

Abstract

Many transition-metal complexes MLn decompose diazo compounds N2=CR1R2 generating metal-carbenes LnM=CR1R2 which transfer the carbene group to other substrates, constituting an important tool in organic synthesis. All previous reports have shown that the CR1R2 fragment at the metal-carbene remains intact from the parent diazo compound. Herein we report the detection and isolation of a monosubstituted copper carbene where the CR1R2 ligand has undergone a modification from the initial diazo reagent. When TpMsCu(THF) (TpMs = hydrotris(3-mesityl)pyrazolylborate ligand) was reacted with N,N-diethyl diazoacetamide [N2=C(H)(CONEt2)], the stable copper carbene TpMsCu=C(H)(NEt2) was isolated, resulting from a decarbonylation process, with carbon monoxide being trapped as TpMsCu(CO). The simultaneous observation of products derived from the intramolecular carbene insertion reaction into C–H bonds demonstrates that the expected TpMsCu=C(H)(CONEt2) complex is also formed. Experimental data, DFT calculations, and microkinetic models allow us to propose that the latter undergoes CO loss en route to the former.
Introduction
More than a century after Buchner postulated the existence of carbene CR2 groups during the thermal decomposition of ethyl diazoacetate,1 the catalytic transfer of such moiety from diazo compounds yet constitutes an area of continuous growth.2,3 Such process consists of the metal-induced decomposition of the diazo reagent in a process in which molecular N2 is extruded and a metallocarbene intermediate MC is formed (Scheme 1a).4 This species is electrophilic5 in nature and reacts with available nucleophiles transferring the carbene group, thus liberating the metal to continue the catalytic cycle. This strategy has been successfully employed in the addition of carbene groups to unsaturated bonds or in its insertion into C–H or other C–X bonds, both intra- and intermolecularly.6−8
Scheme 1. Metal-Catalyzed Carbene Transfer from Diazo Compounds and the Formation of Metalocarbene Intermediates.

With the appropriate tuning of the carbene substituents and the metal precursor, several metallocarbene complexes have been detected, some of them being isolated and structurally characterized. All metals from groups 8 to 11 are known to catalyze the carbene transfer from diazo compounds (Scheme 1b), and at least one detected/isolated example of a metal-carbene intermediate formed from a diazo compound has been reported for each of them.8−18 In all cases, the CR1R2 moiety in the initial diazo compound appears unmodified in the subsequent metal-carbene intermediate (Scheme 1b), from where it is further transferred to the nucleophile. From here, it is assumed that the carbene ligand is always transferred without modification. One of the most popular diazo reagents are diazocarbonyl compounds,4 with a CO group directly bonded to the diazo functionality: the acceptor nature of the −COR group favors the transfer of the carbene group toward the nucleophile.
