

Abstract
Epilepsy is a complex neurological syndrome characterized by seizures resulting from neuronal hyperexcitability and sudden and synchronized bursts of electrical discharges. Impaired astrocyte function that results in glutamate excitotoxicity has been recognized to play a key role in the pathogenesis of epilepsy. While there are 26 drugs marketed as anti-epileptic drugs no current treatments are disease modifying as they only suppress seizures rather than the development and progression of epilepsy. Excitatory amino acid transporters (EAATs) are critical for maintaining low extracellular glutamate concentrations and preventing excitotoxicity. When extracellular glutamate concentrations rise to abnormal levels, glutamate receptor overactivation and the subsequent excessive influx of calcium into the post-synaptic neuron can trigger cell death pathways. In this review we discuss targeting EAAT2, the predominant glutamate transporter in the CNS, as a promising approach for developing therapies for epilepsy. EAAT2 upregulation via transcriptional and translational regulation has proven successful in vivo in reducing spontaneous recurrent seizures and offering neuroprotective effects. Another approach to regulate EAAT2 activity is through positive allosteric modulation (PAM). Novel PAMs of EAAT2 have recently been identified and are under development, representing a promising approach for the advance of novel therapeutics for epilepsy.
Keywords: Astrocytes, EAAT2, epilepsy, excitotoxicity, glutamate, glutamate transporter
Graphical Abstract
Epilepsy
Epilepsy was first described in Mesopotamia in 2000 B.C. Although this syndrome has affected important people in history such as Julius Caesar, Vladimir Lenin, and Fyodor Dostoyevsky, it is still a disabling disease for most patients, so prevention and cure are still great unmet needs [1]. According to CDC data, in 2015 1.2% of the total US population had active epilepsy, meaning approximately 3.4 million people were either currently taking anti-seizure drugs (ASDs) or had a seizure within a year [2]. Worldwide, epilepsy has a prevalence of 1–2 % [3,4].
Epilepsy is a complex neurological syndrome characterized by seizures caused by hyperexcitability of neurons and sudden and synchronized bursts of electrical discharges. These seizures present as peculiar sensations, emotions, behavior, convulsions, muscle spasms, and/ or loss of consciousness [5–7].
Seizures can be classified into partial, or focal, and generalized[9]. Partial epilepsies represent the most common type of adult-onset epilepsy and account for more than 60% of all adult cases of temporal lobe epilepsy (TLE) [10]. For reviews on the pathophysiology of epilepsy, see [7,11,12].
Epileptogenesis
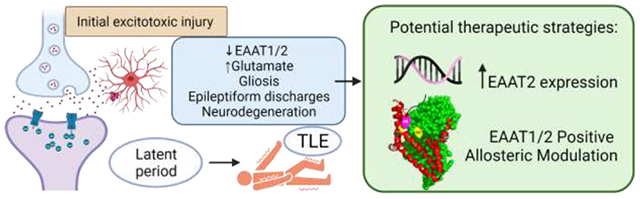
An initial injury leading to seizures may be head trauma, childhood seizures, hypoxia, or status epilepticus (SE), a single seizure that either lasts longer than 5 minutes or is a series of seizures that occur close together with no recovery of consciousness between seizures [13–15]. The intense seizure activity seen in SE causes excessive glutamate release resulting in overstimulation of glutamate receptors leading to massive influxes of Ca2+, subsequently triggering mass neuronal death via glutamate excitotoxicity mechanisms [7,16]. Following the initial insult or injury there is a latent period that may last up to several years in which complex molecular, biochemical, and structural changes occur, including changes in synaptic plasticity and neuronal connectivity reorganization of neuronal networks. Within minutes to days following the initial insult, acute early changes include rapid alterations to ion channel kinetics, post-translational modifications to existing functional proteins, and activation of immediate early genes. Hours to weeks after the insult, subacute changes occur, including neuronal death, activation of inflammatory cascades and several transcriptional events. In the following weeks to months, several chronic changes will take place, including anatomical changes, such as neurogenesis, gliosis, mossy fiber sprouting and network reorganization [17,18]. These changes ultimately lead to neuronal networks being more susceptible to hyperexcitability and synchronous firing of these excitatory neurons, leading to more seizures and eventually spontaneous recurrent seizures [19,20]. This results in the emergence of chronic epilepsies such as TLE [8] (figure 1). Confirming this idea, an important recent study using an in vivo model of pentylenetetrazole (PTZ) insult, established that a single episode of clonic–tonic seizures affected neuronal viability in the hippocampus, up to 1 week after PTZ treatment. This suggests that even a single seizure episode is a powerful event that causes stress to hippocampal neurons and can cause intricate alterations in neuronal plasticity [21].
Figure 1. Representation of a tri-partite glutamatergic synapse, under physiological and excitotoxicity conditions, leading to the development of epilepsy.
