alteration of BMME cells, validating the role of CCL3 in BMME dysfunction. The efficacy of each drug individually in vivo was determined and only MVC resulted in significant reduction of splenic leukemic burden due to more efficient delivery of small molecule drugs to the spleen. Similarly, a previous study showed that MVC-induced apoptosis in leukemia cells in a xenograft model of acute lymphocytic leukemia (ALL).23 Interestingly, MVC treatment did not reduce leukemic load in the BMME, likely due to poor marrow accumulation of MVC.
NP DDS improve drug circulation half-lives, resulting in enhanced therapeutic efficacy in a variety of leukemias.43 Indeed, a liposomal formulation of cytarabine and daunorubicin was approved for AML treatment due to increased residence time in bone marrow owing to the EPR effect.44,45 To further enhance the marrow residence time of MVC beyond EPR, a bone targeting NP DDS was employed. The targeted NP employed here specifically binds to TRAP deposited by osteoclasts during bone resorption30,32 and enhances marrow drug accumulation by twofold compared to untargeted NP controls.30 Thus, TBP-NP-mediated delivery of MVC was hypothesized to facilitate greater accumulation of MVC to cells of the BM to restore the damaged BMME. TBP-NP-mediated delivery of MVC resulted in a reduction in leukemic burden, likely due to prolonged marrow accumulation, as previously documented for other polymeric DDS.46 Along with the leaky vasculature of marrow and binding to TRAP, upon systemic injection of TBP-NP entrapping MVC, albumin and other proteins adsorb onto the NP surface to reduce nonspecific uptake by the mononuclear phagocytic system (MPS), which further promotes marrow accumulation of MVC and overall therapeutic efficacy.47 In the bcCML model, increased frequency of aberrant mesenchymal populations was observed, similar to what has previously been described as a feature of BMME dysfunction, and a myelofibrotic phenotype in myeloid malignancies such as CML.9 The data presented here suggest that TBP-NP-mediated delivery of MVC is required to reduce the level of HSC supportive PααV MSCs, likely due to increased MVC concentrations in the BMME, which modulates a high degree of inhibitor binding that is necessary to block chemokine receptor signaling.48 In particular, TBP-NP-mediated delivery of MVC results in lower data variability (coefficient of variation (CV) of 71% for free MVC vs CV of 23% for TBP-NPMVC), and most strikingly results in a 7-day prolonged survival likely due to sustained release of MVC within the marrow. Despite the improvement in survival of leukemic mice treated with TBP-NPMVC, treatment was not as effective for prolonging survival as genetic loss of CCL3 in the leukemic cells. One possibility to explain this disparity is the fact that start of treatment with MVC is 2 days post-transplant of leukemic cells allowing time for engraftment in the marrow, with genetic loss there is no CCL3 signaling from the leukemic cells at any timepoint. Furthermore, in murine models of CML, CCL3 is required for the maintenance of a leukemic stem cell populations.38 Our results highlight a potentially similar phenomenon as secondary transplants demonstrate no engraftment of CCL3−/− leukemic cells (Figure 1G–H). These results suggest that combining TBP-NPMVC therapy with conventional chemotherapeutics could be more effective than either therapy alone.
While our results strongly suggest CCL3 as a major ligand for CCR5 in the leukemic BMME, CCR5 is a promiscuous receptor with multiple ligands.25 Therefore, our data does not rule out the possibility that by pharmacologically inhibiting CCR5 we are not, in fact, disrupting signaling from a separate CCR5 ligand. This could be a profound strength of the receptor-centric approach as blockade of the receptor will also block signaling of any additional ligands that could prove redundant. Additionally, the promiscuity of CCR5 broadens the potential of this approach. Multiple hematologic malignancies have been demonstrated to produce additional CCR5 ligands such as CCR4 and CCR5.49,50 This suggests that pharmacologic inhibition of CCR5 has therapeutic potential in multiple malignancies that involve multiple ligands.