Herein we report the observation of the unprecedented modification of the carbene unit during the course of a copper-catalyzed transformation. The use of a diazoacetamide compound (Scheme 1c) bearing a CONR2 substituent leads to the formation of the expected, undetected, hot, highly reactive copper carbene MCE (Scheme 1c) which promotes the intramolecular C–H bond functionalization of the ethyl groups of the amide fragment. In a parallel manner, MCE undergoes the loss of CO en route to the formation of the unexpected, stable and isolable, cold, Fischer carbene complex MCU (Scheme 1c), which has been structurally characterized.
Results and Discussion
Reaction of TpMsCu(THF) and N2=C(H)(CONEt2)
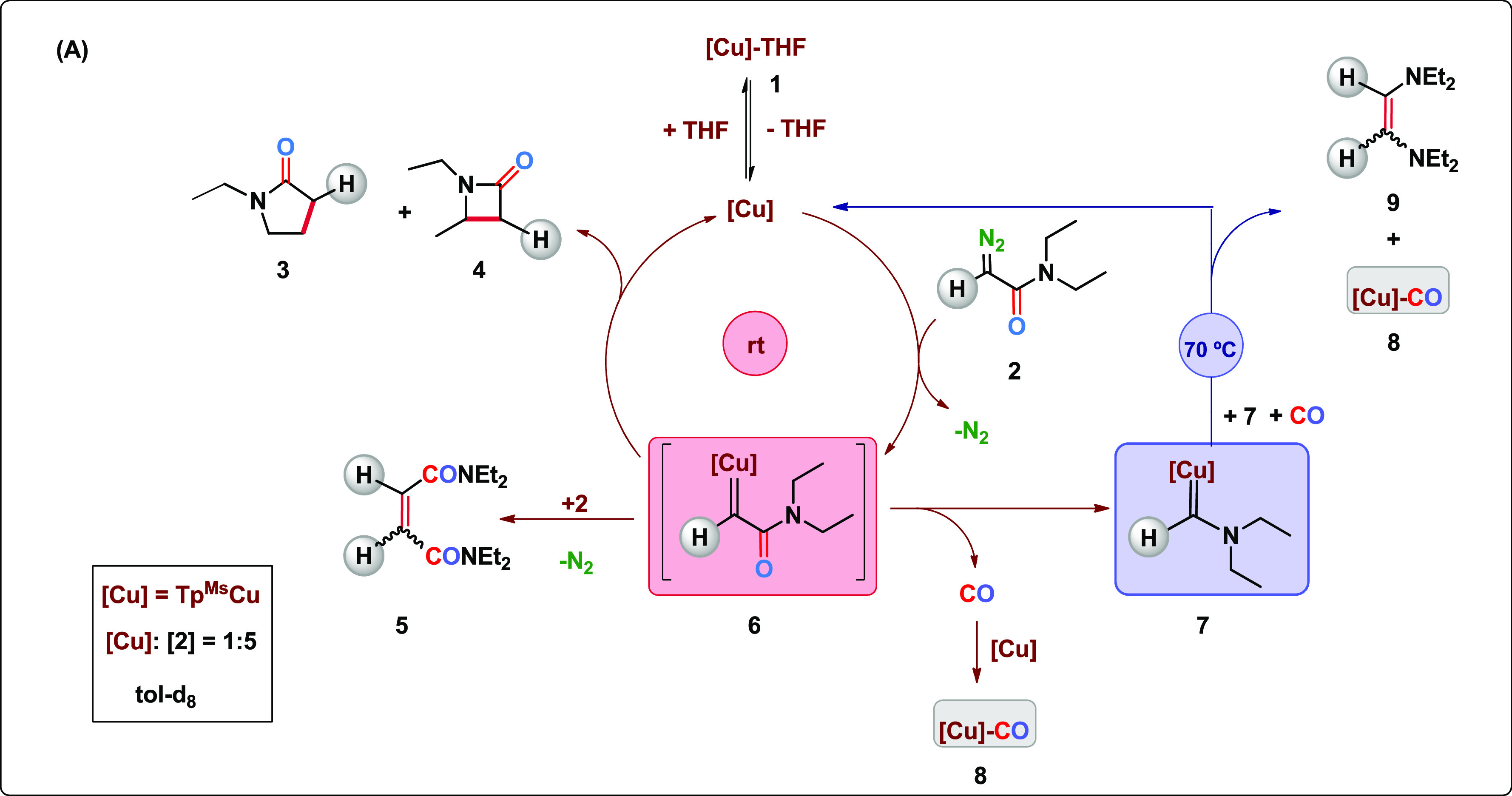
Our group has been involved in the area of carbene transfer from diazo compounds, with emphasis on its application to the functionalization of C–H bonds of unmodified alkanes,6,19 for which the design of very active catalysts precluded the observation of intermediates. However, we recently detected copper-carbene species20 in solution upon reacting TpMsCu(THF) (1) and ethyl phenyldiazoacetate (PheDA), which were stable at temperatures below 10 °C. After those findings, we aimed to detect copper-carbene intermediates with monosubstituted diazo compounds, which yet remains a challenge, particularly with the most popular catalysts within this field, i.e., rhodium and copper, for which only disubstituted carbene species have been detected or isolated.11,13,21 Toward that end we chose TpMsCu(THF)22 as the copper source, since the TpMs ligand provides a high degree of steric protection to the metal center and, when formed, to the carbene ligand. Regarding the diazo reagent, we selected N,N-diethyl diazoacetamide (2), in view of the previously reported23 intramolecular carbene insertion into the C–H bonds of the ethyl N-substituents in the presence of 1 as the catalyst (Scheme 2a), leading to mixtures of lactams 3 and 4. Olefin 5 was also observed from the catalytic reaction of two molecules of 2.24 Since heating at 70 °C was needed to reach efficient ratios, we reasoned that perhaps the copper-carbene intermediate expected from the reaction of 1 and 2, TpMsCu=C(H)(CONEt2) (6), could be stable enough at room temperature to be detected. Based on this idea, we started this study carrying out the reaction of TpMsCu(THF) (1) with 5 equiv of N,N-diethyl diazoacetamide (2*), 100% enriched in 13C at the 13C=N2 site, in toluene-d8 (Scheme 2b). Monitoring of the reaction by 13C{1H} NMR spectroscopy showed the appearance of a resonance centered at 236.6 ppm. This is within the typical region of Cu=C moieties reported by Hofmann25 for the complexes [tBu2P(NSiMe3)2-κ2N]Cu=C(Ar)C(O)R) (235.8–219.0 ppm), by Warren13 for [β-diketiminate]Cu=CPh2 (253.1 ppm), or by our group for complexes TpxCu=C(Ph)(CO2Et) (Tpx = TpiPr2, Tp*, TpMs; 233.9, 236.8, and 248.5 ppm, respectively).20 Under these conditions, the resonances for 3, 4, and 5 were also observed, the latter being the major product. The reaction was immediate, and no free diazo compound 2* was observed after 5 min. Successive registration of 13C{1H} NMR spectra for hours showed no change in the composition of the mixture.
