

Abstract
Autophagy is a multi-step process regulated in part by AMP-activated protein kinase (AMPK). Phosphorylation of threonine 172 on the AMPK α-subunit enhances AMPK kinase activity, resulting in activation of downstream signaling. Integrin-mediated cell adhesion activates Src/Focal Adhesion Kinase (FAK) signaling complex, which regulates multiple cellular processes including cell survival. We show here that Src signaling leads to direct phosphorylation of the AMPK-α subunit on a novel site, tyrosine 179, resulting in suppression of AMPK-T172 phosphorylation and autophagy upon integrin-mediated cell adhesion. By using chemical inhibitors, genetic cell models and targeted mutagenesis, we confirm an important role for Src and FAK in suppressing AMPK signaling and autophagy induced by various additional stimuli, including glucose starvation. Furthermore, we found that autophagy suppression by hydroxychloroquine promotes apoptosis in a cancer cell model that had been treated with Src inhibitors. Our findings reveal a link between the Src/ FAK complex and AMPK/ autophagy regulation, which may play an important role in the maintenance of normal cellular homeostasis and tumor progression.
Keywords: cell attachment, tyrosine phosphorylation, Src, AMPK, autophagy
1. Introduction
Integrins are a large family of heterodimeric receptors that interact with the extracellular matrix (ECM) to mediate cell adhesion and subsequent signaling events (1). The interaction between ECM and integrins is essential for adhesion-mediated cell survival, and also plays an important role in various other cellular events including cell migration and proliferation (2). Detachment of cells from ECM disrupts integrin engagement and can trigger a form of programmed cell death termed anoikis (3-5). However, cell detachment can also result in activation of autophagy, which has been proposed to enhance cell survival and provide protection against anoikis (6, 7).
Cancer cells must survive altered environmental conditions that include restricted amounts of nutrients and aberrant ECM composition. As a critical feature of invasion and metastasis, cancer cells develop the ability to survive after they detach and escape from the primary tumor environment and become exposed to distinct ECM components (8). Activation of autophagy plays an important role in this process by enabling cancer cells to evade anoikis (6, 7, 9). After cancer cells settle at secondary tissue sites, prolonged and excessive autophagy could have a deleterious effect, such that deactivation of autophagy is likely needed for cellular homeostasis. Although it has been established that matrix deprivation can lead to both anoikis and activation of autophagy vis a vis loss of integrin engagement, the signaling mechanisms for the cross-talk between these competing mechanisms have not been explored in detail (9).
Integrin engagement initiates assembly of multiprotein complexes at sites termed focal adhesions, which translate extracellular changes to intracellular signaling in adherent cells (2, 10). Non-receptor tyrosine kinase Src and Focal Adhesion Kinase (FAK) are major components of integrin-mediated signaling. The mutually activated Src/FAK complex can recruit, phosphorylate and activate multiple downstream signaling pathways (11). Recently, several known mediators of integrin signaling, such as the Src family kinase Fyn (12), FAK (13), p130Cas (14) and Paxillin (13, 15), have been reported to be involved in autophagy regulation. Specifically, Fyn was found to negatively regulate the activity of liver kinase B1 (LKB1, also known as STK11) and its target, AMP-activated protein kinase (AMPK) (12, 16, 17). AMPK is a serine/ threonine kinase composed of a catalytic α subunit and two regulatory subunits, β and γ. The α-subunit of AMPK contains a conserved threonine residue (T172), phosphorylation of which by LKB1 results in AMPK activation (18-20). AMPK is a master regulator of cell metabolism, playing a key role in the maintenance of energy homeostasis (21). It also plays a major role in promoting autophagy, by inactivating the target of rapamycin complex 1 (TORC1) kinase pathway and by phosphorylating and activating the Unc-51 like autophagy activating kinase (ULK1) (22, 23).
Here we report that Src activity is required for adhesion-mediated suppression of autophagy. A novel Src phosphorylation site on the AMPK-α subunit is identified, and this tyrosine phosphorylation site, Y179, is adjacent to the regulatory T172 activation site on AMPK-α. Our studies show that Src suppresses AMPK signaling by direct phosphorylation of AMPK-Y179, and 3-dimensional modelling studies predict that post-translational modification of Y179 would disturb the structure of the active site of AMPK and thereby reduce or abolish its catalytic activity and its ability to initiate downstream signaling. Consistent with these observations, suppression of autophagy by hydroxychloroquine (HCQ) or by deletion of the autophagy genes (ATG5 or ATG7) further promotes apoptosis in cells where Src is inhibited. Considering the current exploration of Src (24), FAK (25), and autophagy inhibitors, as well as AMPK activators as cancer therapeutics, the signaling crosstalk identified here is likely to be of interest in oncology therapeutics’ development.
2. Materials and methods
2.1. Antibodies and reagents
Anti-phospho-AMPK(T172), anti-AMPK, anti-phospho-ACC, anti-phospho-ULK1(S555), anti-ULK1, anti-phospho-Src(Y416) and anti-Src antibodies were from Cell Signaling Technologies (Beverly, MA); anti-LC3 and anti-ACC antibodies were from EMD Millipore (Temecula, CA); anti-HA was from Roche Diagnostics GmbH (Mannheim, Germany); anti-phospho-FAK(Y576), anti-FAK and HRP-conjugated anti-phospho-tyrosine (PY20) monoclonal antibodies used in immunoblot analysis were from Transduction Laboratories (Lexington, KY); anti-Flag, anti-GFP and anti-β-actin antibodies were from Sigma (St. Louis, MO). Fibronectin was from Invitrogen (Carlsbad, CA). Phenformin and 5-aminoimidazole-4-carboxamide ribofuranoside (AICAR), A-769662 and Bafilomycin A1 were from Sigma. Hydroxychloroquine (HCQ) and Src inhibitors Bosutinib, Dasatinib and Saracatinib were from Selleck Chemicals LLC (Houston, TX).
2.2. DNA constructs and proteins
Homo Sapiens AMPK-α2 cDNA was a gift from Stefan Riedl (Sanford Burnham Prebys Medical Discovery Institute, originally from Dharmacon, Lafayette, CO), and was constructed into pcDNA3.1-HA and pEGFP-N3 (Invitrogen) vectors to express HA- or GFP-tagged AMPK-α2. AMPK-β2-Flag (Addgene plasmid # 40603) and AMPK-γ1-HA (Addgene plasmid # 40605) (26) were gifts from Harvey Lodish (MIT, Cambridge, MA). Wild-type (WT) Src, kinase-dead (KD) Src (K295N), active Src (Y527F), HA-FAK and various FAK mutants have been described (27). Phospho-mimetic mutant AMPK-α2-Y179E and phosphorylation-resistant mutant AMPK-α2-Y179F were created by using the Quick Change Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). Recombinant AMPK-α/β/γ proteins were from EMD Millipore; recombinant GST-tagged active Src kinase was from (R&D Systems Inc., Minneapolis, MN).
2.3. Cell culture and transfection
293T cells, SYF fibroblasts derived from mouse embryos lacking three Src-family kinases (Src, Fyn, and Yes) and Src++ MEF cells (SYF cells with Src-expression reconstituted) (28), FAK −/− (29) and FAK +/+ cells were from ATCC. AMPK-α1/2-DKO MEFs (30) and 293A AMPK-α1/2-DKO cells (31) have been described before. Py230 cell were isolated from a MMTV-PyMT tumor, and were provided by Leslie Ellies (University of California, San Diego). Atg5−/−, Atg5+/+ MEFs (32), Atg7−/− and Atg7+/+ MEFs (SV40Tag immortalized) (33) were generously provided by Jayanta Debnath (University of California, San Francisco) (34), with permissions from Noboru Mizushima (University of Tokyo, Japan) and Masaaki Komatsu (Juntendo University School of Medicine, Japan), respectively. All cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and penicillin/streptomycin/L-glutamine (Omega Scientific Inc., Tarzana, CA). After detachment with cell dissociation buffer (Invitrogen), cells were resuspended in culture medium for 1-2 hr to induce activation of the autophagy pathway. In some experiments, the suspended cells were then allowed to re-attach to fibronectin-coated Ultra-Low Attachment dishes (REF#3261, Corning, Kennebunk, ME) to deactivate autophagy. Cells were transiently transfected using FuGENE HD reagent (Promega) as indicated, and 24-48 hr later, the transfected cells were subjected to the various assays as described here.
2.4. Immunofluorescence microscopy and statistical analysis
Experiments were carried out as described previously (14). In brief, MEFs grown on a Lab-Tek® II Chamber Slide System (Nalge Nunc International, Naperville, IL) were kept in normal medium or treated as indicated. Cells were fixed with zinc formalin and permeabilized in 0.2% Triton X-100 for 10 min, then were blocked in 2% BSA for 1 hr, followed by incubation with indicated antibodies at 4°C overnight, and Alexa 488- or Alexa 565-conjugated secondary antibody at room temperature for 1 hr. Slides were coverslipped using FluorSave mounting medium containing DAPI and sealed with nail polish. Images were taken under a Zeiss fluorescence microscope, and were analyzed using Image J. Representative images were quantitated and statistical analyses were performed using Student's t-test with Graphpad Prism software, *p <0.05 (n = 3), **p <0.01 (n = 3).