The findings of these studies define the BMME in the bcCML model and highlight the role of leukemic cell CCL3 signaling on disease progression. A significant reduction in leukemic burden and partial restoration of the BMME and hematopoietic cells in marrow was observed via NP delivery of a CCL3 signaling inhibitor, thus validating our approach of focusing on mechanisms of BMME dysfunction in a leukemic microenvironment. Additional investigation is warranted to understand how inhibiting additional CCL3 signaling receptors will affect the AML BMME.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr James McGrath for access to the DLS and plate reader and flow cytometry core facility for assistance with flow cytometry. This work was supported by grants from the University of Rochester, University Research Award (BJF, DSWB), the Wilmot Cancer Institute Research Development Funding Program (BJF), National Institute of Health: National Cancer Institute (F32 CA180615 (BJF), and R01 CA166280 to LMC), (P30 AR069655, R01 AR064200, R01 AR056696 [DSWB]), the National Science Foundation Career Award (CBET1450987 [DSWB]), and the University of Rochester CTSA award number UL1 TR002001 (BJF, DSWB) from the National Center for Advancing Translational Sciences.
Funding information
HHS | NIH | National Cancer Institute (NCI), Grant/Award Number: CA180615; HHS | NIH | National Cancer Institute (NCI), Grant/Award Number: CA166280; HHS | NIH | National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), Grant/Award Number: AR069655; HHS | NIH | National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), Grant/Award Number: AR064200; HHS | NIH | National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), Grant/Award Number: AR056696; National Science Foundation (NSF), Grant/Award Number: CBET1450987; University of Rochester (UR), Grant/Award Number: UL1 TR002001
Abbreviations:
- ALL
acute lymphocytic leukemia
- AML
acute myeloid leukemia
- bcCML
blast crisis chronic myelogenous leukemia
- BMME
bone marrow microenvironment
- CCL3
C-chemokine (C-C motif) ligand 3CCL3−/−, C-chemokine (C-C motif) ligand 3 knockout
- CCR1
4, 5, C-C chemokine receptor type 1, 4, 5
- CML
chronic myeloid leukemia
- DDS
drug delivery system
- EPR
enhanced permeability and retention effect
- FMOC
fluorenylmethyloxycarbonyl chloride
- HSC
hematopoietic stem cells
- HSPC
hematopoietic stem progenitor cells
- JMML
juvenile myelomonocytic leukemia
- LSCs
leukemic stem cells
- LSK
lineage-negative, sca-1+, c-kit+ cells
- LT/ST-HSCs
long-term/short-term hematopoietic stem cells
- MA
maleic anhydride
- MLL-AF9
Mixed lineage leukemia-AF9
- MPP
multi-potent progenitor cells
- MPS
mononuclear phagocytic system
- MSCV-BCR/ABL-IRES
recombinant murine stem cell virus retroviral expression of breakpoint cluster region-abelson murine leukemia – internal ribosome entry site
- MSCV-Nup98/HoxA9
recombinant murine stem cell virus retroviral expression of nucleoporin 98/homeodomain-containing transcription factor
- MVC
Maraviroc
- NP
nanoparticle
- PSMA-b-PS
poly(styrene-alt-maleic anhydride)-b-poly(styrene)
- RAFT
reversible addition-fragmentation chain transfer agent
- STY
styrene
- TBP
tartrate-resistant acid phosphatase binding peptide
- TRAP
tartrate-resistant acid phosphatase
- YFP/GFP
yellow/green fluorescent proteins
Footnotes
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Cancer Stat Facts: Acute Myeloid Leukemia (AML) [Internet]. NCI - surveillance, epidemiology, and end results program. 2017. [Google Scholar]
- 2.Jain P, Kantarjian HM, Ghorab A, et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: Cohort study of 477 patients. Cancer. 2017;123(22):4391–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roboz GJ. Current treatment of acute myeloid leukemia. Curr Opin Oncol. 2012;24(6):711–719. [DOI] [PubMed] [Google Scholar]
- 4.Hoffmann VS, Baccarani M, Hasford J, et al. The EUTOS population-based registry: incidence and clinical characteristics of 2904 CML patients in 20 European Countries. Leukemia. 2015;29(6):1336–1343. [DOI] [PubMed] [Google Scholar]