Scheme 2. (a) Copper-Catalyzed Intramolecular C–H Bond Functionalization of N–N-Diethyldiazoacetamide; (b) 13C{1H} NMR Spectrum of the Reaction of Copper Complex 1 and 13C-Labeled Diazoacetamide (2*).

In view of the apparent stability of the species bearing the Cu=C unit, we repeated the reaction at a bench scale (see Supporting Information (SI) for optimization conditions). When 0.45 g of 1 were reacted with 5 equiv of diazo 2 in toluene at room temperature for 1 h, a yellowish solution was formed from which, after workup, the new compound 7 was isolated as crystalline material in 35% yield. The 1H NMR spectrum of 7 showed three equivalent pyrazolyl rings, two inequivalent ethyl groups, and a singlet at 8.02 ppm (Scheme 2c), which corresponds to one proton that correlates in the 2D-HSQC experiment with the aforementioned signal at 236.6 ppm (Scheme 2d). In the sample derived from the 13C-labeled diazo compound, the singlet at 8.02 in the 1H NMR spectrum split into a doublet with JC–H = 115 Hz (Scheme 2c).
Albeit the above data could support the assignment of complex 7 as the pursued TpMsCu=C(H)(CONEt2), some other experimental data were not in agreement with that proposal: neither the FT-IR spectrum showed the expected absorption for the CO group nor the 13C{1H} NMR spectrum displayed any resonance within the carbonyl region. We suspected that a decarbonylation process could have occurred, an idea that was confirmed when single crystals of this complex were grown, and the molecular structure determined by X-ray studies showed the formulation TpMsCu=C(H)(NEt2) (7).26 As shown in Figure 1, this complex contains the TpMs ligand bonded to copper in a κ3-fashion, and a carbene ligand with two substituents: a hydrogen and a diethylamido group, the latter resulting from the loss of the CO present in the initial diazo compound. The distance Cu1–C1, 1858(5) Å, is similar to that reported by Warren for [β-diketiminate]Cu=CPh2 (1.834(3) Å).13 Complex 7 constitutes the first example of a metal-carbene complex formed from a diazo compound in which the CR1R2 moiety in the latter is different from that in the former. Additionally, it is also the first example of a monosubstituted copper carbene complex.27
Figure 1.

Molecular structure of the molecules for complex 7. Hydrogens have been omitted for clarity.
Further investigation of the 1H NMR spectrum of the experiment carried out with a 1:5 ratio (excess of diazo is employed since C–H bond insertion also occurs in a catalytic manner to some extent) of 1 and 2* showed that, in addition to 7, another TpMsCu-containing species was formed, both accounting for all detectable TpMsCu cores. Such compound has been identified as TpMsCu(CO) (8) (Scheme 3), as the result of the trapping of carbon monoxide by 1.22 To gain further information, we have prepared the doubly isotopically enriched N2=13C(H)(13CONEt2) (2**) diazo compound and monitored its reaction with complex 1, observing the resonances of carbene and carbonyl ligands of 7 and 8, respectively (see Supporting Information also). Thus, this observation unambiguously demonstrates the existence of a decarbonylation process.
Scheme 3. Reaction of TpMsCu(THF) with Doubly Isotopically Labeled 2** and Region of the 13C{1H} NMR Spectrum Showing the Labeled Carbene and Carbonyl Groups of 7* and 8*.

The yield in complex 7 is dramatically affected by temperature, as shown in Figure 2a. The reaction of 1 and 2 (1:5 ratio) has been performed within the −30 to +70 °C range, showing an increase in the reaction yield from 2% to 78%, respectively, in 1 h time experiments. The high yield preparation and isolation of 7 is better performed at 70 °C (see Supporting Information). We have also monitored the concentration of complex 7 with time at three different temperatures (Figure 2b) and have found that at 0 or 25 °C such concentration remains constant after 24 h. However, at 70 °C, a process involving the disappearance of 7 takes place with time in such a way that after 12 h it cannot be detected in solution.
Figure 2.

(a) Plot of the yields into complex 7 vs temperature (°C). (b) Variation of the yield of complex 7 with time at different temperatures.