2.5. Immunoblotting, densitometric quantitation and statistical analysis
Experiments were performed as described previously (27). Briefly, quiescent monolayer cells were treated as described in the figure legends. Whole cell lysates were obtained in modified RIPA lysis buffer (25 mM Tris-HCl, pH7.4, 10% glycerol, 0.2% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA) containing a protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN), and clarified by centrifugation. Equal amounts of protein were separated by SDS-PAGE and electrotransferred to nitrocellulose membranes. After blocking with 3% dry milk (Bio-Rad, Hercules, CA) in TBS-Tween-20, protein-bound membranes typically were cut into several pieces based on the molecular weight of the antigen to be detected, and each filter was then exposed to respective primary antibodies as described for each experiment. Antibody binding was detected using horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse secondary antibodies (Sigma) and enhanced chemiluminescence (GE Healthcare Life Sciences, Pittsburgh, PA), β-actin was used as a loading control for every sample. Densitometric quantitation of specific bands were performed by using Image Studio Lite software. LC3-II/actin, phosphorylated AMPK/total AMPK, phosphorylated ULK1/total ULK1, phosphorylated Src/total Src, and phosphorylated FAK/total FAK were expressed as fold changes to the controls and shown under respective blots. Representative blots are shown from at least three independent experiments. In addition, statistical analyses were performed on the independent blots for experiments shown in Figs. 1A, C, D, F; 2A, B, C; 3A, B, C; 4A, B, C; 5A, B, E; and 6C, D, F, G. Statistical significance was determined using the Student's t-test, and the results were considered statistically significant at p < 0.05 (*, P < 0.05; **, P < 0.01; ***, P < 0.001). All graphs were made using GraphPad Prism software and are shown in Supplementary Figure 1.
Figure 1. Src inhibits autophagy and AMPK signaling in adherent cells.
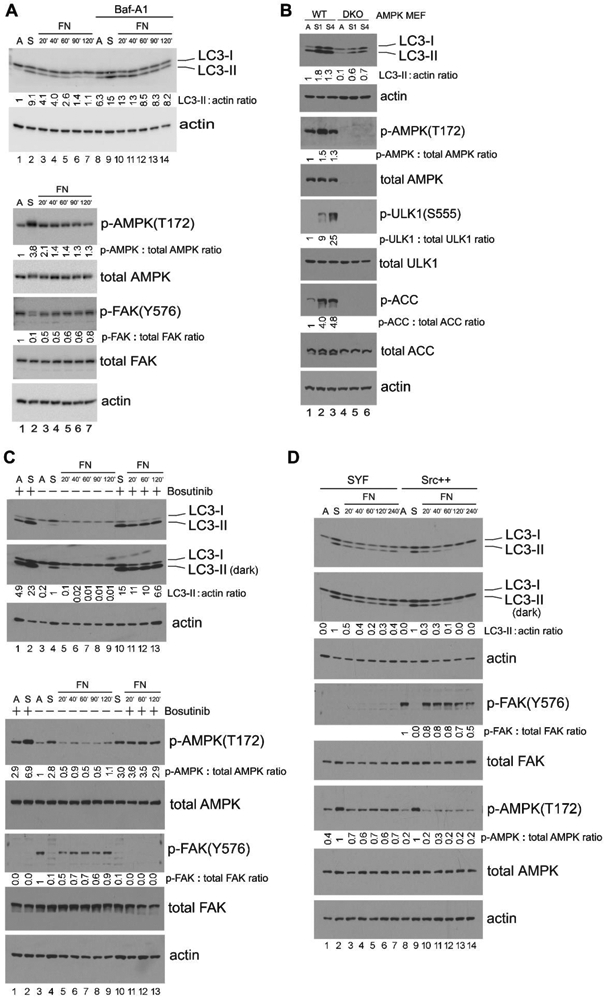

(A) “Detachment-re-attachment” experiment in MEFs. Wild-type (WT) MEFs were kept adherent and treated in the presence or absence of 100 nM of Bafilomycin A1 (Baf-A1) for 1 hr. Cells were then either lysed for analysis (A = Adherent) or detached and kept in suspension with or without Bafilomycin A1 for 1 hr. Suspended cells were then either lysed for analysis (S = Suspended), or re-attached to fibronectin (FN)-coated Ultra-Low Attachment dishes for the indicated times in the presence or absence of Bafilomycin A1. Immunoblot analysis with the indicated antibodies was carried out on total cell lysates. Densitometric quantitation of LC3-II/actin, phosphorylated AMPK/total AMPK, and phosphorylated FAK/total FAK is expressed as the fold change to the normalized control (lane 1, adherent cells). In all panels for Fig. 1, results shown are representative of at least three independent experiments with similar results. (B) Role of AMPK in detachment-induced signaling events. Adherent WT and AMPK-DKO MEFs were either lysed for analysis (A = Adherent), or detached and kept in suspension for 1 hr (S1) or 4 hr (S4) followed by cell lysis. Immunoblot analysis of total cell lysates was done with the indicated antibodies and quantified as above (with lane 1, WT adherent cells as the normalized control). (C) Effect of Src inhibition in “detachment-re-attachment” experiment. WT MEFs were detached and kept in suspension with or without 10 μM of the Src inhibitor Bosutinib for 1 hr, and were then either lysed for analysis (S = Suspended), or replated onto FN-coated dishes for the indicated times in the presence or absence of 10 μM Bosutinib. “A” indicates adherent MEFs that had been treated (+) or not (−) with 10 μM Bosutinib for 1 hr and then lysed for analysis. Immunoblot analysis of total cell lysates was done with the indicated antibodies and quantified as above (with lane 4, untreated suspended cells as the normalized control in the upper panels and lane 3, untreated adherent cells as the normalized control in the lower panels). “Dark” indicates 5 times longer exposure compared to the top panel. (D) “Detachment-re-attachment” experiment in SYF and Src++ MEFs. Adherent cells were either lysed for analysis (A = Adherent), or detached and kept in suspension for 1 hr. Suspended cells were then either lysed (S = Suspended) or re-attached to FN-coated dishes for the indicated times, followed by immunoblot analysis and quantification as above. Lane 2, suspended SYF cells and lane 9, suspended Src++ cells were used as the normalized control for lanes 1-7 and 8-14, respectively in blots for LC3-II and p-AMPK(T172), while lane 8, adherent Src++ cells was used as the normalized control for the p-FAK(Y576) blot. “Dark” indicates 5 times longer exposure compared to the top panel. (E) Effect of Src inhibition on AMPK signaling. WT MEFs cultured as an adherent monolayer were treated (+) or not (−) with the indicated concentrations of Src inhibitors Bosutinib, Dasatinib or Saracatinib for 3 hr, and immunoblot analysis of total cell lysates was carried out and quantified as above (lane 1, untreated cells was used as the normalized control in each panel). (F) Activation of AMPK in SYF and Src++ MEFs. Cells were treated (+) or not (−) with the various AMPK agonists (200 μM A-769662, 2 mM Metformin, 1 mM phenformin or 2 mM AICAR, as indicated) for 1 hr, followed by immunoblot analysis and quantification as above (lane 1, untreated SYF was used as the normalized control). For quantification and statistical analysis of the independent blots for Figs. 1A, C, D and F, please see Supplementary Figure 1.
Figure 2. FAK regulates autophagy and AMPK signaling in adherent cells.
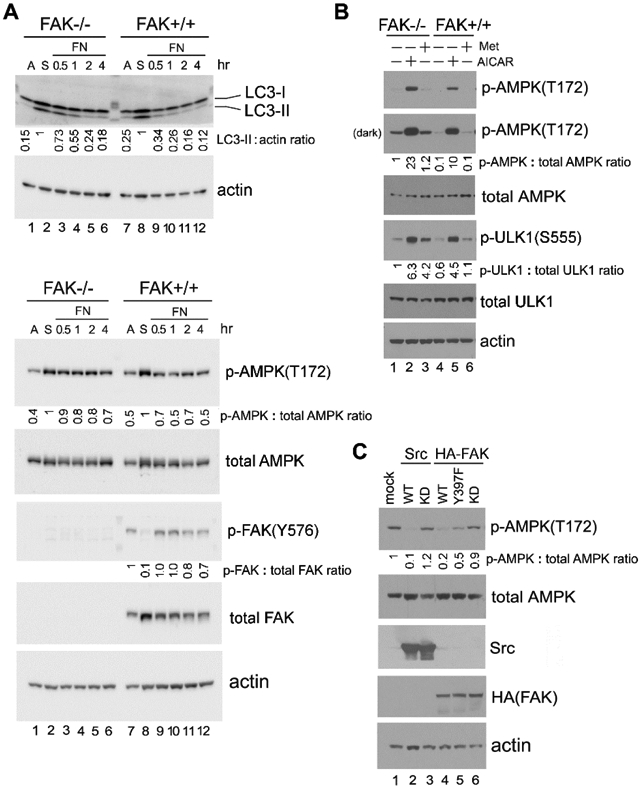
(A) “Detachment-re-attachment” experiment with FAK−/− and FAK+/+ MEFs. Adherent FAK−/− and FAK+/+ MEFs were either lysed for analysis (A = Adherent) or detached and kept in suspension for 1 hr. Suspended cells were then either lysed (S = Suspended) or replated onto FN-coated dishes for the indicated times, and immunoblot analysis was carried out with the indicated antibodies. Densitometric quantitation was performed and expressed as the fold change as in previous figures. Lane 2, suspended FAK−/− cells and lane 8, suspended FAK+/+ cells were used as the normalized control for lanes 1-6 and 7-12, respectively, in blots for LC3-II and p-AMPK(T172). Lane 7, adherent FAK+/+ cells was used as the normalized control for the p-FAK(Y576) blot. In all panels for Fig. 2, results shown are representative of at least three independent experiments with similar results. (B) Activation of AMPK in FAK−/− and FAK+/+ MEFs. Cells were treated (+) or not (−) with the AMPK agonists AICAR (2 mM) or Metformin (Met; 2 mM) for 1 hr, followed by immunoblot analysis and quantification as above (lane 1, untreated FAK −/− cells was used as the normalized control). “Dark” indicates 5 times longer exposure compared to the top panel. (C) Effect of Src or FAK overexpression on AMPK-T172 phosphorylation. 293T cells were transfected or not (mock) to express the various Src and FAK proteins as indicated (WT=wild-type; KD=kinase-dead). Total cell lysates were then subjected to immunoblot analysis with the indicated antibodies. Quantification was performed as above using mock-transfected cells (lane 1) as the normalized control. For quantification and statistical analysis of the independent blots for Figs. 2A, B and C, please see Supplementary Figure 1.
