

Abstract
Aims
In healthy volunteers, the kidney deploys compensatory post-diuretic sodium reabsorption (CPDSR) following loop diuretic-induced natriuresis, minimizing sodium excretion and producing a neutral sodium balance. CPDSR is extrapolated to non-euvolemic populations as a diuretic resistance mechanism; however, its importance in acute decompensated heart failure (ADHF) is unknown.
Methods and results
Patients with ADHF in the Mechanisms of Diuretic Resistance cohort receiving intravenous loop diuretics (462 administrations in 285 patients) underwent supervised urine collections entailing an immediate pre-diuretic spot urine sample, then 6-h (diuretic-induced natriuresis period) and 18-h (post-diuretic period) urine collections. The average spot urine sodium concentration immediately prior to diuretic administration [median 15 h (13–17) after last diuretic] was 64 ± 33 mmol/L with only 4% of patients having low (<20 mmol/L) urine sodium consistent with CPDSR. Paradoxically, greater 6-h diuretic-induced natriuresis was associated with larger 18-h post-diuretic spontaneous natriuresis (r = 0.7, P < 0.001). Higher pre-diuretic urine sodium to creatinine ratio (r = 0.37, P < 0.001) was the strongest predictor of post-diuretic spontaneous natriuresis. In a subgroup of patients (n = 43) randomized to protocol-driven intensified diuretic therapies, the mean diuretic-induced natriuresis increased three-fold. In contrast to the substantial decrease in spontaneous natriuresis predicted by CPDSR, no change in post-diuretic spontaneous natriuresis was observed (P = 0.47).
Conclusion
On a population level, CPDSR was not an important driver of diuretic resistance in hypervolemic ADHF. Contrary to CPDSR, a greater diuretic-induced natriuresis predicted a larger post-diuretic spontaneous natriuresis. Basal sodium avidity, rather than diuretic-induced CPDSR, appears to be the predominant determinate of both diuretic-induced and post-diuretic natriuresis in hypervolemic ADHF.
Keywords: Diuretic, Sodium, Post-diuretic, Heart failure, Acute heart failure, Reabsorption
Graphical Abstract
See page 4478 for the editorial comment for this article ‘Renal sodium avidity, the prevailing renal target in heart failure’, by P. Martens, W.H.W. Tang, and W. Mullens, https://doi.org/10.1093/eurheartj/ehab650.
Introduction
Diuretic resistance in acute decompensated heart failure (ADHF) is common and associated with worse outcomes.1–4 Compensatory post-diuretic sodium reabsorption (CPDSR) is ubiquitously cited as a cause of diuretic resistance in ADHF.1,5–14 More accurately, CPDSR is a mechanism by which, despite an adequate acute diuretic response, patients fail to achieve a negative sodium/fluid balance secondary to an equal amount of compensatory/rebound sodium reabsorption in the post-diuretic period.1,5,7 After the loop diuretic concentration decreases below the diuretic threshold, the concept of CPDSR asserts that the kidneys decrease urinary sodium excretion to very low levels over the following hours to counterbalance the diuretic-induced natriuresis (Figure 1). Therefore, an increase in diuretic response produces an increased CPDSR response, resulting in a neutral 24-h sodium balance. This paradigm arose from research in euvolemic, healthy volunteers and has been extrapolated to patients with ADHF.7,15–19 However, CPDSR has not been studied in contemporary hypervolemic ADHF populations and the failure of therapies such as loop diuretic infusion to improve decongestion raises the question if this physiology is operative in patients with ADHF.20 Our primary objective was to better understand the importance of CPDSR in contemporary hypervolemic ADHF.
Figure 1.
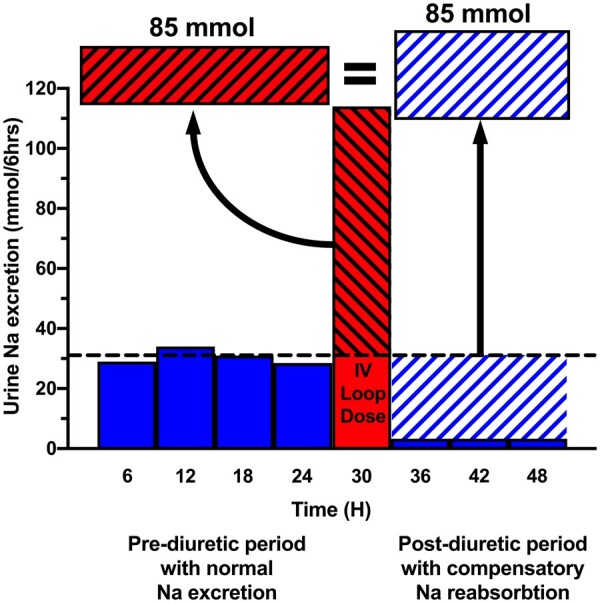
Concept of compensatory post-diuretic sodium reabsorption. The concept of compensatory post-diuretic sodium reabsorption is illustrated by graphing hypothetical urinary sodium excretion over a 48-h period divided into 6-h blocks. A 24-h pre-diuretic period (blue bars) is followed by a 6-h natriuretic period from an IV loop diuretic dose in the red bar and subsequent 18-h post-diuretic period (blue bars). The horizontal dotted black line denotes the average rate of urinary sodium excretion needed every 6 h (31 mmol/6 h) to achieve a net even 24-h sodium balance, where dietary sodium intake (130 mmol) equals urinary sodium excretion (95% of intake = 124 mmol) plus insensible sodium losses (6 mmol). In the pre-diuretic period, the urinary sodium excretion rate equals the sodium intake. The diagonally hashed portion of the red LD bar above the dotted line represents the quantity of natriuresis exceeding the expected rate from dietary intake. In the post-diuretic period, the diagonally hashed blue space indicates the amount of post-diuretic sodium reabsorption, where urinary sodium excretion is depressed following a diuretic period. The concept of compensatory post-diuretic sodium reabsorption asserts the red diagonally hashed area of excess natriuresis will trigger a compensatory post-diuretic sodium reabsorption of equal magnitude, resulting in a net even sodium balance.
Methods
Patients admitted with ADHF to the cardiology service at Yale New Haven Hospital who required treatment with intravenous (IV) loop diuretics (furosemide or bumetanide) were screened for inclusion in the Mechanisms of Diuretic Resistance (MDR) study (NCT02546583). Full study methods have been published previously.21 Briefly, the MDR study was designed to investigate mechanisms of diuretic resistance in a prospective cohort hospitalized with ADHF. The relevance of CPDSR as a diuretic resistance mechanism was an a priori study question, and study methods were designed to investigate CPDSR.21 Patients are followed longitudinally with study visits occurring at several diuretic milestones within the ADHF hospital episode to investigate early and later IV loop diuretic doses. Patients exhibiting diuretic resistance are enrolled in a randomized sub-study comparing increased loop diuretic doses to combination diuretic therapy with chlorothiazide.