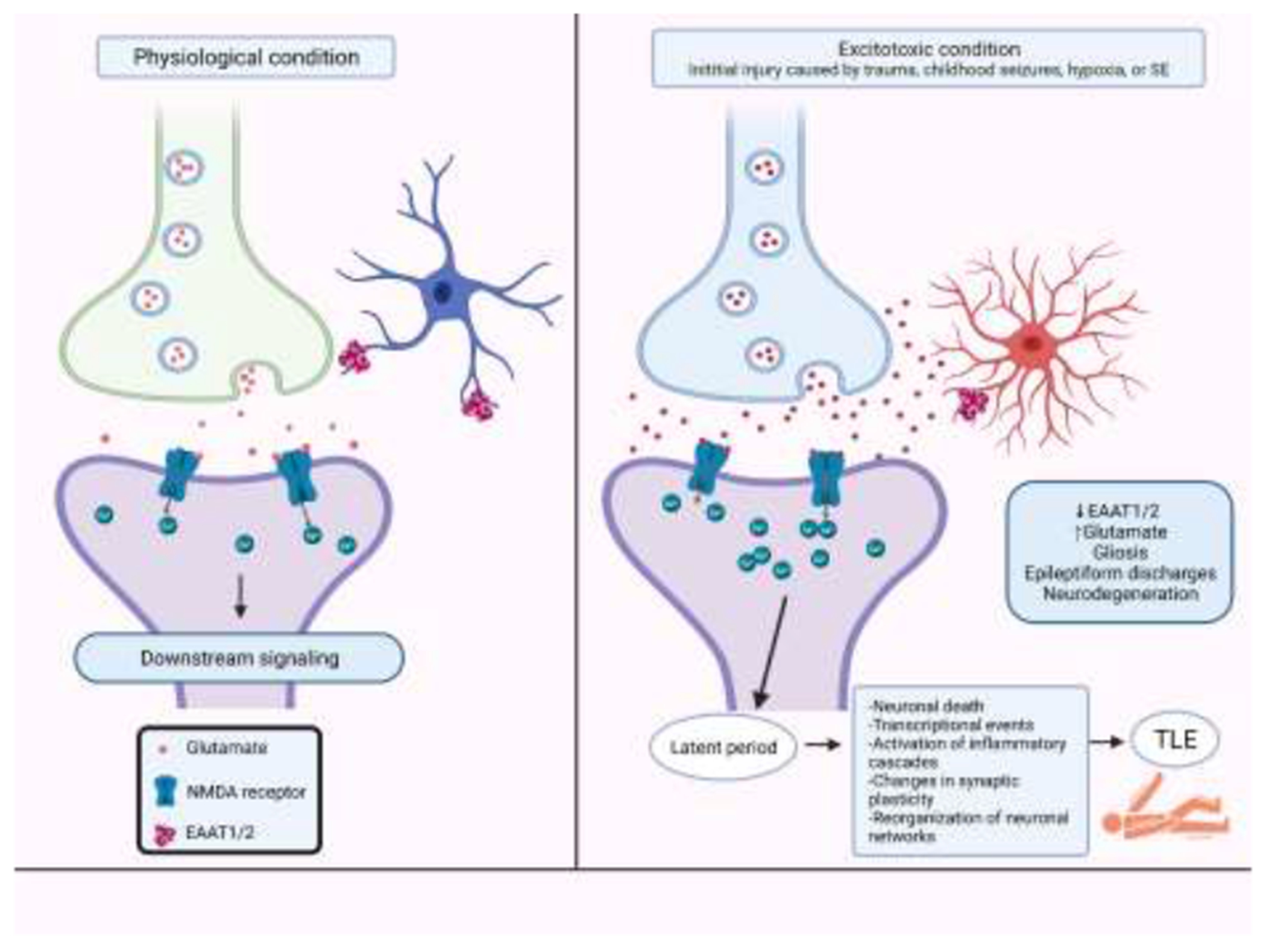
The intense seizure activity seen in SE causes excessive glutamate release resulting in overstimulation of glutamate receptors leading to massive influxes of Ca2+, subsequently triggering mass neuronal death via glutamate excitotoxicity mechanisms. Following the initial insult or injury there is a latent period that may last up to several years in which complex molecular, biochemical, and structural changes occur, including changes in synaptic plasticity and neuronal connectivity reorganization of neuronal networks. Within minutes to days following the initial insult, acute early changes include rapid alterations to ion channel kinetics, post-translational modifications to existing functional proteins, and activation of immediate early genes. Hours to weeks after the insult, subacute changes occur, including transcriptional events, neuronal death, and activation of inflammatory cascades. The chronic changes that follow over weeks to months include anatomical changes, such as neurogenesis, mossy fiber sprouting, network reorganization, and gliosis. These changes ultimately lead to neuronal networks being more susceptible to hyperexcitability and synchronous firing of these excitatory neurons, leading to more seizures and eventually spontaneous recurrent seizures This results in the emergence of chronic epilepsies such as TLE. Figure created with BioRender.com.
A recent study investigated whether glutamate and GABA are linked to the formation of epilepsy networks and the triggering of spontaneous seizures, by infusion of a glutamine synthetase inhibitor into rat hippocampi to create a seizure focus, a translationally rodent model of mTLE. Their results suggest that changes in extracellular brain levels of glutamate and GABA play important roles in epilepsy network formation and in the initiation and propagation of spontaneous seizures [22].