Figure 3. Src/FAK signaling suppresses glucose starvation-induced autophagy and AMPK activation.

(A and B) Glucose starvation of cells. SYF and Src++ MEFs (A), or FAK−/− and FAK+/+ MEFs (B) were cultured in DMEM plus 10% FCS without glucose for the indicated times, and the phosphorylation of AMPK and its downstream targets ULK1 and ACC were detected by immunoblot analysis of total cell lysates. Quantification was performed as in previous figures by using non-starved SYF and FAK−/− cells (lane 1 in each panel), respectively, as the normalized controls. In all panels for Fig. 3, results shown are representative of at least three independent experiments with similar results. (C and D) Induction of autophagy by glucose starvation. SYF and Src++ MEFs (C), or FAK−/− and FAK+/+ MEFs (D) were cultured in DMEM plus 10% FCS without glucose for the indicated times, and LC3-II formation was detected by immunoblot analysis. Quantification was performed by using non-starved SYF and FAK−/− cells (lane 1 in each panel) as the normalized controls, respectively. (E and G) Immunostaining of LC3. SYF and Src++ MEFs (E), or FAK−/− and FAK+/+ MEFs (G) cultured on coverslips were either kept in complete medium (“c”) or glucose-starved (“s”) for 2 hr, and the induction of autophagy was detected by immunofluorescence staining of LC3-II puncta. Quantification of LC3 puncta formation from (E) and (G) is shown in panels (F) and (H), respectively. Statistical analyses were performed using Student's t-test with Graphpad Prism software. *p <0.05 (n = 3), **p <0.01 (n = 3). For quantification and statistical analysis of the independent blots for Figs. 3A, B and C, please see Supplementary Figure 1.
Figure 4. Src phosphorylates AMPK-α on Y179.
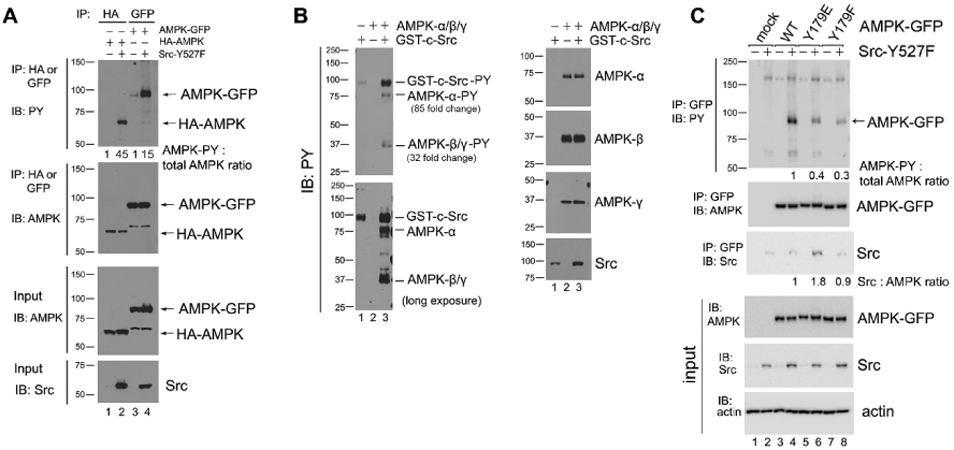

(A) AMPK-α subunit phosphorylation in Src-Y527F transfected cells. Immunoprecipitation with the indicated antibodies was performed in 293T cells that had been co-transfected with either HA-tagged or GFP-tagged AMPK-α2 along with Src-Y527F, and tyrosine-phosphorylated tagged AMPK-α2 was detected by immunoblot with HRP-conjugated anti-PY20 antibody. Quantification was performed as in previous figures by using cells transfected with HA-AMPK (lane 1) or AMPK-GFP (lane 3) in the absence of Src-Y527F as the normalized control, respectively. We surmise that the bands of 65 kD in molecular weight that are visible in anti-AMPK blots in AMPK-GFP lanes are degradation products of the AMPK-GFP protein. In all panels for Fig. 4, results shown are representative of at least three independent experiments with similar results. (B) In vitro phosphorylation of AMPK by Src. Recombinant AMPK-α/β/γ protein complex and GST-tagged Src kinase were incubated together in a kinase buffer and 200 μM ATP at 35°C for 1 hr. Tyrosine phosphorylation of AMPK was examined by immunoblotting using anti-PY20 antibody in the left panel, and total protein amounts were examined by immunoblotting with the indicated antibodies in the right panel. Quantification of tyrosine phosphorylation was performed by using AMPK in the absence of GST-c-Src (lane 2) as the normalized control. The loading amount of Src in lane 1 was 1/3 of that in lane 3. “Long exposure” indicates 10 times longer exposure compared to the top panel. (C) Tyrosine phosphorylation of the AMPK-Y179 mutants in Src-Y527F transfected cells. AMPK-α2-GFP was immunoprecipitated from the extracts of 293T cells that had been transfected or not (mock) with the various GFP-tagged AMPK-α2 constructs along with Src-Y527F as indicated, and tyrosine-phosphorylated AMPK-α2-GFP was detected by immunoblot with HRP-conjugated anti-PY20 antibody in the uppermost panel. Co-precipitation of Src in the various AMPK-Y179 immunoprecipitates was assessed in the third uppermost panel. Quantification in each case was performed by using cells expressing WT-AMPK-GFP in the presence of Src-Y527F (lane 4) as the normalized control. (D) Immunostaining with anti-p-AMPK(Y179) antibody. SYF and Src++ cells were cultured on coverslips, treated with or without 10 μM Bosutinib for 1 hr, and the phosphorylation of AMPK(Y179) and Src(Y416) sites was detected by immunofluorescence with the relevant phospho-specific antibodies. p-Src(Y416) indicates Src activation. DAPI-staining is also shown, along with the three color overlay. (E) Fluorescence intensity of p-AMPK(Y179) staining was determined using ImageJ software. SYF-V and SYF-B, vehicle- and Bosutinib-treated SYF cells, respectively; Src-V and Src-B, vehicle- and Bosutinib-treated Src++ cells, respectively. The statistical analyses were performed using Student's t-test with Graphpad Prism software. *p <0.05 (n = 3). For quantification and statistical analysis of the independent blots for Figs. 4A, B and C, please see Supplementary Figure 1.
Figure 5. The AMPK-Y179 site plays a role in regulating phosphorylation of AMPK at T172.