The key inclusion criteria were (i) use of intermittent IV loop diuretic therapy with projected need of IV diuretics for at least 3 days, (ii) a goal of significant fluid removal (>1 L net fluid loss/day), and (iii) at least one objective sign of volume overload. Key exclusion criteria were significant bladder dysfunction, urinary incontinence, and inability to comply with urine collection procedures. We excluded patients for this analysis of the MDR study cohort that received a thiazide within 24 h of the analysis periods or did not receive a loop diuretic dose prior to the urine collections utilized for post-diuretic assessments. Patients received a 3 g (130 mmol) per day sodium restricted diet. IV diuretic dose was determined by the treating physician. Enrollment could occur at any point during the hospitalization provided the patient met all inclusion and exclusion criteria. All patients provided written informed consent, and the study was approved by the Yale Institutional Review Board.
Study visit protocol
During the post-diuretic period from a prior loop diuretic dose and prior to the morning diuretic dose (Hour 0), a blood sample was obtained, patients were asked to completely empty their bladder, from which a baseline urine sample was obtained, and a bladder scan was performed to quantitate residual volume in the bladder. Following loop diuretic administration, a timed 6-h urine collection with intensive supervision by study staff was performed to ensure all urine was collected. After 6 h, patients were asked to empty their bladder to complete the urine collection and a bladder scan was repeated. Participants then underwent an 18-h timed urine collection (to complete 24 h) which was conducted by the clinical nursing staff. Study visits could be repeated on subsequent hospital days during IV diuretic therapy.
Study cohorts
We analysed two cohorts: healthy volunteers and patients with ADHF in the MDR study (Figure 2). In each cohort, we assessed diuretic-induced natriuresis using the 6-h cumulative urine collection immediately following the diuretic dose. We assessed post-diuretic spontaneous natriuresis using both spot urine collections and the 18-h cumulative urine collection in the post-diuretic period.
Figure 2.

Patient cohort flow diagram. The number of diuretic administrations, unique patients, and purpose for each patient cohort are shown in a flow diagram.
Healthy Volunteer Cohort: We replicated the seminal observations establishing the concept of CPDSR in healthy volunteers to serve as the benchmark for the expected pattern of sodium excretion with a loop diuretic.7 Twenty euvolemic, healthy participants without heart failure or previous loop diuretic therapy received an IV diuretic dose of 40 mg furosemide equivalents in our Clinical Research Center. The methods for collecting the 6-h urine output were similar to those described above. Participants were permitted to consume clear sodium-free liquids and low-sodium snacks after diuretic administration. Following the 6-h urine collection, participants conducted an 18-h cumulative urine collection with guidance to eat salty foods and drink water to replenish intravascular volume from the prior diuretic exposure.22
MDR Study Cohort: Within the MDR study, we examined a Calculated Cohort, Measured Cohort, and Randomized Intervention Cohort. A total of 462 diuretic administrations from 285 unique patients are included in the Calculated Cohort: 130 patients (28%) were included once, 284 were included twice (62%), and 48 were included three times (10%). The Measured Cohort comprises 117 diuretic administrations from 94 unique patients, and the Randomized Intervention Cohort comprises 43 unique patients.
The Measured Cohort serves as the primary cohort and consists of patients that did not receive a diuretic during the 18-h post-diuretic period (i.e. only one diuretic dose in a 24-h period, similar to the cohorts CPDSR was described in). We measured the post-diuretic spontaneous natriuresis directly from the cumulative sodium excretion in the 18-h urine collection (Supplementary material online, Figure S1). The Measured Cohort represents a population without the calculated assumptions of the Calculated Cohort, but with reduced generalizability since most patients with ADHF have more than one diuretic dose per day.
The Calculated Cohort serves as a validation cohort to the Measured Cohort. The majority (75%) of patients received more than one diuretic dose in a day with an additional dose during the 18-h urine collection period. Therefore, in the Calculated Cohort, we calculated the 18-h cumulative sodium excretion from the instantaneous rate of sodium excretion in a spot urine sample obtained in the post-diuretic period after a prior diuretic dose and immediately before the subsequent diuretic dose at Hour 0 (Supplementary material online, Figure S1). The Calculated Cohort is more generalizable and lacks any ‘carryover effect’ of diuretic-induced natriuresis into the post-diuretic period by using a spot urine sample ∼15 h from the last diuretic dose instead of a timed urine collection starting 6 h after the diuretic dose. We calculated the 18-h post-diuretic spontaneous natriuresis using the baseline (Hour 0) spot urine sodium and urine creatinine concentration with the measured 24-h urine creatinine excretion using the equation:
Validation of the calculated 18-h spontaneous natriuresis equation with measured 18-h natriuresis is presented in Supplementary material online, Figure S2.
The Randomized Intervention Cohort measures the intra-patient change in the post-diuretic spontaneous natriuresis following a randomized intervention to increase diuretic-induced natriuresis. Patients with poor diuretic response at the first study visit (6-h cumulative sodium excretion <100 mmol) were included in a randomized, controlled study visit the following day to investigate different diuretic strategies to increase natriuresis. Patients were randomized to either 2.5 X the previous IV loop diuretic or the same IV loop diuretic dose plus IV chlorothiazide as a part of an unrelated study to compare diuretic strategies in diuretic resistance.21 The Randomized Intervention Cohort provides insight into the mechanisms of post-diuretic spontaneous natriuresis by measuring how increased diuretic-induced natriuresis impacts CPDSR and the basal sodium avidity (proclivity of the kidney to excessively retain sodium) independent of diuretic response.
Measurements of natriuresis
For diuretic-induced natriuresis, we defined the 6-h percent expected natriuresis as the percentage of the ADHF patient’s 6-h cumulative sodium output after a diuretic relative to the mean 6-h cumulative sodium output from the Healthy Volunteer Cohort. Diuretic efficiency is expressed as the cumulative 6-h urine sodium excreted per doubling of the diuretic dose as previously described.23
For post-diuretic spontaneous natriuresis, a spot urine sodium concentration <20 mmol/L is a common reference value indicating severe sodium avidity that would be consistent with the CPDSR concept.24–26 From the daily sodium intake of 130 mmol and well-established percentage of dietary sodium excreted renally (∼95%), we calculated the expected 24-h sodium excretion as 124 mmol.27–29 The 18-h percent expected natriuresis is the percentage of the ADHF patient’s 18-h cumulative sodium output relative to the expected 93 mmol over 18 h, calculated as 124 mmol × (18 h/24 h). The 24-h sodium balance is the difference between 124 mmol and the sum of the 6- and 18-h sodium output.