Collectively, these studies suggest that neuronal death due to glutamate excitotoxicity may be both a cause and consequence of epileptic seizures, as abnormal glutamate release by astrocytes is involved in the molecular mechanisms underlying the initial insults during the process of epileptogenesis [23]. Developmental processes play a role in the regulation of epileptogenesis, suggested by differences observed in epileptogenic processes between developing and adult brains. However, age-specific mechanisms of epileptogenesis, as well as specific factors associated with increased seizure susceptibility in the developing brain are not well known. This knowledge is needed for development of proper biomarkers that would allow for the identification of risk factors for developing epilepsy. Further research is also needed for development of therapies for the prevention of epileptogenesis, as well as for monitoring of the progression of epilepsy [24].
Current treatments for epilepsy
Since the early 1990s, many newer ASDs with better tolerability, reduced possibility for drug-drug interactions and satisfactory pharmacokinetics were approved for clinical use [25]. Currently there are 26 drugs marketed as ASDs, with various mechanisms of action including the inhibition of sodium channels to reduce the ability of neurons to fire at high frequencies (phenytoin, carbamazepine, valproate, and lamotrigine, among others), inhibition of T type and L type calcium channels (ethosuximide and gabapentin, respectively), inhibition of voltage-gated potassium channels (Kv7, retigabine/ezogabine) inhibition of glutamate release and receptors (topiramate), and enhancement of GABA action via activation of GABAA receptors (phenobarbital and benzodiazepines), inhibition of GABA transaminase (vigabatrin), and inhibition of GABA reuptake (tiagabine), and modulation of synaptic release, as examples modulators of the α2δ subunit of voltage-gated calcium channels (gabapentin, pregabalin)and the synaptic vesicle protein (SV) 2A (brivaracetam, levetiracetam) [26–28].
Unfortunately, current pharmacotherapies are not efficacious in approximately one third of patients with epilepsy [29]. Additionally, current treatments have proven successful in suppressing seizures, however, they do not treat the underlying epileptic syndrome, i.e., they are not disease modifying as they do not halt the progression of epileptogenesis. Moreover, many of these treatments, especially the GABAA receptor activators, can have serious adverse events and tolerance associated with their use [30–33].
Approximately 20% of epilepsy cases are triggered by acute injuries such as stroke, traumatic brain injury, and bacterial, virus and other types of brain and spinal cord infections. There are several opportunities for pharmacological treatment for epilepsy: the injury onset that triggered the epileptogenic process, medical care at the early phases of presentation, and intervention a latent phase between the injury and the development of clinical epilepsy. Unfortunately, even though there are no available treatments, no extensive clinical research seeking novel therapies has been conducted on the last 20 years. In rare cases, certain “self-limiting” childhood and adolescence epilepsy syndromes have remitted spontaneously. One type of treatment that seems to result in “cure” is resective surgery, however, this is sparsely provided. Some patients benefit of medications or dietary treatments, however, once the treatment is discontinued, the seizures tend to persist. At the present time, there is no enough knowledge on the mechanisms behind such “cures”; impeding the development of translational treatment paradigms to “cure” epilepsy [34].
So, despite being a common neurological disorder, epilepsy remains a serious and unmet medical need. Thus, novel treatments for epileptic syndromes are needed, and research suggests that the glutamatergic system, and in particular the glutamate transporter GLT-1/EAAT2 (rodent/human homologues), may provide novel targets for ASDs. Several studies suggest that dysregulation of astrocytic glutamate transporters could be a factor to the development of epilepsy. The development of approaches that modulate the glial excitatory amino acid transporters EAAT1 and EAAT2, and glutamine synthetase could result in fundamental changes on the way epilepsy is treated, by providing a potential prevention of the epileptogenesis processes [35].
Glutamate transporters
Glutamate is the primary excitatory neurotransmitter in the mammalian CNS and is vital for normal brain functioning [36]. Glutamate is stored in synaptic vesicles at presynaptic terminals, that, in response to an action potential and increase in intracellular Ca2+, fuse with the plasma membrane of the presynaptic neuron and glutamate is released into the synaptic cleft and can then activate these receptors. Glutamate receptors fall into two different classes, ligand gated ion channels (ionotropic receptors) which include α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, kainate receptors, and N-methyl-D-aspartic acid (NMDA) receptors, and G-protein coupled receptors (metabotropic receptors) [37,38]. Upon activation of post-synaptic glutamate receptors, Ca2+ channels are opened and Ca2+ influxes into the post-synaptic neuron trigger an action potential in the post-synaptic neuron and further downstream signaling in physiological conditions.
Glutamate neurotransmission is terminated upon the uptake of extracellular glutamate by a family of five structurally distinct subtypes of high-affinity sodium dependent excitatory amino acid transporters (EAATs, rat/human homologue): GLAST/EAAT1, GLT-1/EAAT2, EAAC1/EAAT3, EAAT4 and EAAT5 [39].