- 5.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861–1865. [DOI] [PubMed] [Google Scholar]
- 6.Kumar B, Garcia M, Weng L, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia. 2018;32(3):575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krevvata M, Silva BC, Manavalan JS, et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood. 2014;124(18):2834–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood. 2012;119(2):540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blau O, Baldus CD, Hofmann WK, et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood. 2011;118(20):5583–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geyh S, Rodriguez-Paredes M, Jager P, et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia. 2016;30(3):683–691. [DOI] [PubMed] [Google Scholar]
- 12.Behrmann L, Wellbrock J, Fiedler W. Acute myeloid leukemia and the bone marrow niche-take a closer look. Front Oncol. 2018;8:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balderman SR, Li AJ, Hoffman CM, et al. Targeting of the bone marrow microenvironment improves outcome in a murine model of myelodysplastic syndrome. Blood. 2016;127(5):616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallet S, Pozzi S, Patel K, et al. A novel role for CCL3 (MIP-1alpha) in myeloma-induced bone disease via osteocalcin down-regulation and inhibition of osteoblast function. Leukemia. 2011;25(7):1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Li YY, Matsushima K, Baba T, Mukaida N. CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J Immunol. 2008;181(9):6384–6393. [DOI] [PubMed] [Google Scholar]
- 16.Zucchetto A, Benedetti D, Tripodo C, et al. CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res. 2009;69(9):4001–4009. [DOI] [PubMed] [Google Scholar]
- 17.Bullinger L, Dohner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350(16):1605–1616. [DOI] [PubMed] [Google Scholar]
- 18.Dong L, Yu WM, Zheng H, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539(7628):304–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Gao A, Zhao H, et al. Leukemia cell infiltration causes defective erythropoiesis partially through MIP-1alpha/CCL3. Leukemia. 2016;30(9):1897–1908. [DOI] [PubMed] [Google Scholar]
- 20.Menu E, De Leenheer E, De Raeve H, et al. Role of CCR1 and CCR5 in homing and growth of multiple myeloma and in the development of osteolytic lesions: a study in the 5TMM model. Clin Exp Metastasis. 2006;23(5–6):291–300. [DOI] [PubMed] [Google Scholar]
- 21.Dairaghi DJ, Oyajobi BO, Gupta A, et al. CCR1 blockade reduces tumor burden and osteolysis in vivo in a mouse model of myeloma bone disease. Blood. 2012;120(7):1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang M, Mallari C, Rosser M, et al. Identification and characterization of a potent, selective, and orally active antagonist of the CC chemokine receptor-1. J Biol Chem. 2000;275(25):19000–19008. [DOI] [PubMed] [Google Scholar]
- 23.Zi J, Yuan S, Qiao J, et al. Treatment with the C-C chemokine receptor type 5 (CCR5)-inhibitor maraviroc suppresses growth and induces apoptosis of acute lymphoblastic leukemia cells. Am J Cancer Res. 2017;7(4):869–880. [PMC free article] [PubMed] [Google Scholar]
- 24.Dorr P, Westby M, Dobbs S, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49(11):4721–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354(6):610–621. [DOI] [PubMed] [Google Scholar]
- 26.Argyle D, Kitamura T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front Immunol. 2018;9:2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baranello MP, Bauer L, Benoit DS. Poly(styrene-alt-maleic anhydride)-based diblock copolymer micelles exhibit versatile hydrophobic drug loading, drug-dependent release, and internalization by multidrug resistant ovarian cancer cells. Biomacromol. 2014;15(7):2629–2641. [DOI] [PubMed] [Google Scholar]
- 29.Baranello MP. Functionalized poly(styrene-alt maleic anhydride)-b-poly(styrene) micelles for targeted delivery of parthenolide to leukemia. Cells. 2015;8(3):455–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Newman MR, Ackun-Farmmer M, et al. Fracture-targeted delivery of beta-catenin agonists via peptide-functionalized nanoparticles augments fracture healing. ACS Nano. 2017;11(9):9445–9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. [DOI] [PubMed] [Google Scholar]