Assays
A Randox Imola automated clinical chemistry analyzer was used to measure concentration of urine or serum chemistry parameters. Serum and urine creatinine and sodium were measured in triplicate, and the average was taken for analysis. The inter-assay coefficient of variation was <3% for all variables. The calibrators, reagents, and urine and serum level 2 and level 3 controls were purchased from Randox Laboratories. All assay measurements were carried out in accordance with the manufacturer’s instructions (Randox Laboratories, UK). Creatinine measurements are standardized to IDMS traceable National Institute of Standards and Technology reference material (SRM 967).
Statistical analysis
Continuous data are reported as mean ± standard deviation for variables with normal distribution, and median (Quartiles 1–3) for variables with skewed distribution. Categorical data are reported as frequency (percentage). Continuous variables were compared between groups with the Student’s t-test or Mann–Whitney U-test in case of normal or skewed distribution, respectively. Categorical variables were compared with the chi-squared test. For comparisons between loop diuretic administrations, which were repeated observations, we accounted for the absence of independence of observations with linear mixed models. We analysed the distribution in 18-h spontaneous natriuresis by deciles of 6-h natriuresis. We also aimed to analyse linear trends of continuous variables across groups of 18-h natriuresis; thus, we used linear mixed models. Associations between continuous variables were analysed with linear mixed models, and the correlations were displayed as the squared root of the R2. The 18-h post-diuretic spontaneous natriuresis was compared against an expected mean of 93 mmol with a one sample t-test. In the randomized intervention cohort, we included unique patients; the comparison between the 18-h natriuresis during the first visit and the 18-h natriuresis during the intervention was done with a paired t-test. A detailed description of the statistical tests used in the manuscript is found in the Supplementary material online. Statistical significance was defined as 2-tailed P < 0.05. Statistical analysis was performed with IBM SPSS Statistics version 26 (IBM Corp, Armonk, NY) and Stata SE version 14.0 (StataCorp, College Station, TX, USA).
Results
Healthy Volunteer Cohort
The Healthy Volunteer Cohort had a mean age of 26 ± 5 years (90% white, 50% male) and a mean weight of 74 ± 13 kg. In the 24 h prior to diuretic therapy, volunteers excreted 149 ± 65 mmol of sodium. Following IV furosemide 40 mg, the mean 6-h sodium excretion was 199 ± 49 mmol. Despite provision and encouragement of the consumption of high sodium foods and fluid intake, the mean cumulative spontaneous natriuresis was only 50 ± 23 mmol total over the subsequent 18 h (Supplementary material online, Figure S3). These results replicate prior observations in non-heart failure populations, indicating that CPDSR had a dominant effect on urine sodium excretion following an acute loop diuretic administration.
ADHF Measured Cohort
Spot urine sodium concentration
The baseline characteristics of the Measured Cohort are provided in Table 1 and Supplementary material online, Table S1. Their mean spot urine sodium concentrations prior to the subsequent diuretic dose at Hour 0 {median time 14.7 h [interquartile range (IQR) 13.2–17.0] from the last diuretic dose} was 64 ± 33 mmol/L and displayed wide variability (Figure 3). Only 4% of patients had a spot urine sodium concentration <20 mmol/L. The baseline characteristics (Supplementary material online, Table S2) and spot urine sodium concentration (mean 62 ± 30 mmol/L) were similar in the Calculated Cohort (Supplementary material online, Figure S4).
Table 1.
Baseline patient characteristics
| Characteristics | Measured cohort (n = 94) | Positive 24-h sodium balance (n = 33, 35%) | Negative 24-h sodium balance (n = 61, 65%) | P-value |
|---|---|---|---|---|
| Age (years) | 65 ± 14 | 69 ± 12 | 63 ± 14 | 0.053 |
| Male sex | 57 (61) | 18 (55) | 39 (64) | 0.39 |
| Race | ||||
| White | 58 (62) | 21 (64) | 37 (61) | 0.83 |
| Black | 28 (30) | 9 (27) | 19 (31) | |
| Others | 8 (7) | 3 (9) | 5 (8) | |
| BMI (kg/m2) | 31.2 [26.8–37.7] | 28.7 [25.3–35.7] | 34.8 [28.5–39.9] | 0.004 |
| LVEF ≤40% | 59 (63) | 20 (61) | 39 (64) | 0.81 |
| LVEF (%) | 42 ± 17 | 43 ± 17 | 41 ± 18 | 0.59 |
| Comorbid conditions | ||||
| Hypertension | 82 (87) | 29 (88) | 53 (87) | 0.89 |
| Diabetes mellitus | 47 (50) | 14 (42) | 33 (54) | 0.39 |
| Vital signs | ||||
| Systolic blood pressure (mmHg) | 116 ± 22 | 106 ± 19 | 121 ± 22 | 0.004 |
| Heart rate (b.p.m.) | 84 ± 19 | 82 ± 18 | 85 ± 20 | 0.49 |
| Medications | ||||
| ACEi or ARB or ARNI | 58 (62) | 20 (61) | 38 (62) | 0.22 |
| Beta-blocker | 63 (67) | 23 (70) | 40 (66) | 0.82 |
| Spironolactone or eplerenone | 20 (21) | 9 (27) | 11 (18) | 0.75 |
| Daily dose (mg) | 25 [25–44] | 25 [25–50] | 25 [25–25] | 0.18 |
| Digoxin | 4 (4) | 3 (9) | 1 (2) | 0.12 |
| Prehospital use of loop diuretic | 72 (77) | 21 (64) | 51 (84) | 0.04 |
| IV loop diuretic dose in furosemide equivalents (mg) | 80 [40–160] | 80 [40–160] | 120 [40–160] | 0.08 |
| Laboratory values | ||||
| Serum sodium (mmol/L) | 136 ± 4 | 135 ± 5 | 136 ± 4 | 0.59 |
| Serum chloride (mmol/L) | 96 ± 4 | 96 ± 5 | 96 ± 4 | 0.97 |
| Serum creatinine (mg/dL) | 1.3 [1.1–1.6] | 1.3 [1.0–1.7] | 1.3 [1.1–1.6] | 0.80 |
| Blood urea nitrogen (mg/dL) | 28 [20–40] | 29 [18–39] | 28 [20–42] | 0.72 |
| eGFR (mL/min/1.73 m2) | 56 ± 19 | 53 ± 21 | 56 ± 18 | 0.51 |
| NT-proBNP (pg/mL) | 3400 [1534–8459] | 4238 [2618–9864] | 3067 [1473–8077] | 0.24 |
| Serum albumin (g/dL) | 3.7 ± 0.4 | 3.8 ± 0.5 | 3.7 ± 0.4 | 0.48 |
Values are presented as mean ± standard deviation, n (%), or median [interquartile range].