The EAATs are secondary-active transporters that couple the movement of one glutamate with the symport of three Na+ ions and one H+ and the counter transport of one K+ [40]. The Na+ gradient generated by Na+ K+- ATPase activity drives glutamate transport under physiological conditions [41]. Glutamate taken up by astrocytes is converted into glutamine via glutamine synthetase and is then transported to the extracellular space, where it is taken up by the presynaptic neuron and converted back into glutamate via glutaminase [42]. Glutamate is then packaged into synaptic vesicles via vesicular glutamate transporters (vGLUTs) [43]. Table 1 shows the differences in the nomenclature of glutamate transporters between human and rodent, their main biological activity and predominant expression in the CNS. This review will focus on EAAT2, the most abundant subtype of EAATs [44], that is expressed throughout the brain and in the spinal cord. EAAT2 is present primarily in astrocytes, and in neurons and oligodendrocytes, accounting for about 95% of the total glutamate transport activity and 1% of total brain protein in the CNS [45–49]. Thus, EAAT2 plays a central role in the CNS for maintaining the homeostasis of glutamate and therefore preventing excitotoxicity [37,50–52].
Table 1.
Nomenclature of glutamate transporters, main biological activity, and predominant expression in the CNS.
| Protein name (human) | Protein name (rodent) | Gene | Main biological activity | Predominant expression in mature brain | References |
|---|---|---|---|---|---|
| EAAT1 | GLAST | SLC1A3 | Glutamate transporter | Astrocytes, olygodendrocytes in cerebellum, cortex, spinal cord (perisynaptic) | [194–196] |
| EAAT2 | GLT-1 | SLC1A2 | Glutamate transporter | Astrocytes (perisynaptic), axon terminals (presynaptic), oligodendrocytes in whole brain and spinal cord | [44,196] |
| EAAT3 | EAAC1 | SLC1A1 | Glutamate and cysteine transporter | Neurons (postsynaptic, cell soma and dendrites) in whole brain | [196,197] |
| EAAT4 | EAAT4 | SLC1A6 | Glutamate transporter, glutamate-gated chloride channel | Neurons (postsynaptic, dendritic spines) in cerebellum | [198] |
| EAAT5 | EAAT5 | SLC1A7 | Glutamate-gated chloride channel | Neurons (presynaptic) in retina | [199] |
| vGLUT1, 2 and 3 | vGLUT1, 2 and 3 | SCL17A7, 6 and 8 | Glutamate transporter | Mainly presynaptic throughout the brain | [200–204] |
| xCT | xCT | SLC7A11 | Cystine-glutamate antiporter | Astrocytes, microglia retinal Muller cells, immature cortical neurons, and glioma in the CNS | [205–207] |
EAATs function is essential for fast removal of glutamate from the extracellular space upon its release into the synaptic cleft. It is key that extracellular glutamate concentrations are kept low (25-600 nM), as elevated concentrations to approximately 2-5 μM result in excitotoxic injury [53–55]. Glutamate excitotoxicity is the process in which failure to remove extracellular glutamate from the synaptic cleft results in sustained extracellular glutamate levels and excessive activation of NMDA receptors [55]. This results in excessive Ca2+ influx into the post-synaptic neuron leading to downstream activation of a cascade of phospholipases, endonucleases, and proteases like calpain that leads to cell apoptosis [56]. Additionally, the excessive Ca2+ influx and subsequent depolarization of the post-synaptic neuron may lead to the efflux of excessive glutamate spillage promoting subsequent excitotoxicity [39]. Thus, the EAATs, and in particular EAAT2, are crucial for maintaining low extracellular glutamate concentrations to prevent NMDA receptor (NMDAR) and AMPA receptor (AMPAR) mediated excitotoxicity. EAATs are also present on GABAergic terminals, where they take up glutamate that is converted into GABA by glutamic decarboxylase (GAD). This function contributes to limiting spill-over of glutamate of active synapses, limiting the activation of extra-synaptic and neighboring synapses glutamate receptors [58].
In addition to the EAAT family of transporters and vesicular glutamate transporters, there are also glutamate-cystine exchangers (SLC7A11, xCT) present in neurons and glia that exchange intracellular glutamate for extracellular cystine to provide a cystine source for the synthesis of glutathione [59]. Extracellular accumulation of glutamate can also cause toxicity through interaction with these exchangers as they become blocked and glutathione cannot be synthesized, leading to oxidative stress and downstream cell death [57]. These exchangers are relevant for epileptic seizures in the context of gliomas, as non-vesicular secretion of glutamate via these exchangers constitutes the main mechanism contributing to high extracellular glutamate concentrations [60].