The reactivity of isolated copper carbene 7 has also been investigated. On one hand, no reaction was observed with styrene, tert-butanol, benzene, or cyclohexane from room temperature to 70 °C. It seems clear that the loss of the CO group modifies the electronic behavior of the carbene C(H)NEt2 ligand decreasing the electrophilicity from TpMsCu=C(H)(CONEt2) (6) to TpMsCu=C(H)NEt2 (7). Interestingly, heating toluene solutions of isolated 7 at 70 °C for hours/days did not lead to any transformation, whereas the same experiment under a CO atmosphere (4 bar) for 6 h induced a clean conversion of 7 into TpMsCu(CO) (8). NMR studies have also shown that the olefin (Et2N)(H)C=C(H)(NEt2) (9) derived from the coupling of two of the carbene units in 7 is formed.This result agrees with the lack of observation of copper-carbene 7 in the reaction of 1 and 2 at 70 °C for several hours, because of the presence of CO from the decarbonylation process.
The reactivity observed from the initial mixture of 1 and 2 (1:5 ratio) is rationalized in Scheme 4. The TpMsCu core is generated upon decoordination of THF and reacts with the diazo compound 2 to generate the transient copper carbene 6. This species undergoes either the intermolecular reaction with a second molecule of 2 or the intramolecular insertion of the carbene group into the secondary or primary C–H bonds of the ethyl groups of the NEt2 fragment, thus yielding lactams 3 and 4, these catalytic cycles taking place at room temperature. Intermediate 6 can alternatively undergo a decarbonylation process which generates the isolable copper carbene 7, a process which occurs, at least, within the −30 to +70 °C interval. Evolved CO can be trapped by TpMsCu cores leading to the formation of TpMsCu(CO) (8). At 70 °C, copper-carbene 7 originates olefin 9 and carbonyl 8 in a process requiring carbon monoxide. This proposal agrees with the initial observation of the need for heating at 70 °C to induce catalysis by 1,23 since the copper–carbonyl 8 under the reaction conditions in the presence of 2 also generates TpMsCu cores to catalyze the C–H bond functionalization reaction.
Scheme 4. General Picture of the Reactivity Derived from the Interaction of TpMsCu(THF) (1) and N2=C(H)CONEt2 (2).
The transformation of 6 into 7 somehow resembles the Wolff rearrangement of diazo compounds (Scheme 5a), which originate short-lived ketenes4,28 which further react with nucleophiles, in a process triggered by light, heating, or silver(I) salts. Grotjahn and co-workers described a ketene iridium complex which undergoes the reversible conversion of the coordinated ketene into a metallocarbene which contains a CO ligand (Scheme 5b).29 Based on these precedents, it could be possible that copper-carbene 6 undergoes a related rearrangement in the coordination sphere of copper, followed by CO extrusion (Scheme 5c). As mentioned above, the reverse reaction is not observed in our case, at least apparently, as inferred from the experiment carried out with 7 and CO.
Scheme 5. (a) Wolff Rearrangement of Diazo Compounds; (b) Reversible Formation of Ketenes from Carbene and CO Ligands Described by Grotjahn; (c) Plausible Intermediate for the Conversion of 6 into 7.

DFT Studies and Microkinetic Model
Given the unexpected formation of 7, we further explored the process with DFT studies (B3LYP-D3 optimizations in solution; see details in the Supporting Information) and microkinetic simulations. Computational results are summarized in Figure 3, showing the free energy profile for the conversion from the starting TpMsCu(THF) complex (1) to compound 7. The initial steps follow the usual mechanism up to the formation of metal-carbene 6, with the highest barrier corresponding to nitrogen extrusion (17.3 kcal/mol–1). Species such as metal-carbene 6 have been previously shown to be very reactive toward homocoupling or C–H activation.19,23,24 The novelty in this system is the availability of an alternative low energy path via decarbonylation. The latter proceeds through a rather stable ketene intermediate INTDCO2, with a linear arrangement between the CO and the former carbene carbon (see Supporting Information for more details). The barrier for the decarbonylation process is low at 298.15 K (13.8 kcal/mol) while the reverse barrier is 28.0 kcal·mol–1, too high to take place significantly at ambient temperature. At 343.15 K the profile is similar, with a barrier of 13.9 kcal/mol for decarbonylation and a barrier of 30.3 kcal/mol for the reverse process. The lack of observation of the reverse pathway when reacting complex 7 and CO at 70 °C is due to the more favorable path of formation of olefin 9 and carbonyl adduct 8 (see below). Further calculations were carried out for the case where the toluene solvent is replaced by dichloromethane, also reproducing the experimental observation of pyrrolidinone (3) and azetidinone (4). The detailed computational results for these cases are given in the Supporting Information.
Figure 3.