BMI, body mass index; LVEF, left ventricular ejection fraction; ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; ARNI, angiotensin receptor–neprilysin inhibitor; eGFR, estimated glomerular filtration rate; NT-proBNP, N-terminal pro-B-type natriuretic peptide.
Figure 3.

Urine sodium concentration in the post-diuretic period. Spot urine sodium concentrations in the post-diuretic period are displayed as a histogram. The dotted line at 20 mmol/L indicates the traditional urine sodium concentration threshold of high sodium avid states.24–26 Only 4% of patients had a urine sodium concentration below this threshold during spontaneous natriuresis in the post-diuretic period.
Relationship between diuretic response and post-diuretic spontaneous natriuresis
We assessed the relationship between the 6-h diuretic-induced natriuresis and the following 18-h post-diuretic spontaneous natriuresis in the Measured Cohort (Figure 4). The mean 6-h diuretic-induced cumulative sodium excretion was 122 ± 90 mmol. The ensuing mean 18-h post-diuretic spontaneous natriuresis was 104 ± 109 mmol, similar to the expected 93 mmol based on dietary intake and counter to the paradigm of CPDSR (P = 0.28, Figure 4A). The post-void urine volume in the bladder was 63 (IQR 14–134) mL at the end of the 6-h urine collection and start of the 18-h urine collection. Patients with a negative 24-h sodium balance had both a greater mean diuretic-induced natriuresis (158 ± 87 mmol) and subsequent post-diuretic spontaneous natriuresis (138 ± 116 mmol) compared to patients with a positive sodium balance, who had a poor diuretic-induced natriuresis (mean 44 ± 33 mmol) followed by a low post-diuretic spontaneous natriuresis (30 ± 18 mmol) (P < 0.0001 for both comparisons) (Figure 4B). These observations were congruent in the Calculated Cohort and when analysed in a sensitivity analysis ‘per patient’ (Supplementary material online, Table S3).
Figure 4.

Diuretic-induced and spontaneous post-diuretic natriuresis by 24-h sodium balance. (A and B) Mean values (SEM) of urine sodium excretion are presented in 6-h intervals as mmol/6 h. The red bar represents the 6-h diuretic-induced cumulative urine sodium excretion following a dose of IV diuretic. The blue bar represents the measured 18-h spontaneous urine sodium excretion divided evenly into three 6-h blocks. The horizontal dotted black line denotes the average rate of urinary sodium excretion needed every 6 h (31 mmol/6 h) to achieve a net even 24-h sodium balance, where dietary sodium intake (130 mmol) equals urinary sodium excretion (95% of intake = 124 mmol) plus insensible sodium losses (6 mmol). (B) The same population as in (A) but stratified by 24-h sodium balance.
When the urine sodium excretions were analysed as percentages of the expected natriuresis, the mean diuretic-induced natriuresis was 61 ± 45% of expected; however, the post-diuretic spontaneous natriuresis exceeded the expected amount (111 ± 116%) (Supplementary material online, Figure S5). Consistent with an overall sodium avid state, those with a positive 24-h sodium balance had both severely depressed diuretic-induced natriuresis (22 ± 11% of expected) and reduced post-diuretic spontaneous natriuresis (32 ± 20% of expected). Paradoxically, greater 6-h diuretic-induced natriuresis was associated with larger 18-h post-diuretic spontaneous natriuresis (r = 0.7, P < 0.001). Across increasing deciles of diuretic-induced natriuresis, the subsequent post-diuretic spontaneous natriuresis increased consistently (P < 0.001) (Figure 5). Similar observations were found in the Calculated Cohort (Supplementary material online, Figure S6).
Figure 5.
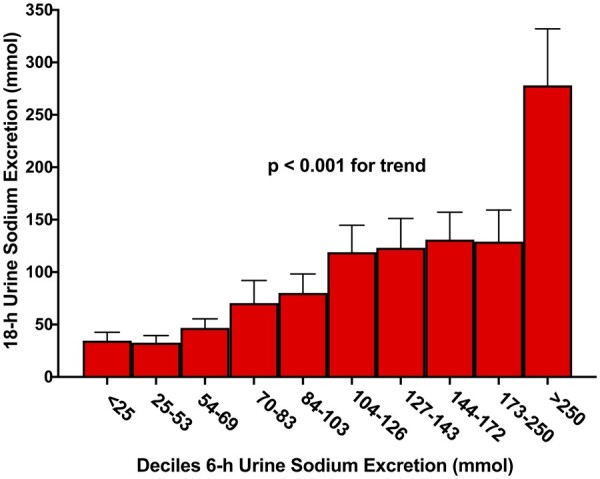
Relationship between diuretic-induced natriuretic response and post-diuretic spontaneous natriuresis. The measured 6-h cumulative sodium excretion is divided into deciles on the x-axis with the corresponding 18-h spontaneous cumulative urine sodium excretion (mean ± SEM) on the y-axis. Contrary to the paradigm of compensatory post-diuretic sodium reabsorption, greater diuretic-induced natriuresis was associated with higher post-diuretic spontaneous natriuresis (P < 0.001).
Variables associated with post-diuretic spontaneous natriuresis
Within the Measured Cohort, we assessed the 18-h post-diuretic spontaneous natriuresis relative to pre-diuretic and diuretic variables to ascertain the drivers of post-diuretic spontaneous natriuresis (Table 2). Neither estimated glomerular filtration rate (P = 0.26) nor blood urea nitrogen (P = 0.89) were associated with post-diuretic spontaneous natriuresis. Indicators of basal sodium avidity prior to the diuretic dose were associated with post-diuretic spontaneous natriuresis, including the pre-diuretic urine sodium concentration (r = 0.30, P < 0.001) and the urine sodium/creatinine ratio (r = 0.37, P < 0.001). Similar observations were found in the Calculated Cohort (Supplementary material online, Table S4).
Table 2.
Characteristics across post-diuretic spontaneous natriuresis
| Characteristics | Measured cohort | 18-h post-diuretic spontaneous natriuresis |
P-value for trend | |||
|---|---|---|---|---|---|---|
| ≥100% expected (≥93 mmol) | 99–50% expecteda (92–47 mmol) | 49–21% expecteda (46–20 mmol) | ≤20% expecteda (<20 mmol) | |||
| N (%) | 117 (100) | 42 (36) | 30 (26) | 31 (26) | 14 (12) | — |
| 18-h urine sodium excretion (mmol) | 69 [30–133] | 210 [117–262] | 70 [53–82] | 30 [25–33] | 15 [10–18] | N/A |
| Pre-diuretic period | ||||||
| eGFR (mL/min/1.73 m2) | 56 ± 19 | 56 ± 17 | 58 ± 24 | 55 ± 20 | 50 ± 13 | 0.259 |
| BUN (mg/dL) | 28 [19–38] | 27 [19–40] | 28 [18–37] | 28 [16–38] | 32 [26–37] | 0.894 |
| Pre-diuretic urine sodium (mmol/L) | 64 ± 33 | 80 ± 36 | 65 ± 31 | 47 ± 22 | 48 ± 26 | <0.001 |
| Pre-diuretic urine sodium/creatinine ratio | 0.07 [0.03–0.13] | 0.12 [0.05–0.19] | 0.07 [0.03–0.13] | 0.03 [0.02–0.08] | 0.04 [0.02–0.07] | <0.001 |
| Diuretic period | ||||||
| 6-h urine sodium excretion (mmol) | 103 [59–158] | 146 [111–243] | 97 [67–170] | 71 [26–109] | 55 [27–75] | <0.001 |
| Diuretic efficiencyb | 41 [20–76] | 56 [33–97] | 56 [36–56] | 32 [19–57] | 30 [10–61] | <0.001 |
Values are presented as n (%), median [interquartile range], or mean ± standard deviation.
eGFR, estimated glomerular filtration rate; BUN, blood urea nitrogen; IV, intravenous; N/A, not applicable.