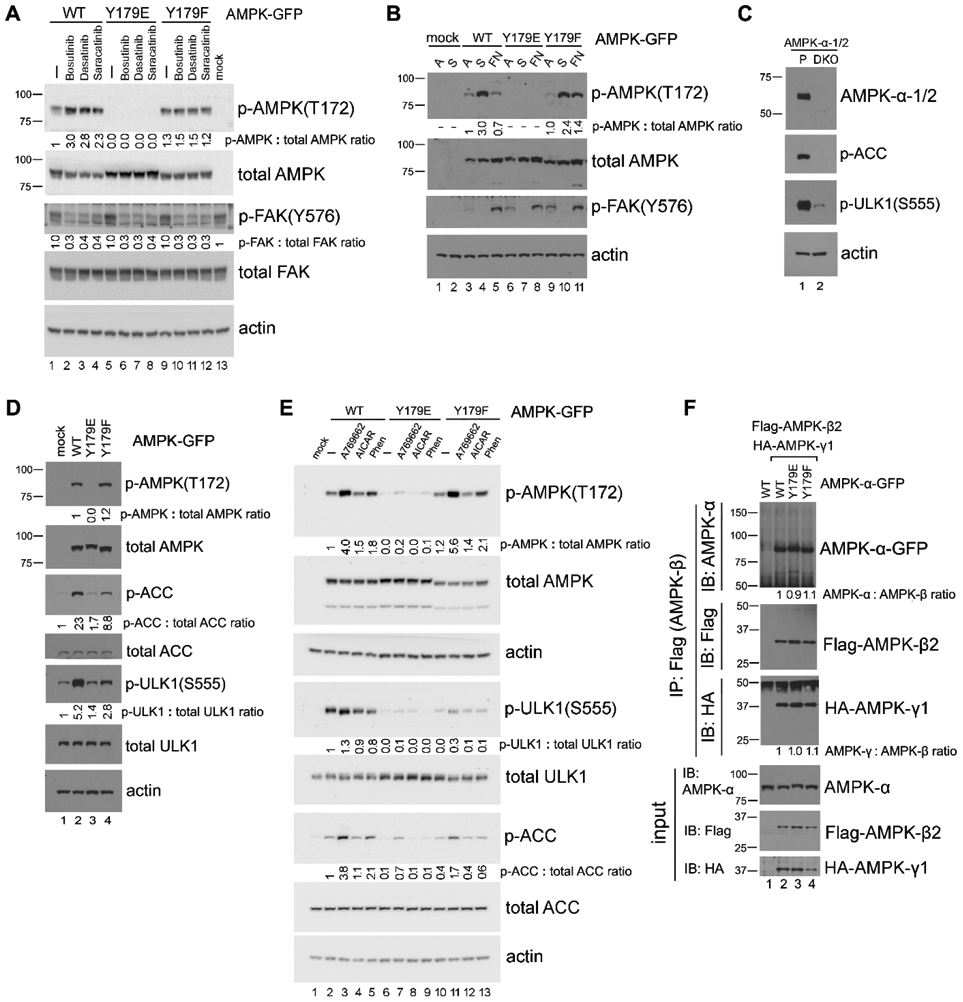
(A) Effect of Src inhibition on AMPK-T172 phosphorylation. 293T cells transfected with the various GFP-tagged AMPK-α2 constructs were treated or not (−) with 10 μM of Src kinase inhibitors Bosutinib, Dasatinib or Saracatinib for 1 hr, and AMPK-α2-GFP phosphorylation on T172 was detected by immunoblot in total cell lysates. Quantification was performed by using untreated WT-AMPK cells (lane 1) as the normalized control. In all panels for Fig. 5, results shown are representative of at least three independent experiments with similar results. (B) “Detachment-re-attachment” experiment. 293T cells transfected with the various GFP-tagged AMPK-α2 constructs were lysed when adherent (A) or kept in suspension (S) for 1 hr before replating of the cells to FN-coated dishes for 2 hr. AMPK-α2-GFP phosphorylation on T172 was detected by immunoblot in total cell lysates as above. Quantification was performed by using adherent WT-AMPK cells (lane 3) as the normalized control. (C) Immunoblot analysis of parental 293A (P) and 293A-AMPK1/2-DKO cells with the indicated antibodies. (D) Expression of various AMPK-Y179 mutant proteins in 293A-AMPK1/2-DKO cells. Cells were transfected with the various GFP-tagged AMPK-α2 constructs, and immunoblot analysis was carried out with the indicated antibodies. Quantification was performed with the WT-AMPK cells (lane 2) as the normalized control for the p-AMPK(T172) blot, and with mock-transfected cells (lane 1) as the normalized control for the p-ACC and p-ULK1(S555) blots. (E) Activation of AMPK in 293A-AMPK1/2-DKO cells transfected with the various GFP-tagged AMPK-α2 constructs. Cells were treated or not (−) with various AMPK agonists (200 μM A-769662, 2 mM AICAR or 1 mM phenformin, as indicated) for 1 hr, followed by immunoblot analysis as above. Fold change of p-AMPK-T172, p-ULK1-S555, and p-ACC was quantified, normalized to that of the total corresponding protein and compared to untreated WT-AMPK-transfected cells (lane 2). (F) Examination of AMPK subunits assembly. Co-IP was performed in lysates from 293T cells that had been co-transfected with Flag-tagged AMPK-β2 and HA-tagged AMPK-γ1, along with GFP-tagged AMPK-α constructs by using anti-Flag antibody, and the associated AMPK subunits were detected by immunoblot with the relevant antibodies. Quantification was performed with WT-AMPK cells (lane 2) as the normalized control. For quantification and statistical analysis of the independent blots for Figs. 5A, B and E, please see Supplementary Figure 1.
Figure 6. Synergistic cell killing effect of Hydroxychloroquine (HCQ) when combined with Src inhibitors.

(A and B) Cell viability assay. Py230 cells cultured in 384-well plates were treated with various concentrations of Src inhibitors Bosutinib (A) or Saracatinib (B) in the presence or absence of 20 μM of HCQ for 48 hr, and cell viability was determined using the CellTiter-Glo Luminescent Cell Viability Assay. The experiments were carried out independently three times in quadruplicate with means and standard deviations calculated with Microsoft Excel before graphing and analyses in GraphPad Prism. Values are normalized to vehicle control. Graphs are nonlinear regression fits of the means (variable slope (four parameters)), and error bars are standard deviations. * = p <0.01. (C and D) Immunoblot analysis of Py230 cells treated (+) or not (−) with 20 μM of HCQ and various concentrations of Bosutinib as indicated for 24 hr. Quantification of the immunoblots was performed as in previous figures by using untreated cells as the normalized control (lane 1). Results shown are representative of three independent experiments with similar results. (E) Py230 cells were kept in suspension and treated with or without 20 μM of HCQ for the indicated times, and cell viability was determined as in (A). The experiment was carried out independently three times in triplicate. (F and G) Immunoblot analysis of Atg5−/− and Atg5+/+ (F) and Atg7−/− and Atg7+/+ MEF cells (G) treated with 5 μM of Bosutinib or Dasatinib for the indicated times. Quantification of the immunoblots was performed as above by using the untreated knock-out cells as the normalized control in each case (lanes 1 and 7). Results shown are representative of three independent experiments with similar results. (H and I) Caspase 3/7 activity assay. Atg5−/− and Atg5+/+ (H), and Atg7−/− and Atg7+/+ MEF cells (I) were treated with 1 or 5 μM of Bosutinib or Dasatinib for 24 hr before Caspase-Glo® 3/7 Assay reagent (Promega Inc.) was added to each well and incubated at room temperature for 30 mins as per manufacturer’s instructions. Sample luminescence intensity was measured and reflected by the number of relative light units (RLU). Data are presented as the mean ± SD, n = 3. * = p <0.01, ** = p <0.001. For quantification and statistical analysis of the independent blots for Figs. 6C, D, F and G, please see Supplementary Figure 1.
2.6. Immunoprecipitation
Experiments were carried out as described previously (27). Cell lysates were prepared in modified RIPA lysis buffer and clarified by centrifugation. After being pre-cleared with protein G beads (GE Healthcare Life Sciences), the lysates were incubated with anti-GFP or anti-HA antibodies in cold room for 4 hr, followed by capture of the immunocomplexes with protein G beads for 2 hr. The beads were washed three times with lysis buffer to remove non-specifically bound proteins, and the immunoprecipitates were analysed by immunoblotting as described above.
2.7. In vitro phosphorylation of AMPK
Purified recombinant AMPK-α/β/γ proteins (EMD Millipore) were incubated with recombinant human active GST-tagged Src kinase (R&D Systems Inc., Minneapolis, MN) in the presence of kinase buffer (Cell Signaling Technologies) and 200 μM ATP at 35°C for 1 hr, and the phosphorylated AMPK was examined by immunoblotting using anti-PY20 antibody.
2.8. Liquid chromatography-coupled tandem mass spectrometry (LC-MS MS) analysis
GFP-tagged AMPK-α-2 expressed in 293T cells along with Src-Y527F was immunoprecipitated using anti-GFP antibody-conjugated protein G beads. The beads then were thoroughly washed with lysis buffer and ammonium bicarbonate to remove non-specific binding proteins. Mass Spectrometry was carried out by Sanford Burnham Prebys’ Proteomics Core facility as described previously (35). Briefly, proteins were digested in 8M urea containing 50 mM ammonium, followed by cysteine alkylation with 15 mM of iodoacetamide. After Trypsin/Lys-C mix digestion (Promega), automated enrichment of phosphorylated peptides was performed through Fe(III)-NTA cartridges using the AssayMAP Bravo Platform (Agilent Technologies), and further analysis was performed by LC-MS/MS using a Proxeon EASY nanoLC system coupled to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Waltham, MA). All mass spectra were analyzed with MaxQuant software version 1.5.5.1.
2.9. Cell viability assay
Cell viability assessments were carried out essentially as described previously (36). Briefly, 25 nL of 1000x 1:1 serially diluted Src inhibitors (Bosutinib or Saracatinib, Selleck Chemicals) were spotted in quadruplicate onto a 384-well tissue culture treated plate (Greiner) using a Labcyte Echo acoustic dispensing device. Py230 cells were resuspended in 20 μM of Hydroxychloroquine (HCQ) or vehicle at 1x105 cells/mL, and 25 μl (2,500 cells) were plated per well. Cells were incubated at 37°C for 48 hr before CellTiterGlo (Promega) was utilized as a proxy for cell viability on a Synergy 2 plate reader using Gen5 software (BioTek, Winooski, VT). Percent viability was calculated by normalizing to vehicle only controls in Microsoft Excel and plotted using GraphPad Prism.
2.10. Caspase-3/7 activity assay
Caspases-3/7 activity was determined by using Caspase-Glo Assay kit (Promega, Madison, WI) following the manufacturer’s instructions (14). 5x103 cells per well were grown in white-wall 96-well plates overnight, then treated with Bosutinib or Dasatinib for 24 hr. Caspase-Glo reagent was added to each well and incubated at room temperature for 30 min. Sample luminescence intensity was measured in a plate-reading luminometer (BioTek). Luminescence activity was reflected by the number of relative light units (RLU) and plotted using GraphPad Prism.
2.11. Antibody development
Mouse polyclonal antibody to phospho-AMPK-α2-Y179 was generated commercially by Precision Antibody (A&G Pharmaceutical, Inc., Columbus, MD). Briefly, 10 mice were immunized with the phospho-peptide CGGAGSSGSPN(pY)AAPE and antibody titer was assessed after 5 and 8 weeks by ELISA to the same peptide and the non-phosphorylated control peptide CFLRTSSGSPNYAAPE. Sera from 9 animals with good titer were pooled and enriched by binding to a column conjugated with the phosphorylated peptide before being depleted by running over a second column conjugated with the non-phosphorylated peptide. Phospho-peptide specificity (>750 fold at 0.1 μg/mL) was confirmed by ELISA using standard protocols.