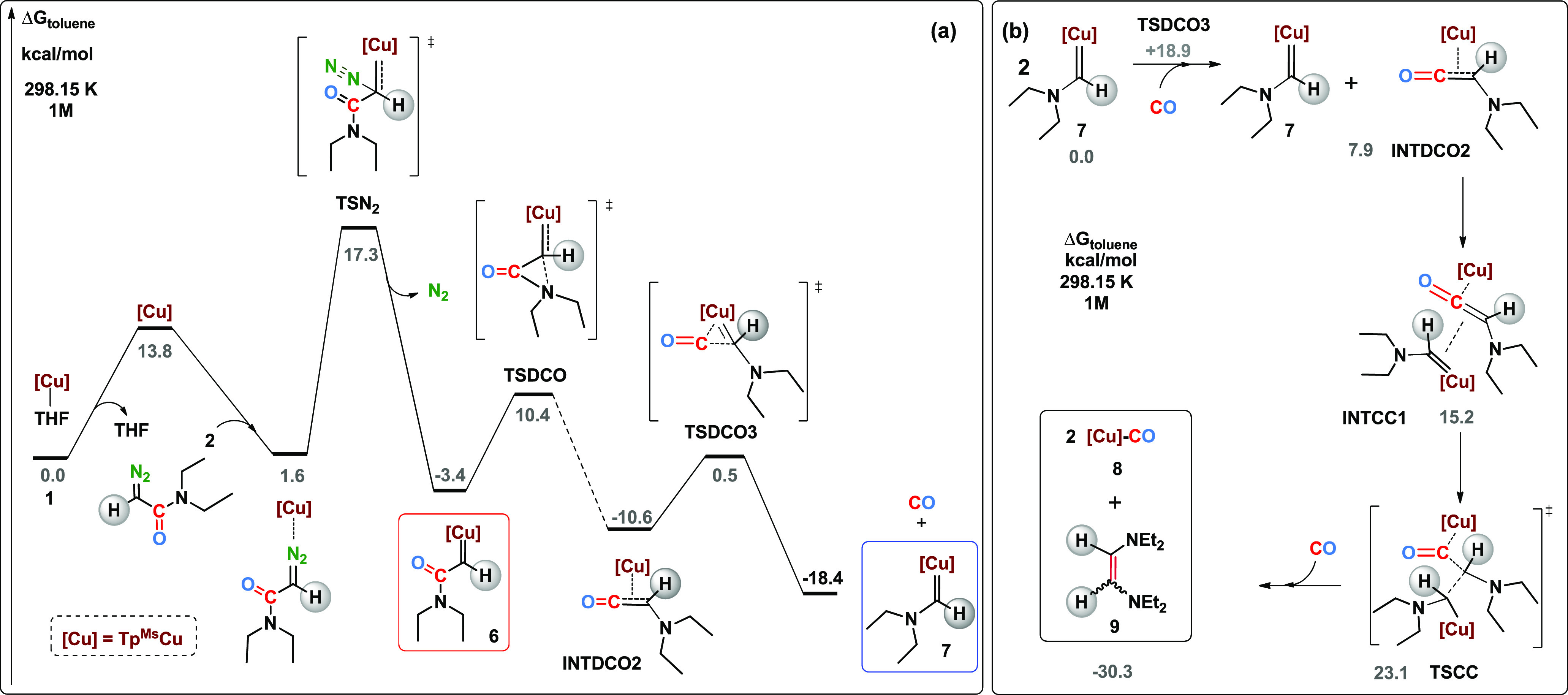
(a) Simplified reaction free energy profile from DFT calculations for the generation of copper carbenes 6 and 7. (b) Simplified reaction mechanisms for the formation of olefin 9.
The formation of 9 has also been investigated. Several paths previously reported for such carbene coupling have been discarded based on their high energy barriers: (a) direct interaction of two molecules of 7;24 (b) carbene C(H)NEt2 dissociation and reaction with 7;24 and (c) coupling of two dissociated carbene units30 (see Supporting Information). At variance with that, we have found an accessible route which involves two molecules of 7 and one of CO. The simplified mechanism for the formation of olefin 9 is depicted in Figure 3b. The mechanism starts with one molecule of the metal-carbene 7 adding a molecule of CO to form the above-mentioned intermediate INTDCO2 which further reacts with a second molecule of 7. The formation of the olefin 9 must overcome a barrier of 23.1 kcal/mol. Thus, albeit olefin 9 is more stable than metal-carbene 7, its formation involves a higher barrier (see Supporting Information for further details).
The third possible product from the interaction between the precursor 1 and N,N-diethyl diazoacetamide is olefin 5, which is also experimentally observed. We also computed the mechanism of its formation from 6, which is detailed in the Supporting Information. The key result is that the free energy barrier for C=C bond formation leading to 5 is 8.9 kcal/mol. This value is significantly lower than the 13.8 kcal/mol reported above for decarbonylation, which from a simplistic view would indicate that decarbonylation should not take place. However, for convoluted mechanisms such as the one present here, it is better to translate the computed free energy barriers to rate constants and use those to estimate reaction times through microkinetic modeling.31 We performed such microkinetic modeling with additional energy adjustment32 (see Supporting Information for details), and we obtained the results shown in Figure 4. The agreement between calculations and experimental data (displayed in Figure 2) is certainly remarkable, and this strongly supports the validity of our mechanistic proposal.
