
Figure 3. Schematic illustration of the possible pathophysiology of aldosterone induced renal injuries.
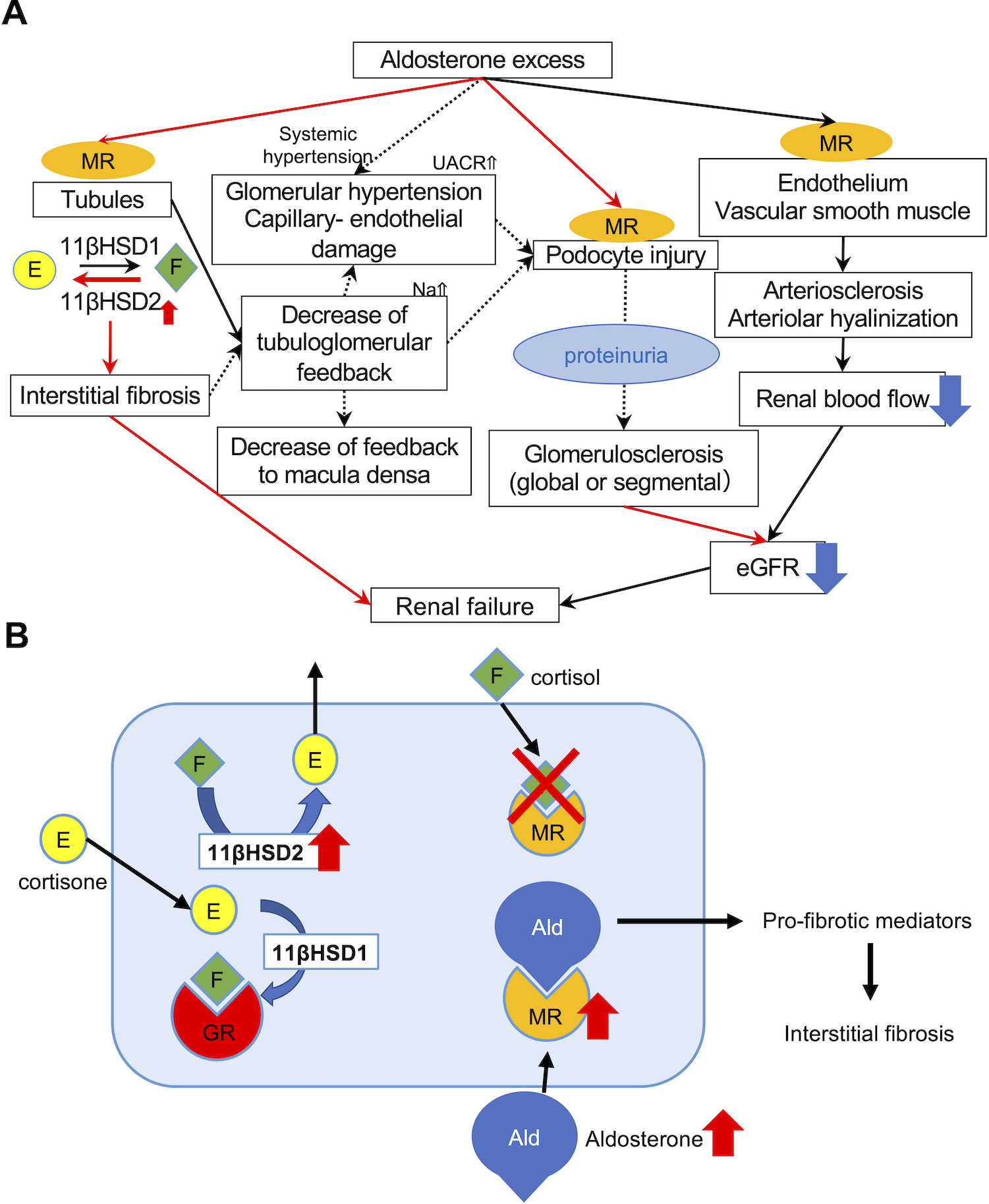
A. MR nuclear immunolocalization in PA was higher than that in EH, demonstrating that MR was more activated and translocated into nucleus as a result of abundant binding with aldosterone. Predominant 11βHSD2 catalytic status detected could compensate for aldosterone excess. Autonomous aldosterone excess (low renin and high aldosterone) could also reduce tubulo-glomerular feedback, resulting in hyperfiltrating status of the remaining glomerulus, histologically recognizable as glomerular hypertrophy. Aldosterone could also directly damage podocytes, and cause foot process effacement. Subsequently induced proteinuria also exacerbated glomerulosclerosis, histologically recognizable as progressive glomerular sclerosis demonstrated by the higher proportion of not only GGS but also SGS. In particular, the prevalence of global glomerulosclerosis accurately reflected the post-operative eGFR after adrenalectomy. Aldosterone could also influence vessels via non-genomic and genomic pathways and cause endothelial swelling and dysfunction, increasing vascular stiffness or peripheral resistance. Therefore, topical renal blood flow could be finally reduced. Therefore, aldosterone-induced renal injury could be systemically induced by multiple pathways. B. 11βHSD2 and MR immunoreactivity was more abundant in PA than EH. 11βHSD2 converted cortisol into cortisone, an inactive form of cortisol. In contrast, 11βHSD1 converted cortisone into cortisol. When 11βHSD2 was not active, MR was occupied by cortisol and aldosterone could not bind with MR. In PA, 11βHSD2-predominant status allowed aldosterone to bind MR and activate pro-fibrotic pathways.