Astrocytes and Epilepsy
Historically, the focus of epilepsy research has been neurocentric, since epilepsy is caused by aberrant synchronized firing of neuronal populations, primarily due to imbalance between excitatory and inhibitory neurotransmission. However, in the past two decades we have seen more research looking further into astrocytes and their role supporting and modulating neuronal activity, which has provided compelling evidence that glial cells are involved in pathophysiology of epilepsy [4]. Astrocytes can release may neuroactive molecules, including glutamate, D-serine, ATP, adenosine, GABA, TNFα (tumour necrosis factor α), prostaglandins, proteins and peptides that may affect neuronal activity and synaptic physiology [61,62]. Astrocytes regulate and respond to extracellular glutamate levels in the CNS via astrocytic glutamate transporters and the metabotropic glutamate receptors (mGluRs) 3 and 5 [63]. In epileptic tissue, reactive astrocytes utilize several mechanisms that regulate seizure development, which include dysregulation of astrocytic transporters and mGluRs [63,64]. Moreover, epileptic seizures cause neuronal cell loss and astrogliosis, a hallmark of epilepsy [65,66]. Inflammatory processes concomitant to SE result in changes in morphology and protein expression in astrocytes, including decreased expression of EAAT proteins, resulting in decreased glutamate uptake [67–69]. These inflammatory changes can also result in TNFαR and AMPA and NMDA receptors activation, further promoting excitotoxicity [70]. IL-1R activation by IL-1β can also promote glutamate release from the astrocytes [71,72].
It is strongly suggested that mTOR signaling is altered in SE [73], but just recently evidence of upregulation of the vascular endothelial growth factor-3 (VEGF-3) -mediated mTOR activation in reactive astrocytes after the onset of SE was associated with the regulation of GLT-1 expression. This suggests that this pathway could be involved in preventing hyperexcitability induced by repeated seizure activity [74].
Another area that has gathered increased recognition in the pathogenesis of epilepsy is energy homeostasis. Proper energy sources available to neurons are needed for excessive neuronal discharges to happen. On the other hand, an endogenous mechanism for seizure termination is energy depletion during seizures. In this sense, astrocytes play a very important role as they can control neuronal energy homeostasis through neurometabolic coupling, hence, astrocyte dysfunction in epilepsy leads to bias of key metabolic and biochemical mechanisms, and evidence suggests that dysfunctional glutamate metabolism in astrocytes contribute directly to neuronal hyperexcitability [75–77]. In early epileptogenesis, closure of astrocyte intercellular gap junction coupling limits activity-dependent trafficking of energy metabolites, resulting in impaired clearance of glutamate and K+ from the extracellular space. Accumulation of extracellular K+ leads to neuronal hyperexcitability, which can result in gap junction uncoupling, a recent finding demonstrated in a study using a transgenic mouse with astrocyte specific expression of a pH sensor (Lck-E2GFP), that established that astrocytes react to epileptiform activity with intracellular alkalization [78]. Additionally, the metabolism of adenosine, a metabolic product of ATP degradation that largely inhibits energy-consuming processes as an evolutionary adaptation to conserve energy, is increased by dysfunctional astrocytes.
Metabolic therapeutic approaches that prevent the utilization of glucose could represent an effective therapy for epilepsy, as astroglia energy homeostasis play a critical role in the control of neuronal excitability. In this regard, high fat low carbohydrate “ketogenic diets”, as well as inhibitors of glycolysis and lactate metabolism are of increasing interest as antiepileptic strategy [76,79].
Additionally, loss of glutamine synthetase in the human epileptogenic hippocampus was suggested as possible mechanism for raised extracellular glutamate in mTLE [80], as well as an important causative factor for glioblastoma-associated epilepsy [81]. Indeed, a recent study reported that selective deletion of gene for glutamine synthetase in the mouse cerebral cortex induces glial dysfunction with decreased levels of EAAT1 and EAAT2, and vascular impairment that precede epilepsy and neurodegeneration. This study suggests that abnormal glutamate metabolism could cause epilepsy by initially affecting glia which then results in disruption of the neurovascular coupling [82].
For further information on the general role of glia and neuron-glia interactions in epilepsy, see [4,63,64,76,83,84].
Glutamate Excitotoxicity and Epilepsy
As described above, epilepsy has been characterized by recurrent spontaneous seizures due to hyperexcitability and hypersynchrony of brain encephalic neurons [4,84]. Seizures occur with a disbalance between excitatory and inhibitory neurotransmission, and glutamate, as the predominant excitatory neurotransmitter in the brain, unquestionably plays a crucial role in both the initiation and proliferation of seizure activity and in epileptogenesis. Studies in the 1980s demonstrated that intracerebral injection of glutamate into experimental animals induced seizures [85], Later, in vivo microdialysis studies revealed increased glutamate levels in the hippocampus following the seizures emergence induced by pilocarpine [86]. Additionally, EEG and microdialysis studies of patients with epilepsy showed significant increases in extracellular glutamate levels in the hippocampus. These studies described excitotoxic glutamate concentrations of 5.9 μM before a seizure, 17.6 μM during a seizure onset, and 11.7 μM up to 16.5 minutes following the termination of a seizure, i.e., 2, 6 and 4-fold increases compared to basal glutamate concentrations, respectively [87,88]. A study observed that SE-induced glutamate release overstimulates glutamate receptors and results in sustained chronic seizure activity and development of seizure-induced brain damage that occurs over a dynamic process [89]. This was further validated in subsequent studies showing that the increase in glutamate concentrations positively correlates with the intensity of epileptic activity [67,90], demonstrating that increased levels of extracellular glutamate play a role in the initiation and spreading of seizures [23]. Other studies established that augmented concentration of glutamate in the synaptic cleft during the initial insult is associated with excitotoxicity and is remains high during epileptogenesis [91,92]. Furthermore, studies demonstrated that, during SE, astroglial cells are activated by the presence of cytokines and reactive oxygen species. This leads to decreased glutamate clearance, resulting in accumulation in the extracellular space and increased risk for neuronal excitotoxicity [71,93]. Beyond that, it was demonstrated that abnormal release of glutamate through a single astrocyte was able to activate increases in intracellular Ca2+ mediated by NMDA receptor in many neighboring neurons [23,67]. This can lead to an increase in intracellular Ca2+ in the astrocytes themselves, resulting in a further intensified release of glutamate that promotes excitotoxicity [67,69,94].