3. Results
3.1. Src inhibits autophagy and AMPK signalling in adherent cells
Cell detachment has been reported to trigger autophagy in multiple cell types (6, 7). Here we used mouse embryonic fibroblasts (MEFs) to study autophagy induction by ECM deprivation. In cells that had been detached and placed in suspension, autophagy was induced as evidenced by elevated conversion of non-lipidated LC3-I to lipidated LC3-II (37) (Fig. 1A upper panel, lane 2)._LC3-II levels were further increased in the presence of Bafilomycin A1, indicating that autophagic flux was truly promoted upon cell detachment (Fig. 1A upper panel, compare lanes 2 and 9, p <0.01, n = 3). This induction of autophagy correlated with activation of AMPK, as measured by phosphorylation on T172, and with suppression of integrin signaling, as indicated by the loss of FAK phosphorylation on Y576 (a known Src phosphorylation site (38)) (Fig. 1A lower panel, lane 2). When suspended cells were re-attached to fibronectin-coated dishes, integrin signaling was re-activated, as evidenced by increased FAK-Y576 phosphorylation, while LC3-II levels and AMPK-T172 phosphorylation were reduced back to basal levels (Fig. 1A, lanes 3-7). Thus, these results demonstrate a correlation between activation of integrin/ Src/ FAK signaling and suppression of AMPK signaling and autophagy.
Next we compared detachment-induced signaling events in wild-type (WT) and AMPK-α1/2-DKO MEFs (which lack both AMPK-α1 and AMPK-α2 expression (30)). As shown in Fig. 1B, neither AMPK expression nor AMPK-T172 phosphorylation was detected in AMPK-α1/2-DKO MEFs, and the AMPK downstream substrates ULK1 (22, 39) and Acetyl-CoA carboxylase (ACC) were not phosphorylated. Moreover, suspension-elicited increases in formation of the autophagy activation marker LC3-II were greatly reduced in the cells that lacked AMPK-α1/2, indicating that AMPK plays a key role in autophagy induction following cell detachment. In this context, it is important to note that AMPK has been shown to negatively regulate the expression of the cytoskeletal scaffold proteins tensin1 and tensin3, which bind to the β1 integrin and support integrin activation (40). Thus, AMPK loss in MEFs activates integrins, which in feed-forward manner may further suppress autophagy in this model system.
When suspended wild-type MEF cells were treated with the Src kinase inhibitor Bosutinib, both LC3-II and AMPK-T172 phosphorylation levels were further elevated (Fig. 1C, compare lanes 2 and 4, p <0.01, n = 3). Similarly, Bosutinib treatment prevented down-regulation of LC3-II and AMPK-T172 phosphorylation in cells that had been re-attached on fibronectin (Fig. 1C, lanes 11-13). As expected, inhibition of Src by Bosutinib also suppressed FAK phosphorylation on Y576 (Fig. 1C lower panel, lanes 11-13). These findings support the notion that Src activity is required for integrin-mediated downregulation of AMPK signaling and autophagy upon cell re-attachment.
In MEF cells genetically deficient in the three Src family kinases, Src, Yes and Fyn (SYF cells (28)), Src/ FAK signaling is undetectable as reflected by lack of FAK-Y576 phosphorylation (Fig. 1D, lanes 1-7). Cell detachment inhibited Src/ FAK signaling (Fig. 1D, lane 9) in Src re-expressing cells (Src++), resulting in elevated LC3-II levels that were comparable to those observed in SYF cells (Fig. 1D, compare lane 2 to lane 9). When suspended cells were re-attached to fibronectin, Src/FAK pathway was re-activated in Src++ cells (as measured by FAK-Y576 phosphorylation) (Fig. 1D, lanes 10-14), but not in SYF cells (Fig. 1D, lanes 3-7). Consistent with this, we observed that autophagy induction (as measured by LC3-II levels) and AMPK activation (as measured by AMPK-T172 phosphorylation) were enhanced and prolonged during the experimental “detachment/ re-attachment” process in SYF cells compared to Src++ cells (Fig. 1D, p <0.01, n = 3). These results add to the conclusion that adhesion mediated Src activation suppresses AMPK signaling and autophagy.
To confirm these observations, we employed a chemical biology approach. Consistent results were obtained when adherent wild-type MEFs were treated with multiple Src inhibitors (Bosutinib, Dasatinib and Saracatinib) (Fig. 1E). These inhibitors effectively blocked Src signaling (as demonstrated by reduced FAK-Y576 phosphorylation), and they enhanced AMPK activation (as measured by AMPK-T172 phosphorylation), as well as autophagy activation (as measured by elevated LC3-II levels) in a concentration-dependent manner.
To further explore the role of Src in AMPK activation, we next utilized several diverse chemical compounds to activate AMPK. The anti-diabetic biguanides Metformin (Met) and Phenformin (Phen) inhibit mitochondrial ATP production and thus activate AMPK indirectly by altering the cellular AMP:ATP ratio (41). The AMP analog 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) is a direct activator of AMPK (42, 43), and A-769662, a specific and direct activator of AMPK, mimics the effects of AMP on AMPK on both allosteric activation and inhibition of dephosphorylation. We treated adherent SYF and Src++ cells with each of these compounds and found that these agents elicited much greater AMPK-T172 and ULK1-S555 phosphorylation in SYF cells compared to the Src-expressing Src++ cells (Fig. 1F). These findings further support the notion that Src has an inhibitory effect on AMPK activation, directly at the level of AMPK.
3.2. FAK regulates autophagy and AMPK signaling
FAK is an important intracellular tyrosine kinase acting downstream of integrin-ECM engagement. Upon cell attachment, FAK is recruited to focal adhesions, and its activation and auto-phosphorylation generates a Src binding site that allows additional Src activation and phosphorylation events to occur (44). To determine if Src may act, at least in part, through FAK in suppressing AMPK activation and autophagy upon cell attachment, we employed FAK null MEFs (FAK−/− cells). Cell detachment was found to activate autophagy in the FAK−/− and FAK+/+ cells approximately to the same extent, as measured by LC3-II levels (Fig. 2A upper panel, compare lanes 2 and 8). Following re-attachment of cells to fibronectin, however, autophagy was rapidly suppressed in FAK+/+ cells, while this suppression was significantly perturbed in FAK−/− cells (Fig. 2A upper panel). Similarly, AMPK phosphorylation on T172 was slightly prolonged in FAK−/− cells compared to FAK+/+ cells after re-attachment (Fig. 2A lower panel).
We additionally found that the AMPK activating compounds AICAR and Metformin (Met) induce higher levels of both AMPK-T172 and ULK1-S555 phosphorylation in FAK−/− cells compared to FAK+/+ cells (Fig. 2B, compare lanes 2 and 5, and lanes 3 and 6). Taken together, these results suggest that FAK also plays a role in autophagy regulation, presumably acting through the AMPK/ ULK1 pathway. The effect of FAK, however, appears to be less pronounced than that of Src. Indeed, although Src and FAK can positively regulate each other, there are other mechanisms to activate them as well (11, 45). Additionally, FAK −/− cells are known to express the related kinase Pyk2 at an elevated level, which can compensate for some FAK functions, and may also contribute to Src signaling (46, 47).
In a reverse experiment, we over-expressed Src or FAK in 293T cells (Fig. 2C). Overexpression of wild-type (WT) Src or FAK, but not of catalytically inactive (kinase-dead, KD) forms of Src or FAK, resulted in suppression of AMPK-T172 phosphorylation (Fig. 2C, compare lanes 2 and 3, and lanes 4 and 6). In addition, the mutant FAK-Y397F protein lacking the auto-phosphorylation and Src-binding site was less effective in suppressing AMPK-T172 phosphorylation compared to wild-type FAK (Fig. 2C, compare lanes 4 and 5). These data are consistent with the notion that the kinase activities of Src and FAK (and, presumably, the formation of the dual Src/ FAK kinase complex) mediate the suppression of AMPK signaling.
3.3. Src/ FAK signaling suppresses glucose starvation-induced autophagy and AMPK activation
Glucose deprivation is known to trigger activation of the AMPK and autophagy pathways (22, 48). To determine if the Src/ FAK complex regulates autophagy and AMPK signaling induced by nutrient deprivation we next studied the effects of glucose starvation. We found that glucose deprivation induced stronger AMPK-T172 phosphorylation in cells lacking Src or FAK, as compared to cells expressing these proteins [SYF vs. Src++ (Fig. 3A), and FAK−/− vs. FAK+/+ MEFs (Fig. 3B)]. Also, the phosphorylation levels of AMPK downstream targets ACC and ULK1, were significantly higher in glucose-starved Src-deficient cells compared to Src++ cells (Fig. 3A). Similar results were observed when glucose-starved FAK−/− and FAK+/+ cells were compared to each other (Fig. 3B). In a like manner, the autophagy activation marker LC3-II was significantly elevated in glucose-deprived SYF (p <0.01, n = 3) and FAK−/− MEFs (p <0.05, n = 3) as compared to cells expressing Src or FAK, respectively (Fig. 3, C and D). These observations were confirmed by microscopic analyses of autophagosome formation as indicated by increased LC3 puncta. Specifically, cells lacking Src or FAK showed increased autophagosome formation in response to glucose deprivation compared to cells expressing these kinases (Fig. 3, E-H).