Expected 18-h urinary sodium excretion is 93 mmol based on 124 mmol/day from 130 mmol dietary sodium intake.
Diuretic efficiency calculated as 6-h cumulative urine sodium excretion (mmol) per doubling of the IV furosemide equivalent dose (furosemide 40 mg IV = bumetanide 1 mg IV).
Effect of increasing diuretic response on post-diuretic spontaneous natriuresis
While the cross-sectional observations above suggest CPDSR is not a primary determinant of sodium excretion on a population level in ADHF, they do not address if CPDSR is relevant in poor diuretic responders forced to have an adequate acute natriuretic response with escalation of diuretic therapies. To assess the intra-patient change in post-diuretic spontaneous natriuresis following increased diuretic intensity and diuretic-induced natriuresis, patients in the Randomized Intervention Cohort (n = 43) received either an increased IV loop diuretic dose (n = 18) or IV loop with IV chlorothiazide (n = 25). Baseline characteristic are presented in Supplementary material online, Table S5. The 6-h diuretic-induced natriuresis increased in 65% of patients from a mean 6-h urine sodium excretion of 55 ± 24 to 155 ± 95 mmol (Figure 6). Despite a three-fold increase in diuretic-induced natriuresis, the corresponding mean 18-h post-diuretic spontaneous natriuresis did not change significantly from 46 ± 47 to 54 ± 50 mmol (P = 0.47), in contrast to the substantial decrease the CPDSR paradigm would predict.
Figure 6.
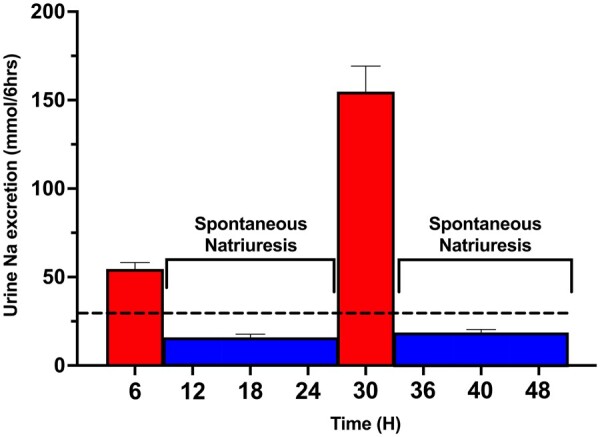
Influence of increased diuretic-induced natriuresis on post-diuretic spontaneous natriuresis. In the Randomized Intervention Cohort, the initial poor diuretic response (first red bar) was followed by poor 18-h spontaneous natriuresis (blue bars), plotted as mean ± SEM. The 18-h spontaneous natriuresis is shown evenly distributed across three 6-h bars, producing identical means and standard errors. Following an intensified diuretic regimen the following day, the diuretic-induced natriuresis increased (second red bar) and the subsequent post-diuretic spontaneous natriuresis did not decrease due to CPDSR but instead increased slightly with increasing diuretic-induced natriuresis.
Discussion
In contrast to the prevailing wisdom, the primary observation in this study is that, on a population level, CPDSR did not appear to be a meaningful contributor of diuretic resistance in hypervolemic ADHF. In contrast, we observed that larger diuretic-induced natriuresis was actually followed by greater spontaneous natriuresis in the post-diuretic period, the inverse pattern predicted by CPDSR. Even with protocol-driven intensified diuretic therapy, which resulted in a three-fold increase in diuretic-induced natriuresis, we found no change in post-diuretic spontaneous natriuresis. Overall, these findings indicate that, in the volume expanded setting of ADHF, CPDSR is not a relevant mechanism leading to blunted renal sodium excretion at the population level. Rather, a basal and relatively fixed degree of sodium avidity (proclivity of the kidney to excessively retain sodium) intrinsic to each patient appears to drive both the diuretic-induced and post-diuretic spontaneous natriuresis (Graphical abstract).

The concept of CPDSR was established in healthy volunteers on a high sodium diet7 and has been extrapolated to be a significant mechanism of diuretic resistance in ADHF. While often simplified as only compensatory to the preceding diuresis, both volume-dependent and volume-independent mechanisms cause intense compensatory renal sodium retention in the post-diuretic period.9,10,18 Although the expanded extracellular volume in patients with ADHF may eliminate volume-dependent CPDSR mechanisms, mechanisms independent of volume and diuretic therapy can determine basal sodium avidity and therefore post-diuretic sodium excretion.9,30 According to the traditional paradigm, CPDSR can be eliminated and decongestion significantly improved by provision of a very low-sodium diet, diminishing available sodium for the kidney to reabsorb, or continuous infusion of loop diuretic.6 However, a multitude of studies have demonstrated a lack of meaningful increase in decongestion with continuous infusion relative to bolus dosing at an equivalent total daily dose.11,20,31 Furthermore, randomization to intense sodium restriction (0.8 g/day) during ADHF did not improve weight loss, decongestion, or time to oral diuretic transition compared to an unrestricted sodium intake (3–5 g/day).32 Our findings provide a mechanism to explain why these strategies did not result in measurable clinical improvement in hypervolemic ADHF populations. As profound differences in renal sodium handling exist between patients with and without heart failure, historically accepted diuretic concepts founded in euvolemic healthy controls and hypertensive patients need to be validated in ADHF populations.