Non-NMDA receptor-mediated excitotoxicity is also relevant in the context of glutamate transporter suppression. Studies in cultured organotypic spinal cord slices were very useful to study this matter. In early studies, slow toxicity was achieved by inhibiting glutamate transport continuously, raising the concentration of glutamate concentration in the medium and resulting in degeneration of motor neurons over several weeks. This degeneration was prevented by non-NMDA receptor antagonists, glutamate synthesis or release inhibitors, but not by NMDA receptor antagonists [95,96]. Another study identified motor neurons to be selective sensible to calcium- permeable AMPA/kainate receptors [97]. Other study investigated topiramate, an anti-convulsant compound that has anti-excitotoxic properties by blocking AMPA- receptor evoked currents, for potential treatment of ALS. The study revealed that topiramate prevented motor neuron degeneration, but it did not increase survival in G93A SOD1 transgenic mice, an animal model of ALS, suggesting that it could be useful as a neuroprotectant, but were not effective in more complex motor injury paradigms such as the mouse model of ALS [98]. Importantly, AMPA receptors mediate fast synaptic excitation in brain regions pertinent to epilepsy, and AMPA receptor antagonists, such as perampanel, have been shown to decrease epileptiform activity in in vitro and in vivo studies [99].
Several studies have also indicated a causal role for excessive glutamate release by astrocytes and activation of mGluRs in the excitotoxic events leading to the synchronous firing of large populations of neurons during seizures [19,64,71,100,101].
In addition to observing that increased extracellular levels of glutamate have been associated with seizures and epilepsy, it is also noted that initiation of glutamatergic hyperactivity and seizures are facilitated by increased expression and activity of NMDA and AMPA receptors [8,102]. A study found that glutamate binding is altered in hippocampus and cortex of rats after pilocarpine-induced SE [103], and a later study using the same model expanded these findings and found an increase in NMDA receptor expression and altered AMPA receptor activity in the epileptic areas of the hippocampus [104]. Other studies demonstrated that an early event in SE in rats leads to an increase in surface expression of the GluN1 subunits of NMDA receptors along with an increase of NMDA synaptic and extra-synaptic currents in both kainate and pilocarpine induced models of epilepsy [102,105]. Additionally, autopsy samples from TLE patients have also shown that there is increased expression of NMDA, AMPA, and kainate receptors in the epileptic foci [106]. To further support this, increased NMDA channel activation in the epileptic region in the frontal cortex and temporal lobe in patients with epilepsy were revealed by positron emission tomography (PET) analysis in situ using an NMDA tracer [105,107]. Genome wide screens have also shown that p38 MAPK, which is activated upon the binding of glutamate to the NMDA receptor and can induce apoptosis [108], is overexpressed in the hippocampus of epileptic rats [109]. Prolonged SE as an initial insult has been shown to result in the internalization of GABA receptors and migration of NMDARs to neuronal synapses [110] resulting in further reduced inhibition and hyperexcitability [23]. Additionally, it has been found that SE induces posttranscriptional modifications and epigenetic changes in NMDAR subunits, which contribute to long-lasting changes in circuit connectivity [111].
In this context, the development of therapeutic approaches targeting NMDARs to reducing glutamate excitotoxicity have been proposed to prevent seizure-induced neuronal death, epileptogenesis, and subsequently spontaneous recurrent seizures. This potential strategy is reasonable, as much of the excitotoxic damage and molecular and synaptic changes that further propagate epileptogenesis are mediated by NMDARs [5,7,102]. Early studies demonstrated that NMDAR antagonist AP-V ((2R)-amino-5-phosphonovaleric acid (AP-V) inhibits epileptogenesis in in vitro models of epilepsy including the three-chamber and low magnesium models [112]. Additionally, treatment with MK-801, another NMDAR antagonist, was shown to impair epileptogenesis in the kindling model of epilepsy, in which animals are repeatedly stimulated with electrodes to activate neural pathways inducing increased susceptibility to evoked seizures, resulting in the progression to spontaneous recurrent seizures [113]. Similar results with MK-801 have also been observed in the kainic acid model of epilepsy [114]. Also, this treatment was shown to impair mossy fiber sprouting induction, a structural change to neuronal circuits that occurs during the latent period that promotes epileptogenesis [17]. MK-801 and ketamine treatments have also been shown to suppress seizures and be neuroprotective following prolonged SE in preclinical studies [17,115,116].