- 32.Sheu TJ, Schwarz EM, O’Keefe RJ, Rosier RN, Puzas JE. Use of a phage display technique to identify potential osteoblast binding sites within osteoclast lacunae. J Bone Miner Res. 2002;17(5):915–922. [DOI] [PubMed] [Google Scholar]
- 33.Newman MR, Russell SG, Schmitt CS, et al. Multivalent presentation of peptide targeting groups alters polymer biodistribution to target tissues. Biomacromol. 2018;19(1):71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye H, Adane B, Khan N, et al. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer Cell. 2018;34(4):659–73 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neering SJ, Bushnell T, Sozer S, et al. Leukemia stem cells in a genetically defined murine model of blast-crisis CML. Blood. 2007;110(7):2578–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Somervaille TCP, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10(4):257–268. [DOI] [PubMed] [Google Scholar]
- 37.Frisch BJ, Hoffman CM, Latchney SE, et al. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via Interleukin1B. JCI Insight. 2019;5:e124213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baba T, Naka K, Morishita S, Komatsu N, Hirao A, Mukaida N. MIP-1alpha/CCL3-mediated maintenance of leukemia-initiating cells in the initiation process of chronic myeloid leukemia. J Exp Med. 2013;210(12):2661–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morikawa S, Mabuchi Y, Kubota Y, et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med. 2009;206(11):2483–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinho S, Lacombe J, Hanoun M, et al. PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med. 2013;210(7):1351–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim S, Lin L, Brown GAJ, Hosaka K, Scott EW. Extended time-lapse in vivo imaging of tibia bone marrow to visualize dynamic hematopoietic stem cell engraftment. Leukemia. 2017;31(7):1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Staversky RJ, Byun DK, Georger MA, et al. The chemokine CCL3 Regulates Myeloid Differentiation And Hematopoietic Stem Cell Numbers. Sci Rep. 2018;8(1):14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deshantri AK, Varela Moreira A, Ecker V, et al. Nanomedicines for the treatment of hematological malignancies. J Control Release. 2018;287:194–215. [DOI] [PubMed] [Google Scholar]
- 44.Lim WS, Tardi PG, Dos Santos N, et al. Leukemia-selective uptake and cytotoxicity of CPX-351, a synergistic fixed-ratio cytar-abine:daunorubicin formulation, in bone marrow xenografts. Leuk Res. 2010;34(9):1214–1223. [DOI] [PubMed] [Google Scholar]
- 45.Feldman EJ, Lancet JE, Kolitz JE, et al. First-in-man study of CPX-351: a liposomal carrier containing cytarabine and daunorubicin in a fixed 5:1 molar ratio for the treatment of relapsed and refractory acute myeloid leukemia. J Clin Oncol. 2011;29(8):979–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalela M, Shrivastav TG, Kharbanda S, Singh H. pH-sensitive bio-compatible nanoparticles of paclitaxel-conjugated poly(styrene-co-maleic acid) for anticancer drug delivery in solid tumors of syngeneic mice. ACS Appl Mater Interfaces. 2015;7(48):26530–26548. [DOI] [PubMed] [Google Scholar]
- 47.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oyajobi BO, Williams PJ, Pulkrabek D, Franchin G, Sherry B, Mundy GR. In vivo osteoclastogenic effects of the C-C chemokine macrophage inflammatory protein-1 alpha. Bone. 2001;28(5):S81. [Google Scholar]
- 49.Yazdani Z, Mousavi Z, Ghasemimehr N, et al. Differential regulatory effects of chemotherapeutic protocol on CCL3_CCL4_CCL5/CCR5 axes in acute myeloid leukemia patients with monocytic lineage. Life Sci. 2020;240:117071. [DOI] [PubMed] [Google Scholar]
- 50.Chen YL, Tang C, Zhang MY, et al. Blocking ATM-dependent NF-kappaB pathway overcomes niche protection and improves chemotherapy response in acute lymphoblastic leukemia. Leukemia. 2019;33(10):2365–2378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.