3.4. Src phosphorylates AMPK-α subunit in vivo and in vitro on Y179
Src is a well characterized tyrosine kinase that phosphorylates multiple substrates and thus regulates numerous signaling pathways. Previously, the Src kinase family member Fyn was reported to phosphorylate AMPK-α2 at site Y436 both in vitro and in vivo in skeletal muscle, resulting in inhibition of TNFα-induced activation of AMPK and autophagy (12). Here we show that co-expression of a constitutively active form of Src (Src-Y527F) and AMPK-α in 293T cells results in robust tyrosine phosphorylation of AMPK (Fig. 4A, lanes 2 and 4). Furthermore, in an in vitro kinase reaction we demonstrate that incubation of purified recombinant Src and the AMPK heterotrimeric complex results in tyrosine phosphorylation of the AMPK-α subunit (as well as of the β and γ subunits), thus demonstrating that AMPK is a direct target for Src kinase activity (Fig. 4B).
In order to ascertain the phosphorylation site(s) on AMPK upon exposure to Src, we subjected the immunoprecipitated AMPK-α co-expressed with Src-Y527F to a mass spectrometric analysis as described in the Material and Methods. Results from these studies showed that one of the Src phosphorylation sites on the AMPK-α subunit is Y179 (Supplementary Fig. 2). Y179 resides in the activation loop of AMPK, and in close proximity to the regulatory amino acid T172, phosphorylation of which is crucial for AMPK activation (49). We generated two AMPK-α mutants to study the function of the Y179 site in AMPK signalling: a phospho-mimetic form (tyrosine residue changed to glutamic acid, Y179E) and an uncharged non-phosphorylatable form (tyrosine changed to phenylalanine, Y179F). Both of these mutant forms of AMPK demonstrated reduced overall tyrosine phosphorylation levels (down to 40% and 30% respectively, p<0.001, n=4) compared to wild-type AMPK when co-expressed with active Src (Fig. 4C). Residual tyrosine phosphorylation of the AMPK-Y179E and AMPK-Y179F proteins observed is consistent with our mass spectrometry findings, in that additional phosphorylation sites appear be present in AMPK (data not shown). Of note, we also observed co-immunoprecipitation of AMPK, especially the AMPK-Y179E mutant, with the Src-Y527F protein (Fig. 4C), consistent with active Src protein binding irreversibly to its (non-hydrolyzable) pseudo-product.
To further evaluate Src phosphorylation of AMPK on Y179, we developed a custom antibody against AMPK-phospho-Y179. As shown in Fig. 4D, the phosphorylation level of AMPK-Y179 as observed by immunofluorescence in Src null (SYF) cells was very low. This phosphorylation was substantially increased in cells where Src was re-expressed (Src++ cells). Treatment of the Src++ cells with the Src inhibitor Bosutinib in turn suppressed AMPK-Y179 phosphorylation (Fig. 4, D and E).
3.5. Src phosphorylation of AMPK-Y179 inhibits AMPK-T172 phosphorylation and AMPK activation
Our results so far demonstrate that Src activity suppresses autophagy and AMPK signaling, as measured by decreased LC3-II, reduced AMPK-T172 phosphorylation and by reduced phosphorylation of AMPK substrates ACC and ULK1. We further showed that Src can directly phosphorylate AMPK on Y179. Next, we investigated the possible connection between Src-mediated phosphorylation at the AMPK-Y179 site and suppression of AMPK signaling.
Upon expression in 293T cells, the AMPK-Y179F protein demonstrated a slightly increased phosphorylation on AMPK-T172 compared to wild-type AMPK (Fig. 5A, compare lanes 1 and 9). In contrast, no AMPK-T172 phosphorylation was detected on the phospho-mimetic AMPK-Y179E protein (Fig. 5A, lane 5). These results suggest that phosphorylation on AMPK-Y179 (as mimicked by the Y179E mutation) has a negative effect on AMPK-T172 phosphorylation status. Consistent with this notion, we found that treatment of the AMPK-transfected cells with either of the Src kinase inhibitors Bosutinib, Dasatinib or Saracatinib, significantly enhanced AMPK-T172 phosphorylation on wild-type AMPK (compare lane 1 to lanes 2-4), but not on the AMPK-Y179F (compare lane 9 to lanes 10-12) or AMPK-Y179E mutant proteins (compare lane 5 to lanes 6-8).
We next examined AMPK-T172 phosphorylation on exogenously expressed wild-type AMPK, AMPK-Y179F and AMPK-Y179E proteins in cellular “detachment/ re-attachment” experiments in order to first perturb, and then activate Src signaling (Fig. 5B). As observed previously, cell detachment-induced activation of wild-type AMPK (as measured by AMPK-T172 phosphorylation) was reduced back to basal levels upon cell re-attachment on fibronectin (Fig. 5B, lanes 3-5). For the AMPK-Y179F mutant, AMPK-T172 phosphorylation was also induced upon cell detachment but remained elevated compared to the wild-type AMPK protein upon cell re-attachment [Fig. 5B, compare lanes 11 and 5; 58% vs 23% of the detached sample, respectively; p<0.001, (n=3)]. Once again, the phospho-mimetic mutant AMPK-Y179E was devoid of any AMPK-T172 phosphorylation (Fig. 5B, lanes 6-8) consistent with our previous observations that Src phosphorylation of Y179 inhibits phosphorylation of AMPK at the T172 site.
To eliminate the potential masking effect of endogenous AMPK, we next employed AMPK-α null 293A cells (AMPK-α-1/2-DKO cells) (31), and these cells were transfected to express either wild-type AMPK or the AMPK-Y179 mutants. No endogenous AMPK expression or phosphorylation was detected in the DKO cells (Fig. 5C). When re-expressed in these cells, wild-type AMPK showed strong phosphorylation on T172, and promoted robust phosphorylation of downstream substrates ULK1 and ACC (Fig. 5D). By contrast, the phospho-mimetic AMPK-Y179E mutant was not phosphorylated on T172, and it failed to activate downstream signaling events, as measured by ACC and ULK1 phosphorylation (Fig. 5D, compare lanes 1 and 3). While the tyrosine-phosphorylation deficient AMPK-Y179F mutant showed (as we had observed above) a similar or slightly elevated level of phosphorylation on AMPK-T172 compared to wild-type AMPK, its ability to activate downstream signaling was nevertheless slightly reduced compared to wild-type AMPK, as demonstrated by low phosphorylation levels of ULK1 and ACC (Fig. 5D, compare lanes 2 and 4). We surmise that the Y179F mutation has a limited effect on the structure of the activation loop and its ability to be phosphorylated at T172; but, given the proximity of the mutation to the active site (in the context of the closed kinase conformation), the mutation leads to a more significant reduction in AMPK catalytic activity. This notion is compatible with a previous report that cellular AMPK signaling can be independent of AMPK-T172 phosphorylation (50).
We subsequently treated the various AMPK-transfected cells with A-769662, Phenformin, or AICAR to activate AMPK. Consistent with the results obtained above, these agents elicited a stronger AMPK-T172 phosphorylation on the AMPK-Y179F mutant compared to wild-type AMPK. We again observed that the ability of the AMPK-Y179F mutant to induce phosphorylation of the downstream substrates ULK1 and ACC was somewhat reduced compared to wild-type AMPK (see discussion above) (Fig. 5E, compare lanes 3-5 and 11-13). As expected, the response on the phosphomimetic mutant AMPK-Y179E was barely detectable. These findings are consistent with the notion that Y179 phosphorylation by Src has an inhibitory effect on AMPK activation, directly at the level of AMPK.
Since AMPK is a heterotrimeric enzyme, we also examined if Y179 perturbation affected assembly of the AMPK complex, potentially resulting in modified downstream signaling. As shown in Fig. 5F, levels of AMPK heterotrimeric complex assembly were not affected by Y179 mutations when all the three AMPK subunits were co-expressed. This suggests that AMPK activity regulated by tyrosine phosphorylation of Y179 on AMPK-α subunit is primarily mediated via perturbation of the T172 phosphorylation site.
3.6. Simultaneous inhibition of Src and autophagy synergizes to kill cancer cells
Aberrantly high Src activity has been noted in diverse cancers, and it is thought to play a role in cancer progression. Thus Src itself is an important potential target in cancer treatment (51). MMTV-PyMT transgenic mouse is one of the most studied models of breast cancer, and interaction of the polyomavirus middle T-antigen (PyMT) with the Src family kinases is a key for its transforming function (52). In order to gain preliminary insight into the potential role of autophagy regulation by Src in tumorigenesis, we decided to employ the PyMT-driven murine mammary Py230 tumor cell model (53).
We first established that treatment with several Src inhibitors suppressed the proliferation of murine mammary Py230 tumor cells in a dose-dependent manner (Fig. 6, A and B). Consistent with our studies above, we also observed an increase in autophagy, as measured by LC3-II levels, in the Bosutinib-treated Py230 cells (Fig. 6C). We then co-treated the cells with hydroxychloroquine (HCQ), which is known to prevent autophagy by blocking autophagosome fusion and degradation (54). Further increase in LC3-II levels was observed in the presence of HCQ, indicating that blockade of Src signaling in these cells leads to an increased autophagic flux (Fig. 6C).
It has been recently shown that HCQ synergistically kills certain cancer cells when combined with kinase inhibitors, including MEK1/2 inhibition (55). We reasoned that combining Src inhibitors similarly with HCQ could result in synergistic cancer cell killing. As shown in Figs. 6A and 6B, combination treatment with HCQ significantly sensitized Py230 cells to Src inhibition by both Bosutinib and Saracatinib. The synergistic effect of Bosutinib and HCQ combination on cell apoptosis was confirmed by immunoblot, with the detected cleavage of the apoptosis markers caspase-3 and PARP shown in Fig. 6D. Notably, we also observed that cell detachment and HCQ treatment synergized to induce cell killing, as shown in Fig. 6E.