Another potential strategy to target NMDARs was demonstrated by inhibition of brain-specific microRNA-134, which is necessary for NMDAR-dependent spine remodeling [117]. This resulted in very efficacious prevention of spontaneous recurrent seizures months after the initial SE event in mice [17,118].
However, NMDA receptor antagonists have been found to lead to on-target side effects such as hallucinations and dissociative effects, due to the spectrum of activities that are mediated by excitatory signaling and NMDA activation [119–121]. Additionally, blockage of AMPA-receptor activity also yield in considerable adverse events, such as CNS-depressant side effects [122]. This suggests that other approaches for reducing glutamate-mediated excitotoxicity and prevent epileptogenesis should be explored.
EAAT2 and epilepsy
Many studies provide ample evidence that epileptic seizures influence the expression levels of EAATs (table 2). It is suggested that enhanced glutamate release and neuronal activity during epilepsy may result in modulation of EAATs expression. The regulatory processes of EAATs during epilepsy are not fully understood, but they seem to occur over a dynamic process that involves several factors, including compensatory mechanisms and changes in ion gradients and therefore electrochemical environment associated with epileptic hyperactivity. Despite decades of research, there still much to know about the functional characteristics of glutamate transporters and their relationship with neuropathology and behavior of epilepsy [123].
Table 2.
Involvement of astroglial transporters in genetic/pharmacological models of epilepsy syndromes. GLAST and GLT-1 are the rodent homologues, EAAT1 and EAAT2 are the human homologues.
| Transporter | Model/epilepsy syndrome | Effects | References |
|---|---|---|---|
| Knock down studies | |||
| GLAST and GLT-1 | Knock down rats | Increased extracellular glutamate
concentrations Neuronal degeneration Progressive paralysis |
[124] |
| GLT-1 | Knockout mice | Spontaneous lethal seizures at 6
weeks Increased susceptibility to acute cortical injury Increased glutamate synaptic concentrations Neurodegeneration in the hippocampus CA1 |
[46] |
| GLT-1 | Conditional deletion of astrocytic or neuronal GLT-1 in mice | Astrocytic GLT-1
deletion: - 80% loss of GLT-1 protein and of glutamate uptake activity - excess mortality - lower body weight\ - seizures Neuronal GLT-1 deletion: - normal survival - weight gain - no seizures - reduction of synaptosomal glutamate uptake capacity |
[125] |
| GLT-1 | Region-specific GLT-1 knockout mice | Selective deletion of GLT-1 in the
diencephalon, brainstem and spinal cord reproduced global GLT-1 null
mice phenotypes: - excess mortality - decreased body weight - lethal spontaneous seizure Dorsal forebrain-specific GLT-1 knockout mice resulted in nonlethal complex seizures: - myoclonic jerks - hyperkinetic running - spasm - clonic convulsion |
[126] |
| System xc− | Genetic knockout of xCT in murine model of viral-induced epilepsy | Slight decrease in neuronal
injury Increase in astrogliosis in the hippocampal CA1 Increase in GLT-1 expression in the cortex |
[127] |
| No changes in expression | |||
| GLT-1 | Rodent kindling model | GLT-1 levels remain unchanged | [128] |
| EAAT2 | Resected tissue from mTLE patients | EAAT2 levels remain unchanged | [80,129] |
| Increased expression | |||
| GLT-1 | Kainic acid-induced seizures and hippocampal organotypic slice cultures | ↑ GLT-1 expression ↓ EAAC1 expression |
[130] |
| GLAST and GLT-1 | Chronic model of epilepsy with PTZ kindling | ↑ expression of EAAT1 and EAAT2 in the
hippocampus within 24 h of PTZ kindling Levels returned to basal levels 30 days after the last seizure |
[131] |
| GLT-1 | SE model with intrahippocampal kainic acid | ↑ GLT-1 levels within 24h
post-SE At later timepoints, GLT-1 levels dropped to lower levels than in control brains |
[35,65] |
| GLT-1 | Mouse model of Lafora disease | ↑ GLT-1b associated with compensatory mechanisms activated in response to neuronal disturbance | [132] |
| Decreased expression | |||
| EAAT1 and EAAT2 | Epileptogenic hippocampus of mTLE patients | ↓ levels of EAAT1 and