The global observation in this study is that post-diuretic spontaneous natriuresis was positively correlated to the amount of diuretic-induced natriuresis (i.e. opposite of the CPDSR concept), providing strong evidence that there is not a robust renal compensation to diuretic-induced sodium losses in volume expanded ADHF. Among these study observations, the most compelling CPDSR refutation is the lack of intra-patient change in post-diuretic spontaneous natriuresis following diuretic therapy escalation in patients with a poor initial diuretic response. Despite an aggressive diuretic intervention resulting in a three-fold increase in diuretic-induced natriuresis, there was no change in post-diuretic spontaneous natriuresis. Presumably patients had similar basal sodium avidity on both days, but via substantial diuretic escalation (addition of 500 mg IV chlorothiazide or 2.5 × the loop dose) we were able achieve an adequate acute diuretic response. However, consistent with post-diuretic natriuresis dependence on the basal intrinsic sodium avidity of the patient, the post-diuretic spontaneous natriuresis did not increase or decrease.
These data suggest that the most effective strategies to improve decongestion in ADHF would focus on modulating the basal sodium avidity substrate of the patient, which could improve both diuretic response and spontaneous natriuresis. However, an important observation is that even in a high disease severity tertiary referral center population such as this, the sodium avidity physiology was quite variable and many patients did not have high basal avidity. To that end, two-thirds of the Measured Cohort was able to achieve a negative sodium balance with only once daily and relatively low-dose loop diuretic therapy. There has been a multitude of ‘failed’ cardio-renal therapeutics in ADHF populations such as nesiritide, ularitide, low-dose dopamine, and high-dose spironolactone.33–36 Many of these interventions would not be expected to provide measurable benefit in patients without significant sodium avidity. The current findings indicate this substrate is not present in all ADHF patients and raises the question if some of these ‘failed’ therapies may in fact have significant value in selected populations with high basal sodium avidity.33,34,37
Our findings have important implications for the care of patients with ADHF. Heart failure guidelines state frequent/short interval diuretic dosing or continuous infusions may be needed as sodium reabsorption will occur once the tubular concentration of diuretics decline.6 However, we found in many patients a good diuretic response was followed by correspondingly good spontaneous natriuresis that ultimately contributed meaningfully to achieving a negative sodium balance. Notably, in the measured cohort almost equal quantities of the total sodium excretion occurred in the 18-h post-diuretic period as the 6-h diuretic period. As such, patients with a good diuretic-induced natriuretic response will often continue to have significant natriuresis in the post-diuretic period. These patients may be ideally treated with once or twice daily IV loop diuretic dosing which will avoid unnecessary diuretic exposure.
However, patients with a poor acute diuretic response are in double jeopardy with limited sodium excretion in the post-diuretic period. Although we found that substantially increasing diuretic intensity rectified the diuretic-induced natriuresis, the subsequent spontaneous natriuresis remained low. Given the ceiling of sodium excretion with a single dose of diuretic, diuretic resistant patients need both higher doses and also more frequent dosing/continuous infusion of those higher doses. Importantly, continuous infusion has not shown meaningful superiority over bolus diuretic administration when administered at equivalent total daily doses.20 However, the use of a significantly higher total daily loop diuretic dose administered as an infusion, could theoretically be the preferred strategy for patients with a highly sodium avid substrate. Thus, patients with significant diuretic resistance may be optimally treated with both higher diuretic doses and frequent dosing/continuous infusion, not because of CPDSR but due to high basal sodium avidity.
Several limitations warrant discussion. The gold standard approach to performing sodium balance studies is to confine participants in a metabolic ward and provide a precise metabolic diet with well quantitated sodium content. This is obviously not feasible in a large ADHF study of real-world hospitalized patients. Rather, we constructed two cohorts to buttress each cohort's limitations in ascertaining sodium balance. The Measured Cohort is devoid of the calculated assumptions of the Calculated Cohort but is limited by potential selection bias from including patients who only received once daily IV loop diuretics, missing urine voids during the 18-h urine collection performed by clinical nursing staff, and the diuretic effect potentially continuing into the post-diuretic period. The Calculated Cohort lacks these limitations but has limitations arising from calculation assumptions. The limitations of the Measured and Calculated cohorts are mitigated by the strengths of the other, and the findings from each cohort were mutually reinforcing. Both cohorts are from two hospitals in a tertiary care medical center which could introduce selection bias, although the high variation in population basal sodium avidity aids generalizability. Additional limitations relate to the prescribed 3 g sodium diet as the assumed dietary sodium intake. Although the above limitations may reduce the quantitative precision of the conclusions, it is highly unlikely that the qualitative conclusion that CPDSR is not a dominant driver of diuretic resistance would be altered by more rigorous sodium balance methodology.
In conclusion, sodium excretion in the post-diuretic period is highly variable in a contemporary ADHF cohort. In contrast to the current paradigm of compensatory post-diuretic sodium reabsorption, increasing diuretic response was associated with subsequent increasing spontaneous natriuresis. Basal sodium avidity and not compensatory post-diuretic sodium reabsorption is the predominant stimulus for post-diuretic spontaneous natriuresis in most patients with ADHF.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This publication was made possible by the National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute Grant R01HL139629, R21HL143092, R01HL128973, and R01HL148354 (to Dr. Testani), Award P30DK079310 from the National Institute of Diabetes and Digestive and Kidney Disease, and CTSA Grant Number UL1 TR000142 from the National Center for Advancing Translational Science, a component of the NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH. The normal healthy volunteer cohort was supported by a research grant from Bristol Myers Squib.
Conflict of interest: J.M.T. reports grants and/or personal fees from 3ive labs, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Astra Zeneca, Novartis, Cardionomic, MagentaMed, Reprieve inc., FIRE1, W.L. Gore, Sanofi, Sequana Medical, Otsuka, Abbott, Merck, Windtree Therapeutics, Lexicon pharmaceuticals, Precardia, Relypsa, Regeneron, BD, Edwards life sciences, and Lilly. In addition, J.M.T. has a patent Treatment of diuretic resistance issued to Yale and Corvidia Therapeutics Inc, a patent Methods for measuring renalase issued to Yale, and a patent Treatment of diuretic resistance pending with Reprieve inc. V.S.R. has a patent Treatment of diuretic resistance US20200079846A1 issued to Yale and Corvidia Therapeutics Inc. with royalties paid to Yale University, V.S.R. and J.M.T. and a patent Methods for measuring renalase WO2019133665A2 issued to Yale. V.S.R. reports personal fees from Translational Catalyst. Z.L.C. reports grants from AstraZeneca.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
Supplementary Material
Contributor Information
Zachary L Cox, Department of Pharmacy Practice, Lipscomb University College of Pharmacy, 1 University Park Drive, Nashville, TN 37204, USA; Department of Pharmacy, Vanderbilt University Medical Center, 1211 Medical Center Drive, Nashville, TN 37232, USA.
Veena S Rao, Department of Internal Medicine, Section of Cardiovascular Medicine, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA.
Juan B Ivey-Miranda, Department of Internal Medicine, Section of Cardiovascular Medicine, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA; Hospital de Cardiologia, Instituto Mexicano del Seguro Social, 330 Cuauhtemoc Avenue. Cuauhtemoc, Mexico City 06720, Mexico.