Figure 4.

(a) Computed yield (%) of metallocarbene 7 at 1 h of reaction for different reaction temperatures (K). (b) Evolution over time (h) of the yield (%) of metallocarbene 7 at different temperatures (continuous line obtained from DFT and microkinetic simulations; dots obtained from experiment).
Conclusion
We herein report the first example of the modification of a carbene group during its metal-catalyzed transfer from a diazo functionality, a discovery that should result in further consideration from the community involved in this area. From now on, the belief that the carbene moiety remains undisturbed in this type of processes must be reconsidered when analyzing reaction outcomes, particularly in those catalytic systems which either seem deactivated or require anomalous heating. Additionally, the chemistry of these isolated metal-carbenes is yet to be developed and could find important applications as surrogates of well-known midtransition metal Fischer carbene complexes
Experimental Section
Synthesis and Characterization of Complexes 7 and 7*
In a 250 mL Schlenk flask, TpMsCu(THF) (1, 0.450 g, 0.64 mmol) was dissolved in 75 mL of toluene, and a solution of 2-diazo-N,N-diethylacetamide (2, 0.451 g, 3.2 mmol) in the same solvent (37 mL) was added via canula. The mixture was stirred at 70 °C for 1 h before the volatiles were removed under reduced pressure. The residue was washed with dry acetone (5 mL) and cold Et2O (3 × 5 mL) and dried under vacuum, to afford 0.35 g of a yellow solid corresponding, according to NMR studies, to a mixture of complexes 7 and 8 (yields of 47% and 10% referred to initial 1). Crystallization from a mixture of toluene/hexane (4:4 mL) at −30 °C led to the isolation of complex 7 as yellow crystals in 35% yield (combined crops). Analytically calculated for C41H51BCuN7 (1): C, 68.75; H, 7.18; N, 13.69%. Found: C, 69.41; H, 7.12; N, 13.66%. Complex 7* was prepared following the same procedure, with the corresponding labeled diazo compound (see SI) with an isolated yield of 28%.
1H NMR (C6D6, 500 MHz): δ 0.28 (t, JHH = 7.3 Hz, 3H, NCH2CH3), 0.44 (t, JHH = 7.3 Hz, 3H, NCH2CH3), 2.12 (s, 27H, CH3), 2.23 (q, JHH = 7.3 Hz, 2H, NCH2CH3), 2.41 (q, JHH = 7.3 Hz, 2H, NCH2CH3), 6.01 (d, JHH = 2.0 Hz, 3H, CH), 6.71 (s, 6H, CH), 7.80 (d, JHH = 2.0 Hz, 3H, CH), 8.02 (s, 1H, Cu=CH). 13C{1H} NMR (C6D6, 125 MHz): δ 12.9 (CH3), 13.6 (CH3), 20.9 (CH3,Ms), 21.1 (CH3,Ms), 53.1 (NCH2), 54.9 (NCH2), 104.5 (CHpz), 127.8 (CHMs), 133.6 (Cq,Ms), 134.9 (CHpz), 136.2 (Cq,Ms), 138.3 (Cq,Ms), 150.9 (Cq,pz), 236.6 (Cu=C).
Acknowledgments
We dedicate this work to the memory of Professor Victor Riera, one of the pioneers in Organometallic Chemistry in Spain. We acknowledge financial support from the Ministerio de Ciencia e Innovación (CTQ2017-82893-C2-1-R, CTQ2017-87792-R, P. O. Feder UHU-1260216 and P. O. Feder UHU-1254043, and Red Intecat CTQ2016-81923-REDC).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c01483.