To further confirm these findings, we utilized genetic knockout model systems. When treated with Src inhibitors Bosutinib or Dasatinib, autophagy-deficient Atg5−/− and Atg7−/− MEFs exhibited increased cleavage of PARP and caspase-3 (Fig. 6, F and G), and higher levels of caspases-3/7 activity (Fig. 6, H and I) as compared to the WT MEF controls, indicating that Src inhibitor-induced apoptosis was enhanced in these cells when autophagy was impaired.
Taken together, our results suggest that suppression of Src activity (either upon treatment of cells with Src inhibitors or upon cell detachment) leads to autophagy activation, which in turn plays a protective role in Src inhibitor-induced apoptosis and detachment-induced cell death (anoikis). Combining Src inhibition and autophagy inhibition therefore appears to result in synergistic cell killing, and additional studies are warranted to explore the potential therapeutic utility of this observation further.
4. Discussion
In this study, we show that the AMPK-autophagy pathway is activated upon cell detachment and becomes rapidly inactivated upon fibronectin-mediated cell re-attachment. Similarly, chemical inhibition of the ECM-integrin signaling mediators Src and FAK also results in autophagy induction. Furthermore, cells genetically deficient for Src or FAK show increased autophagy in response to ECM or glucose deprivation. Thus, we provide a substantial body of evidence that adhesion-mediated signaling via Src and FAK is a potent and relevant autophagy suppressive signal. Furthermore, we demonstrate that Src directly phosphorylates AMPK on a novel tyrosine residue (Y179), rendering AMPK inactive (and/ or un-activatable). We further show that this post-translational modification plays an important role in Src-mediated suppression of AMPK activity and autophagy. Inhibition of Src activity, in turn, was shown to result in activation of autophagy in various cell types, including cancer cells. Induction of autophagy is known to be an important protective mechanism for cell survival, and as a corollary, inhibition of this protective effect could be exploited in cancer therapeutics. Simultaneous use of Src inhibitors and inhibitors of autophagy could therefore provide a potentially useful therapeutic intervention point in selected cancers (55).
Notably, autophagy in cancer cells can be a double-edged sword due to its possible opposing functions on primary tumor growth versus metastasis. Although autophagy is typically considered to promote metastasis by enhancing tumor cell fitness (56), Marsh et al. (57) uncovered a novel role for autophagy in suppressing metastasis by degradation of the autophagy cargo receptor, Neighbor to BRCA1 (NBR1); and Flynn et al. (58) reported that autophagy inhibition elicits metastatic dormancy from breast cancer stem cells. These findings highlight the context-dependent role of autophagy, and more importantly, they suggest that Src inhibition-mediated autophagy induction could contribute to cancer treatment under certain circumstances. Thus, a better understanding of these underlying mechanisms would be of great importance when assessing Src inhibitors’ application in cancer treatment.
Matrix attachment leads to Src/ FAK pathway activation through integrin signaling. We demonstrated that Src interacts with the AMPK α-subunit and phosphorylates it on the activation loop at Y179 (Figs. 4, A-C and Supplementary Fig. 3). Our data further suggest that AMPK-Y179 plays an important role in the regulation of AMPK activation and therefore downstream signaling. A phospho-mimetic mutant of AMPK (Y179E) is completely devoid of T172 phosphorylation and fails to activate downstream signaling pathways. The non-phosphorylatable AMPK mutant (Y179F) shows higher baseline phosphorylation on the activation site of T172 and is “resistant” to further activation upon inhibition of Src activity (Fig. 5A). In reverse, this non-inhibitable mutant retains its suspension-induced activation for a prolonged time period after cell re-attachment compared to wild-type AMPK (Fig. 5B). Since Y179 is within the AMPK-α activation loop, one would assume that Y179 plays an important role in maintaining the structural integrity of the environment adjacent to the active site (T172). In the closed, active structure, Y179 is almost entirely buried in a hydrophobic and acidic environment, and phosphorylation of Y179 should strongly promote (or maintain) the open, inactive conformation (Supplementary Fig. 3). The state of activation of AMPK-α (T172 phosphorylation) is regulated through the balance between phosphorylation by upstream kinase(s) and dephosphorylation by phosphatase(s). Src modulation of AMPK activity could be through regulation of either or both of these events, but culminating at T172 phosphorylation status. That is, phosphorylation of AMPK-α-Y179 by Src may induce a conformational change resulting in a vulnerable exposure to a phosphatase, and/ or impaired access for upstream kinases of AMPK-T172, such as LKB1.
As the phospho-mimetic mutant Y179E has lower levels of pan-tyrosine phosphorylation, with concomitant loss of T172 phosphorylation, it could be predicted to migrate faster than wild-type AMPK under SDS-gel electrophoresis. However, we found that the Y179E mutant actually showed reduced migration (Fig. 4C and Fig 5, A, B, D, E and F); this could be a consequence of the default “open” conformation expected for the kinase domain of this mutant (which may affect how it binds to SDS), and/or of additional post-translational modification(s).
Our data also revealed an association of Src with AMPK in co-immunoprecipitation experiments, and this interaction is negatively affected by AMPK activity since Src preferentially binds to the inactive phospho-mimetic Y179E mutant (Fig. 4C). These data suggest Src may become trapped in a complex with inactive AMPK where the glutamate of AMPK-Y179E inhibits Src by acting as a non-hydrolyzable product mimetic (59). It may also suggest that Src could be an AMPK substrate to be phosphorylated before Src is targeted to autophagosomes for degradation (60). In this case mutual phosphorylation of the two kinases may take place.
Various stress signals can induce autophagy, such as starvation, oxidative stress, protein aggregation, hypoxia, and endoplasmic reticulum (ER) stress (61). In this study, we found that Src plays an inhibitory role on AMPK activation in various signaling conditions. Thus, Src is not only required for attachment-mediated AMPK suppression (Fig. 1, C and E), but also has an inhibitory effect on the induction of AMPK activation by the agonists A-769662, AICAR and Phenformin (Fig. 1F), as well as by glucose deprivation (Fig. 3A).
Glucose deprivation promotes formation of a complex containing the large adapter protein Axin, and kinases LKB1 and AMPK. The resulting proximity of LKB1 and AMPK leads to AMPK-T172 phosphorylation and thus activation of the latter (62, 63). It remains to be determined if Y179 phosphorylation has an inhibitory effect on LKB1-AMPK interaction in these signaling conditions. AMP in turn binds to the AMPK γ-subunit to trigger upstream kinase phosphorylation on AMPK-T172 and we demonstrated that the AMP analogue AICAR induces stronger AMPK activation in the absence of Src (Fig. 1F) or FAK (Fig. 2B), as well as on the phosphorylation-resistant AMPK-Y179F mutant (Fig. 5E). The exact mechanism of how Src/ FAK suppression of direct AMPK activation by AMP occurs remains to be investigated, but our results suggest that Y179 phosphorylation by Src and subsequent effects on T172 phosphorylation and AMPK activation play a major role.
Src activity is frequently upregulated in various cancers and inhibition of Src is an approach under investigation for anti-cancer therapy. Here, all the three tested Src inhibitors (Bosutinib, Dasatinib and Saracatinib) showed similar effects on release of Src inhibition of AMPK and autophagy. In a recent publication (64), another Src kinase inhibitor (SU6656) was found to promote AMPK-T172 phosphorylation and activation by the upstream kinase LKB1. On one hand, these authors demonstrated that SU6656 directly binds to the AMPK catalytic site, resulting in AMPK inhibition. On the other hand, they showed that such binding induced a conformation change that promoted AMPK-T172 phosphorylation and activation by LKB1. In the same study, the multi-kinase inhibitor Sorafenib (which also inhibits Src) was found to activate AMPK by inhibiting mitochondrial metabolism, which increased the cellular AMP: ATP and ADP:ATP ratios. Indeed, we found that SU6656 and Sorafenib induce AMPK activation in SYF cells lacking Src (data not shown). Thus, inhibitors such as SU6656 and Sorafenib can have an effect on the regulation of AMPK activation in both Src-dependent and independent manner.
In our proposed model (Fig. 7), upon cell attachment, interaction of AMPK with activated Src/ FAK complex leads to tyrosine phosphorylation of AMPK-α on Y179 resulting in AMPK inactivation. Upon cell detachment, the Src/ FAK signaling pathway becomes inactive, and AMPK is dephosphorylated on Y179. AMPK-T172 in the activation loop becomes phosphorylated, resulting in activation of AMPK and downstream signaling events, such as autophagy. Induction of autophagy in turn may play a crucial role for protecting cells from detachment-induced cell death (anoikis). In addition, the mammalian target of rapamycin complex 1 (mTORC1) can phosphorylate ULK1 and prevent its association and activation by AMPK (22, 65). Recently, mTORC1 has been reported to be activated in focal adhesions, and focal adhesions in turn are required for mTORC1 signaling (66). Thus, cell detachment-mediated disruption of focal adhesions can contribute to autophagy induction. Furthermore, mTORC1 could be involved in adhesion-mediated autophagy regulation at the level of ULK1. Importantly, Src can regulate mTORC1 (67), suggesting Src may play a central role in regulation of autophagy activation at multiple levels. As such, our findings reveal a link between the Src/ FAK complex and AMPK/ autophagy regulation, which may play an important role in the maintenance of normal cellular homeostasis and tumor progression (Fig. 7).
Figure 7. Schematic representation of AMPK phosphorylation regulated by Src.

Upon cell attachment, activated Src/ FAK complex interacts with AMPK, resulting in tyrosine phosphorylation on AMPK-α-Y179 and AMPK inactivation. Upon cell detachment from ECM, the Src/ FAK complex is inactivated, AMPK is dephosphorylated on Y179, and AMPK becomes activated by phosphorylation on the activation loop at T172. Active AMPK signaling in turn leads to ULK-autophagy activation.