Julieta Moreno-Villagomez, Department of Internal Medicine, Section of Cardiovascular Medicine, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA; Universidad Nacional Autónoma de México, Avenida Insurgentes Sur, Mexico City 3000, Mexico.
Devin Mahoney, Department of Internal Medicine, Section of Cardiovascular Medicine, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA.
Piotr Ponikowski, Department of Heart Diseases, Wrocław Medical University, Rektorat, wybrzeże Ludwika Pasteura 1, Wroclaw 50-367, Poland.
Jan Biegus, Clinical Military Hospital, Weigla 5, Wroclaw 50-981, Poland.
Jeffrey M Turner, Department of Medicine, Division of Nephrology, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA.
Christopher Maulion, Department of Internal Medicine, Section of Cardiovascular Medicine, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA.
Lavanya Bellumkonda, Department of Internal Medicine, Section of Cardiovascular Medicine, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA.
Jennifer L Asher, Department of Comparative Medicine, Yale University School of Medicine, 310 Cedar Street, New Haven, CT 06520, USA.
Helen Parise, Department of Internal Medicine, Section of Cardiovascular Medicine, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA.
Perry F Wilson, Clinical and Translational Research Accelerator, Yale University School of Medicine, 60 Temple Street, New Haven, CT 06520, USA.
David H Ellison, Oregon Clinical and Translational Research Institute, Oregon Health and Science University and the Veterans Affairs Portland Health Care System, 3181 S.W. Sam Jackson Park Road Portland, OR 97239, USA.
Christopher S Wilcox, Division of Nephrology and Hypertension and Hypertension Center, Georgetown University, 3800 Reservoir Road, N.W., Washington, DC 20007, USA.
Jeffrey M Testani, Department of Internal Medicine, Section of Cardiovascular Medicine, Yale University School of Medicine, 135 College Street, Suite 230, New Haven, CT 06510, USA.
References
- 1. Felker GM, Ellison DH, Mullens W, Cox ZL, Testani JM. Diuretic therapy for patients with heart failure: JACC state-of-the-art review. J Am Coll Cardiol 2020;75:1178–1195. [DOI] [PubMed] [Google Scholar]
- 2. Testani JM, Brisco MA, Turner JM, Spatz ES, Bellumkonda L, Parikh CR, Tang WH. Loop diuretic efficiency: a metric of diuretic responsiveness with prognostic importance in acute decompensated heart failure. Circ Heart Fail 2014;7:261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kiernan MS, Stevens SR, Tang WHW, Butler J, Anstrom KJ, Birati EY, Grodin JL, Gupta D, Margulies KB, LaRue S, Dávila-Román VG, Hernandez AF, de las Fuentes L; NHLBI Heart Failure Clinical Network Investigators. Determinants of diuretic responsiveness and associated outcomes during acute heart failure hospitalization: an analysis from the NHLBI Heart Failure Network Clinical Trials. J Card Fail 2018;24:428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Valente MA, Voors AA, Damman K, Van Veldhuisen DJ, Massie BM, O'Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, Davison B, Cleland JG, Givertz MM, Bloomfield DM, Fiuzat M, Dittrich HC, Hillege HL. Diuretic response in acute heart failure: clinical characteristics and prognostic significance. Eur Heart J 2014;35:1284–1293. [DOI] [PubMed] [Google Scholar]
- 5. Ellison DH, Felker GM. Diuretic treatment in heart failure. N Engl J Med 2017;377:1964–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yancy CW, Jessup M, Bozkurt B, Masoudi FA, Butler J, McBride PE, Casey DE Jr., McMurray JJ, Drazner MH, Mitchell JE, Fonarow GC, Peterson PN, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. ACCF/AHA Guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62:1495–1539. 2013; [DOI] [PubMed] [Google Scholar]
- 7. Wilcox CS, Mitch WE, Kelly RA, Skorecki K, Meyer TW, Friedman PA, Souney PF. Response of the kidney to furosemide. I. Effects of salt intake and renal compensation. J Lab Clin Med 1983;102:450–458. [PubMed] [Google Scholar]
- 8. Wilcox CS, Testani JM, Pitt B. Pathophysiology of diuretic resistance and its implications for the management of chronic heart failure. Hypertension 2020;76:1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ellison DH. Diuretic therapy and resistance in congestive heart failure. Cardiology 2001;96:132–143. [DOI] [PubMed] [Google Scholar]
- 10. Ellison DH. The physiologic basis of diuretic synergism: its role in treating diuretic resistance. Ann Intern Med 1991;114:886–894. [DOI] [PubMed] [Google Scholar]
- 11. Felker GM, O'Connor CM, Braunwald E; Heart Failure Clinical Research Network Investigators. Loop diuretics in acute decompensated heart failure: necessary? Evil? A necessary evil? Circ Heart Fail 2009;2:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Felker GM, Mentz RJ. Diuretics and ultrafiltration in acute decompensated heart failure. J Am Coll Cardiol 2012;59:2145–2153. [DOI] [PubMed] [Google Scholar]
- 13. Mullens W, Damman K, Harjola VP, Mebazaa A, Brunner-La Rocca HP, Martens P, Testani JM, Tang WHW, Orso F, Rossignol P, Metra M, Filippatos G, Seferovic PM, Ruschitzka F, Coats AJ. The use of diuretics in heart failure with congestion—a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2019;21:137–155. [DOI] [PubMed] [Google Scholar]
- 14. Cox ZL, Testani JM. Loop diuretic resistance complicating acute heart failure. Heart Fail Rev 2020;25:133–145. [DOI] [PubMed] [Google Scholar]
- 15. Wilcox CS, Guzman NJ, Mitch WE, Kelly RA, Maroni BJ, Souney PF, Rayment CM, Braun L, Colucci R, Loon NR. Na+, K+, and BP homeostasis in man during furosemide: effects of prazosin and captopril. Kidney Int 1987;31:135–141. [DOI] [PubMed] [Google Scholar]
- 16. Kelly RA, Wilcox CS, Mitch WE, Meyer TW, Souney PF, Rayment CM, Friedman PA, Swartz SL. Response of the kidney to furosemide. II. Effect of captopril on sodium balance. Kidney Int 1983;24:233–239. [DOI] [PubMed] [Google Scholar]
- 17. Hammarlund MM, Odlind B, Paalzow LK. Acute tolerance to furosemide diuresis in humans. Pharmacokinetic-pharmacodynamic modeling. J Pharmacol Exp Ther 1985;233:447–453. [PubMed] [Google Scholar]
- 18. Almeshari K, Ahlstrom NG, Capraro FE, Wilcox CS. A volume-independent component to postdiuretic sodium retention in humans. J Am Soc Nephrol 1993;3:1878–1883. [DOI] [PubMed] [Google Scholar]
- 19. Cook JA, Smith DE. Development of acute tolerance to bumetanide: constant-rate infusion studies. Pharm Res 1988;5:86–91. [DOI] [PubMed] [Google Scholar]
- 20. Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter MM, Deswal A, Rouleau JL, Ofili EO, Anstrom KJ, Hernandez AF, McNulty SE, Velazquez EJ, Kfoury AG, Chen HH, Givertz MM, Semigran MJ, Bart BA, Mascette AM, Braunwald E, O'Connor CM; NHLBI Heart Failure Clinical Research Network. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 2011;364:797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox ZL, Fleming J, Ivey-Miranda J, Griffin M, Mahoney D, Jackson K, Hodson DZ, Thomas D Jr., Gomez N, Rao VS, Testani JM. Mechanisms of Diuretic Resistance Study: design and rationale. ESC Heart Fail 2020;7:4458–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shah S, Pitt B, Brater DC, Feig PU, Shen W, Khwaja FS, Wilcox CS. Sodium and fluid excretion with torsemide in healthy subjects is limited by the short duration of diuretic action. J Am Heart Assoc 2017;6:e006135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rao VS, Planavsky N, Hanberg JS, Ahmad T, Brisco-Bacik MA, Wilson FP, Jacoby D, Chen M, Tang WHW, Cherney DZI, Ellison DH, Testani JM. Compensatory distal reabsorption drives diuretic resistance in human heart failure. J Am Soc Nephrol 2017;28:3414–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skorecki K, Chertow GM, Marsden PA, Taal MW, Yu ASL, Brenner & Rector's the Kidney, 10th ed. Philadelphia, PA: Elsevier; 2016. [Google Scholar]
- 25.KDIGO Clinical Practice Guideline for Acute Kidney Injury. Online Appendices A-F, March 2012. https://kdigo.org/wp-content/uploads/2016/10/KDIGO-AKI-Suppl-Appendices-A-F_March2012.pdf. Date accessed 31 August 2021.