All procedures and characterization data, computational data, and Cartesian coordinates of the optimized structures (PDF)
Accession Codes
CCDC 2057177 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Buchner E.; Curtius T. Ueber die Einwirkung von Diazoessigäther auf aromatische Kohlenwasserstoffe. Ber. Dtsch. Chem. Ges. 1885, 18, 2377–2379. 10.1002/cber.188501802119. [DOI] [Google Scholar]
- Doyle M. P.; Duffy R.; Ratnikov M.; Zhou L. Catalytic Carbene Insertion into C–H Bonds. Chem. Rev. 2010, 110, 704–724. 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Morton D. Guiding principles for site selective and stereoselective intermolecular C–H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869. 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Mckervey M. A.; Ye T.. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998; pp 61–99. [Google Scholar]
- Cheng Q.-Q.; Doyle M. P. The Selection of Catalysts for Metal Carbene Transformations. Adv. Organomet. Chem. 2016, 66, 1–31. 10.1016/bs.adomc.2016.07.002. [DOI] [Google Scholar]
- Caballero A.; Díaz-Requejo M. M.; Fructos M. R.; Olmos A.; Urbano J.; Pérez P. J. Catalytic Functionalization of Low Reactive C(sp3)–H and C(sp2)–H Bonds of Alkanes and Arenes by Carbene Transfer from Diazo Compounds. Dalton Trans. 2015, 44, 20295–20307. 10.1039/C5DT03450G. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Qiu D.; Wang J. Transition-Metal-Catalyzed Cross-Couplings through Carbene Migratory Insertion. Chem. Rev. 2017, 117, 13810–13889. 10.1021/acs.chemrev.7b00382. [DOI] [PubMed] [Google Scholar]
- Li Y.; Huang J.; Zhou Z.; Che C.; You X. Remarkably Stable Iron Porphyrins Bearing Nonheteroatom-Stabilized Carbene or (Alkoxycarbonyl)carbenes: Isolation, X-ray Crystal Structures, and Carbon Atom Transfer Reactions with Hydrocarbons. J. Am. Chem. Soc. 2002, 124, 13185–13193. 10.1021/ja020391c. [DOI] [PubMed] [Google Scholar]
- Collman J. P.; Rose E.; Venburg G. D. Reactivity of ruthenium 5,10,15,20-tetramesitylporphyrin towards diazoesters: formation of olefins. J. Chem. Soc., Chem. Commun. 1993, 934–935. 10.1039/c39930000934. [DOI] [Google Scholar]
- Djukic J.-P.; Smith D. A.; Young V. G. Jr; Woo L. K. Properties and Molecular Structures of Osmium(II) Porphyrin Carbene Complexes: (5,10,15,20-tetra-p-tolylporphyrinato)osmium Di-p-tolylmethylidene and (5,10,15,20-tetra-p-tolylporphyrinato)osmium (Trimethylsilyl)methylidene. Organometallics 1994, 13, 3020–3026. 10.1021/om00020a018. [DOI] [Google Scholar]
- a Werle C.; Goddard R.; Fürstner A. The First Crystal Structure of a Reactive Dirhodium Carbene Complex and a Versatile Method for the Preparation of Gold Carbenes by Rhodium-to-Gold Transmetalation. Angew. Chem., Int. Ed. 2015, 54, 15452–15456. 10.1002/anie.201506902. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Werle C.; Goddard R.; Philipps P.; Farès C.; Fürstner A. Stabilization of a Chiral Dirhodium Carbene by Encapsulation and a Discussion of the Stereochemical Implications. Angew. Chem., Int. Ed. 2016, 55, 10760–10765. 10.1002/anie.201605502. [DOI] [PubMed] [Google Scholar]
- Mindiola D. J.; Hillhouse G. L. Synthesis, structure, and reactions of a three-coordinate nickel-carbene complex, {1,2-bis(di-tert-butylphosphino)ethane}Ni = CPh2. J. Am. Chem. Soc. 2002, 124, 9976–9977. 10.1021/ja0269183. [DOI] [PubMed] [Google Scholar]
- Dai X.; Warren T. H. Discrete Bridging and Terminal Copper Carbenes in Copper-Catalyzed Cyclopropanation. J. Am. Chem. Soc. 2004, 126, 10085–10094. 10.1021/ja047935q. [DOI] [PubMed] [Google Scholar]
- Hussong M. W.; Hoffmeister W. T.; Rominger F.; Straub B. F. Copper and Silver Carbene Complexes without Heteroatom-Stabilization: Structure, Spectroscopy, and Relativistic Effects. Angew. Chem., Int. Ed. 2015, 54, 10331–10335. 10.1002/anie.201504117. [DOI] [PubMed] [Google Scholar]
- Hussong M. W.; Rominger F.; Kramer P.; Straub B. F. Isolation of a Non-Heteroatom-Stabilized Gold–Carbene Complex. Angew. Chem., Int. Ed. 2014, 53, 9372–9375. 10.1002/anie.201404032. [DOI] [PubMed] [Google Scholar]