Supplementary Material
Highlights.
Src inhibition promotes autophagy and AMPK signaling.
Src phosphorylates AMPK-α subunit on Y179 to suppress downstream signaling.
Simultaneous inhibition of Src and autophagy synergizes to kill cancer cells.
Acknowledgements:
We thank Dr. Ericka Eggleston for technical help, and Sanford Burnham Prebys’ Proteomics Core facility for assistance on mass spectrometry analysis. The Core is supported by SBP’s NCI Cancer Center Support Grant P30 CA030199.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The authors declare no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References
- 1.Hynes RO, Integrins: A family of cell surface receptors. Cell 48, 549–554 (1987). [DOI] [PubMed] [Google Scholar]
- 2.Miranti CK, Brugge JS, Sensing the environment: a historical perspective on integrin signal transduction. Nature Cell Biology 4, E83 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Frisch S, Francis H, Disruption of epithelial cell-matrix interactions induces apoptosis. The Journal of Cell Biology 124, 619–626 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frisch SM, Ruoslahti E, Integrins and anoikis. Current Opinion in Cell Biology 9, 701–706 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Gilmore AP, Anoikis. Cell Death Differ 12, 1473–1477 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Fung C, Lock R, Gao S, Salas E, Debnath J, Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Molecular Biology of the Cell 19, 797–806 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen N, Debnath J, IκB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy 9, 1214–1227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Debnath J, Detachment-induced autophagy during anoikis and lumen formation in epithelial acini. Autophagy 4, 351–353 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Vlahakis A, Debnath J, The Interconnections between Autophagy and Integrin-Mediated Cell Adhesion. Journal of Molecular Biology 429, 515–530 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burridge K, Fath K, Kelly T, Nuckolls G, Turner C, Focal Adhesions: Transmembrane Junctions Between the Extracellular Matrix and the Cytoskeleton. Annual Review of Cell Biology 4, 487–525 (1988). [DOI] [PubMed] [Google Scholar]
- 11.Zhao X, Guan J-L, Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Advanced Drug Delivery Reviews 63, 610–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamada E et al. , Fyn phosphorylates AMPK to inhibit AMPK activity and AMP-dependent activation of autophagy. Oncotarget 7, 74612–74629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharifi Marina N. et al. , Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Reports 15, 1660–1672 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao M, Vuori K, The docking protein p130Cas regulates cell sensitivity to proteasome inhibition. BMC Biology 9, 73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen G-C et al. , Genetic interactions between Drosophila melanogaster Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy 4, 37–45 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Yamada E, Bastie CC, Disruption of Fyn SH3 Domain Interaction with a Proline-Rich Motif in Liver Kinase B1 Results in Activation of AMP-Activated Protein Kinase. PLOS ONE 9, e89604 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada E, Pessin JE, Kurland IJ, Schwartz GJ, Bastie CC, Fyn-Dependent Regulation of Energy Expenditure and Body Weight Is Mediated by Tyrosine Phosphorylation of LKB1. Cell Metabolism 11, 113–124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hawley SA et al. , Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. Journal of Biology 2, 28 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woods A et al. , LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Current Biology 13, 2004–2008 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Shaw RJ et al. , The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proceedings of the National Academy of Sciences of the United States of America 101, 3329–3335 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardie DG, Alessi DR, LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biology 11, 36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Kundu M, Viollet B, Guan K-L, AMPK and mTOR regulate autophagy through direct phosphorylation of ULK1. Nature Cell Biology 13, 132 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egan DF et al. , Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 331, 456–461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roskoski R Jr., Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res 94, 9–25 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Dawson JC, Serrels A, Stupack DG, Schlaepfer DD, Frame MC, Targeting FAK in anticancer combination therapies. Nature Reviews Cancer 21, 313–324 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong KA, Lodish HF, A Revised Model for AMP-activated Protein Kinase Structure: The α-subunit binds to both the β- and γ-subunits although there is no direct binding between the β- and γ-subunits. Journal of Biological Chemistry 281, 36434–36442 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Zhao M, Finlay D, Zharkikh I, Vuori K, Novel Role of Src in Priming Pyk2 Phosphorylation. PLOS ONE 11, e0149231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P, Src family kinases are required for integrin but not PDGFR signal transduction. The EMBO Journal 18, 2459–2471 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ilić D et al. , Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377, 539 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Laderoute KR et al. , 5′-AMP-Activated Protein Kinase (AMPK) Is Induced by Low-Oxygen and Glucose Deprivation Conditions Found in Solid-Tumor Microenvironments. Molecular and Cellular Biology 26, 5336–5347 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konagaya Y et al. , A Highly Sensitive FRET Biosensor for AMPK Exhibits Heterogeneous AMPK Responses among Cells and Organs. Cell Reports 21, 2628–2638 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Kuma A et al. , The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Komatsu M et al. , Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. Journal of Cell Biology 169, 425–434 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malhotra R, Warne JP, Salas E, Xu AW, Debnath J, Loss of Atg12, but not Atg5, in pro-opiomelanocortin neurons exacerbates diet-induced obesity. Autophagy 11, 145–154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brudvig JJ et al. , MARCKS regulates neuritogenesis and interacts with a CDC42 signaling network. Scientific Reports 8, 13278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bell JB et al. , Differential Response of Glioma Stem Cells to Arsenic Trioxide Therapy Is Regulated by MNK1 and mRNA Translation. Molecular Cancer Research 16, 32–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G, Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8, 741–752 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Calalb MB, Polte TR, Hanks SK, Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Molecular and Cellular Biology 15, 954–963 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bach M, Larance M, James DE, Ramm G, The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochemical Journal 440, 283–291 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Georgiadou M et al. , AMPK negatively regulates tensin-dependent integrin activity. Journal of Cell Biology 216, 1107–1121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hawley SA et al. , Use of Cells Expressing γ Subunit Variants to Identify Diverse Mechanisms of AMPK Activation. Cell Metabolism 11, 554–565 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sullivan JE et al. , Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Letters 353, 33–36 (1994). [DOI] [PubMed] [Google Scholar]
- 43.Corton JM, Gillespie JG, Hawley SA, Hardie DG, 5-Aminoimidazole-4-Carboxamide Ribonucleoside. European Journal of Biochemistry 229, 558–565 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Schaller MD et al. , Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Molecular and Cellular Biology 14, 1680–1688 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitra SK, Schlaepfer DD, Integrin-regulated FAK–Src signaling in normal and cancer cells. Current Opinion in Cell Biology 18, 516–523 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Weis SM et al. , Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. Journal of Cell Biology 181, 43–50 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan H, Guan J-L, Compensatory Function of Pyk2 Protein in the Promotion of Focal Adhesion Kinase (FAK)-null Mammary Cancer Stem Cell Tumorigenicity and Metastatic Activity. Journal of Biological Chemistry 286, 18573–18582 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.SALT IP, JOHNSON G, ASHCROFT SJH, HARDIE DG, AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic β cells, and may regulate insulin release. Biochemical Journal 335, 533–539 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hawley SA et al. , Characterization of the AMP-activated Protein Kinase Kinase from Rat Liver and Identification of Threonine 172 as the Major Site at Which It Phosphorylates AMP-activated Protein Kinase. Journal of Biological Chemistry 271, 27879–27887 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Dite TA et al. , The autophagy initiator ULK1 sensitizes AMPK to allosteric drugs. Nature Communications 8, 571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeatman TJ, A renaissance for SRC. Nature Reviews Cancer 4, 470–480 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Dilworth SM et al. , Transformation by polyoma virus middle T-antigen involves the binding and tyrosine phosphorylation of Shc. Nature 367, 87–90 (1994). [DOI] [PubMed] [Google Scholar]
- 53.Guy CT, Cardiff RD, Muller WJ, Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Molecular and Cellular Biology 12, 954–961 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solitro AR, MacKeigan JP, Leaving the lysosome behind: novel developments in autophagy inhibition. Future Medicinal Chemistry 8, 73–86 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kinsey CG et al. , Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nature Medicine 25, 620–627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kenific CM, Thorburn A, Debnath J, Autophagy and metastasis: another double-edged sword. Current Opinion in Cell Biology 22, 241–245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marsh T et al. , Autophagic Degradation of NBR1 Restricts Metastatic Outgrowth during Mammary Tumor Progression. Developmental Cell 52, 591–604.e596 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.La Belle Flynn A et al. , Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nature Communications 10, 3668 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flint AJ, Tiganis T, Barford D, Tonks NK, Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proceedings of the National Academy of Sciences 94, 1680–1685 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sandilands E et al. , Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 14, 51–60 (2012). [DOI] [PubMed] [Google Scholar]
- 61.He C, Klionsky DJ, Regulation Mechanisms and Signaling Pathways of Autophagy. Annual review of genetics 43, 67–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang C-S et al. , Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hardie DG, Lin S-C, AMP-activated protein kinase – not just an energy sensor. F1000Research 6, 1724 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ross FA et al. , Mechanisms of Paradoxical Activation of AMPK by the Kinase Inhibitors SU6656 and Sorafenib. Cell Chemical Biology 24, 813–824.e814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hosokawa N et al. , Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Molecular Biology of the Cell 20, 1981–1991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rabanal-Ruiz Y et al. , mTORC1 activity is supported by spatial association with focal adhesions. Journal of Cell Biology 220, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pal R et al. , Src regulates amino acid-mediated mTORC1 activation by disrupting GATOR1-Rag GTPase interaction. Nature Communications 9, 4351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.