- 26. Palmer BF, Clegg DJ. The use of selected urine chemistries in the diagnosis of kidney disorders. Clin J Am Soc Nephrol 2019;14:306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.World Health Organization. Strategies to Monitor and Evaluate Population Sodium Consumption and Sources of Sodium in the Diet: Report of a Joint Technical Meeting Convened by WHO and the Government of Canada. Geneva: WHO; 2011.
- 28. Colin-Ramirez E, Arcand J, Ezekowitz JA. Estimates of dietary sodium consumption in patients with chronic heart failure. J Card Fail 2015;21:981–988. [DOI] [PubMed] [Google Scholar]
- 29. Cogswell ME, Loria CM, Terry AL, Zhao L, Wang CY, Chen TC, Wright JD, Pfeiffer CM, Merritt R, Moy CS, Appel LJ. Estimated 24-hour urinary sodium and potassium excretion in US adults. JAMA 2018;319:1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martens P, Dupont M, Verbrugge FH, Damman K, Degryse N, Nijst P, Reynders C, Penders J, Tang WHW, Testani J, Mullens W. Urinary sodium profiling in chronic heart failure to detect development of acute decompensated heart failure. JACC Heart Fail 2019;7:404–414. [DOI] [PubMed] [Google Scholar]
- 31. Salvador DR, Rey NR, Ramos GC, Punzalan FE. Continuous infusion versus bolus injection of loop diuretics in congestive heart failure. Cochrane Database Syst Rev 2005;CD003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aliti GB, Rabelo ER, Clausell N, Rohde LE, Biolo A, Beck-da-Silva L. Aggressive fluid and sodium restriction in acute decompensated heart failure: a randomized clinical trial. JAMA Intern Med 2013;173:1058–1064. [DOI] [PubMed] [Google Scholar]
- 33. Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigran MJ, Goldsmith SR, Bart BA, Bull DA, Stehlik J, LeWinter MM, Konstam MA, Huggins GS, Rouleau JL, O'Meara E, Tang WH, Starling RC, Butler J, Deswal A, Felker GM, O'Connor CM, Bonita RE, Margulies KB, Cappola TP, Ofili EO, Mann DL, Davila-Roman VG, McNulty SE, Borlaug BA, Velazquez EJ, Lee KL, Shah MR, Hernandez AF, Braunwald E, Redfield MM; NHLBI Heart Failure Clinical Research Network. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013;310:2533–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Butler J, Anstrom KJ, Felker GM, Givertz MM, Kalogeropoulos AP, Konstam MA, Mann DL, Margulies KB, McNulty SE, Mentz RJ, Redfield MM, Tang WHW, Whellan DJ, Shah M, Desvigne-Nickens P, Hernandez AF, Braunwald E; National Heart Lung and Blood Institute Heart Failure Clinical Research Network. Efficacy and safety of spironolactone in acute heart failure: the ATHENA-HF Randomized Clinical Trial. JAMA Cardiol 2017;2:950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O'Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJV, Nieminen MS, Reist CJ, Rouleau JL, Swedberg K, Adams KF, Anker SD, Atar D, Battler A, Botero R, Bohidar NR, Butler J, Clausell N, Corbalán R, Costanzo MR, Dahlstrom U, Deckelbaum LI, Diaz R, Dunlap ME, Ezekowitz JA, Feldman D, Felker GM, Fonarow GC, Gennevois D, Gottlieb SS, Hill JA, Hollander JE, Howlett JG, Hudson MP, Kociol RD, Krum H, Laucevicius A, Levy WC, Méndez GF, Metra M, Mittal S, Oh B-H, Pereira NL, Ponikowski P, Tang WHW, Wilson WH, Tanomsup S, Teerlink JR, Triposkiadis F, Troughton RW, Voors AA, Whellan DJ, Zannad F, Califf RM. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med 2011;365:32–43. [DOI] [PubMed] [Google Scholar]
- 36. Packer M, O'Connor C, McMurray JJV, Wittes J, Abraham WT, Anker SD, Dickstein K, Filippatos G, Holcomb R, Krum H, Maggioni AP, Mebazaa A, Peacock WF, Petrie MC, Ponikowski P, Ruschitzka F, van Veldhuisen DJ, Kowarski LS, Schactman M, Holzmeister J; TRUE-AHF Investigators. Effect of ularitide on cardiovascular mortality in acute heart failure. N Engl J Med 2017;376:1956–1964. [DOI] [PubMed] [Google Scholar]
- 37. Wan SH, Stevens SR, Borlaug BA, Anstrom KJ, Deswal A, Felker GM, Givertz MM, Bart BA, Tang WH, Redfield MM, Chen HH. Differential response to low-dose dopamine or low-dose nesiritide in acute heart failure with reduced or preserved ejection fraction: results from the ROSE AHF Trial (Renal Optimization Strategies Evaluation in Acute Heart Failure). Circ Heart Fail 2016;9:e002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.