- Bellow J. A.; Stoian S. A.; van Tol J.; Ozarowski A.; Lord R. L.; Groysman S. Synthesis and Characterization of a Stable High-Valent Cobalt Carbene Complex. J. Am. Chem. Soc. 2016, 138, 5531–5534. 10.1021/jacs.6b02747. [DOI] [PubMed] [Google Scholar]
- Barrett B. J.; Iluc V. M. An Adaptable Chelating Diphosphine Ligand for the Stabilization of Palladium and Platinum Carbenes. Organometallics 2017, 36, 730–741. 10.1021/acs.organomet.6b00924. [DOI] [Google Scholar]
- Tindall D. J.; Werlé C.; Goddard R.; Philipps P.; Farès C.; Fürstner A. Structure and Reactivity of Half-Sandwich Rh(+3) and Ir(+3) Carbene Complexes. Catalytic Metathesis of Azobenzene Derivatives. J. Am. Chem. Soc. 2018, 140, 1884–1893. 10.1021/jacs.7b12673. [DOI] [PubMed] [Google Scholar]
- Caballero A.; Despagnet-Ayoub E.; Díaz-Requejo M. M.; Díaz-Rodríguez A.; González-Núñez M. E.; Mello R.; Muñoz B. K.; Ojo W.-S.; Asensio G.; Etienne M.; Pérez P. J. Silver-Catalyzed C–C Bond Formation between Methane and Ethyl Diazoacetate in Supercritical CO2. Science 2011, 332, 835–838. 10.1126/science.1204131. [DOI] [PubMed] [Google Scholar]
- Pereira A.; Champouret Y.; Martín C.; Álvarez E.; Etienne M.; Belderrain T. R.; Pérez P. J. Copper–Carbene Intermediates in the Copper-Catalyzed Functionalization of O-H Bonds. Chem. - Eur. J. 2015, 21, 9769–9775. 10.1002/chem.201500776. [DOI] [PubMed] [Google Scholar]
- Kornecki K. P.; Briones J. F.; Boyarskikh V.; Fullilove F.; Autschbach J.; Schrote K. E.; Lancaster K. M.; Davies H. M. L.; Berry J. F. Direct Spectroscopic Characterization of a Transitory Dirhodium Donor-Acceptor Carbene Complex. Science 2013, 342, 351–354. 10.1126/science.1243200. [DOI] [PubMed] [Google Scholar]
- Schneider J. L.; Carrier S. M.; Ruggiero C. E.; Young V. G. Jr.; Tolman W. B. Influences of Ligand Environment on the Spectroscopic Properties and Disproportionation Reactivity of Copper–Nitrosyl Complexes. J. Am. Chem. Soc. 1998, 120, 11408–11418. 10.1021/ja982172q. [DOI] [Google Scholar]
- Martín C.; Belderraín T. R.; Pérez P. Rediscovering copper-based catalysts for intramolecular carbon–hydrogen bond functionalization by carbene insertion. Org. Biomol. Chem. 2009, 7, 4777–4781. 10.1039/b911589g. [DOI] [PubMed] [Google Scholar]
- Rivilla I.; Sameera W. M. C.; Álvarez E.; Díaz-Requejo M. M.; Maseras F.; Pérez P. J. Catalytic cross-coupling of diazo compounds with coinage metal-based catalysts: an experimental and theoretical study. Dalton Trans. 2013, 42, 4132–4138. 10.1039/c2dt32439c. [DOI] [PubMed] [Google Scholar]
- Straub B. F.; Hofmann P. Copper(I) Carbenes: The Synthesis of Active Intermediates in Copper-Catalyzed Cyclopropanation. Angew. Chem., Int. Ed. 2001, 40, 1288–1290. . [DOI] [PubMed] [Google Scholar]
- CCDC 2057177 contains the supplementary crystallographic data for this compound. www.ccdc.cam.ac.uk/structures (accessed 2021/03/02).
- Peloso R.; Carmona E. Non-heteroatom-substituted alkylidene complexes of groups 10 and 11. Coord. Chem. Rev. 2018, 355, 116–132. 10.1016/j.ccr.2017.07.018. [DOI] [Google Scholar]
- Wolff L. Ueber Diazoanhydride. Liebigs Ann. Chem. 1902, 325, 129–195. 10.1002/jlac.19023250202. [DOI] [Google Scholar]
- Grotjahn D. B.; Bikzhanova G. A.; Collins L. S. B.; Concolino T.; Lam K.-C.; Rheingold A. L. Controlled, Reversible Conversion of a Ketene Ligand to Carbene and CO Ligands on a Single Metal Center. J. Am. Chem. Soc. 2000, 122, 5222–5223. 10.1021/ja000243r. [DOI] [Google Scholar]
- Soleilhavoup M.; Bertrand G. Stable carbenes, nitrenes, phosphinidenes, and borylenes: Past and future. Chem. 2020, 6, 1275–1282. 10.1016/j.chempr.2020.04.015. [DOI] [Google Scholar]
- Besora M.; Maseras F. Microkinetic Modeling in Homogeneous Catalysis. WIREs: Comput. Mol. Sci. 2018, 8, e1372. [Google Scholar]
- Pérez-Soto R.; Besora M.; Maseras F. The Challenge of Reproducing with Calculations Raw Experimental Kinetic Data for an Organic Reaction. Org. Lett. 2020, 22, 2873–2877. 10.1021/acs.orglett.0c00367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.