EAAT2 Increased extracellular glutamate levels Neuron loss observed during and after seizures |
[67,133,134] |
| EAAT2 | Epileptogenic hippocampus of mTLE patients | ↓ levels of EAAT2 in sclerotic, but not the intact parts of the hippocampus in TLE | [135] |
| EAAT1 and EAAT2 | Temporal lobe and hippocampus in intractable temporal lobe epilepsy | ↓ expression of EAAT1 and EAAT2 in
hippocampal CA1 Neuronal death |
[67] |
| EAAT1 and EAAT2 | Epileptogenic hippocampus of mTLE patients | ↓ EAAT1 and EAAT2 expression in sclerotic hippocampi with neuronal loss in CA1 | [136] |
| EAAT2, EAAT3 and EAAT4 | Human focal epileptic brain | ↓ EAAT3 and EAAT4 mRNAs and
protein No changes in EAAT2 mRNA, but ↓ EAAT2 protein at epileptic foci |
[137] |
| EAAT2 | Whole exome sequencing followed by transfection in HEK293 cells | Identification of a recurrent de
novo Leu85Pro EAAT2 variant which
causes: - severe, early onset developmental epileptic encephalopathy via a dominant negative mechanism that modulates EAAT2 localization and function |
[146] |
| GLAST and GLT-1 | Chronic seizures induced by FeCl3 amygdalar injection in rats | ↓ GLAST and GLT-1 at 60 days in the hippocampal tissue | [148] |
| GLAST | Amygdala-kindling in rats | ↓ GLAST in the piriform cortex/amygdala
region as early as 24 h after one stage 3 seizure and persisting through
multiple stage 5 seizures ↑ EAAC1 in same region and hippocampus once animals reached stage 5 level GLT-1 levels remained unchanged |
[149] |
| GLT-1 | Induction of SE by i.p. injection of pilocarpine and kainic acid in rats | ↓ GLT-1, correlated with thalamic neuronal death | [104,150] |
| GLT-1 | Intrahippocampal kainic acid model of SE in rats | ↑ GLT-1 immunoreactivity in the
hippocampus (initially) ↓ GLT-1 at 4- and 7-days post-insult |
[65,81] |
| GLT-1 | Microarray analysis on differentially expressed genes in the pilocarpine induced mTLE model in rats | ↓ GLT-1 in the hippocampus | [151] |
| GLAST and GLT-1 | Tuberous sclerosis mouse model | ↓ GLAST and GLT-1 expression and
function: - increases in extracellular glutamate levels - excitotoxic neuronal cell death |
[152,153] |
| GLT-1 | TBI model in rats | ↓ GLT-1 expression | [154] |
| GLAST and GLT-1 | Spontaneously epileptic rat (SER) | ↓ GLAST and mGluR1 in the
hippocampus ↑ GLT-1, associated with: - tonic convulsions - absence-like seizures from 8 weeks |
[155] |
| GLAST and GLT-1 | Conditional knock-out of mGluR5 from astrocytes during epilepsy development | ↓ glutamate uptake mediated by GLAST and GLT-1 | [156] |
| GLT-1 | BBB-breakdown with deoxycholic acid and serum-derived albumin exposure to neocortex in rats | ↓ GLT-1, with reduced clearance
capacity for both extracellular glutamate and potassium ↓ GFAP, followed by delayed development of an epileptic focus |
[157] |
| GLT-1 | Endothelial Cdk5-deficient mouse model | ↓ GLT-1, decreased glutamate reuptake, increased glutamate synaptic function and spontaneous seizures | [159] |
| GLAST and GLT-1 | Pilocarpine-induced-SE in male and female mice | ↑ GLAST and GLT-1 in
females ↑ GAT-3 in males, accompanied by more hippocampal neuronal death, glial activation, higher levels of inflammatory markers, shorter latency to seizures and higher prevalence of SE in males |
[160] |
Below we discuss knockdown studies that were pivotal for our current understand of the role of EAATs in epilepsy studies, as well as studies demonstrating changes in EAATs expression in several animal models and human epilepsies, including temporal and developmental differences. Table 2 summarizes these findings, and figure 2 depicts the human mutation studied thus far
Figure 2.
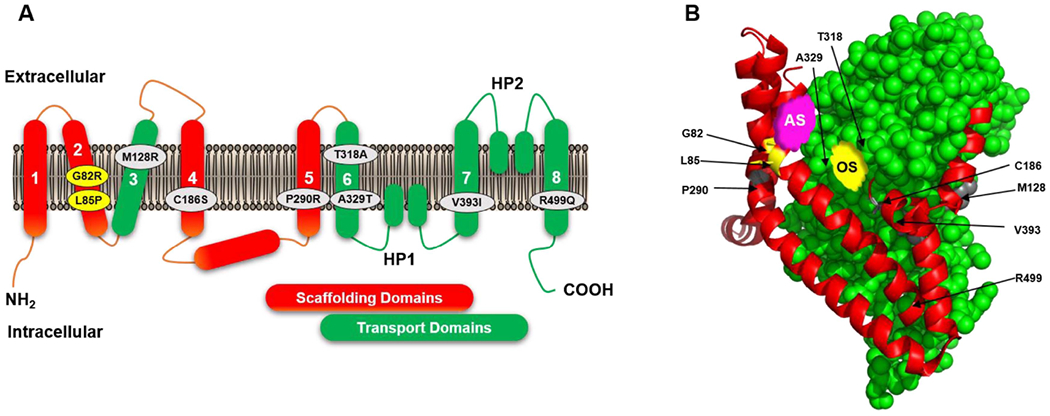
A. Two-dimensional membrane topology diagram of a single protomer of glutamate transporter, depicting eight transmembrane domains and hairpin loops, and showing the spatial distribution of EA6 and epileptic encephalopathies associated missense mutations: M128R, C186S, P290R, T3128A, A329T, V393I, R499Q in EAAT1 (in grey circles) and G28R and L85P in EAAT2 (in yellow circles). The scaffolding and transport domains are shown in red and green, respectively.
B. The tertiary structure of a single glutamate transporter protomer shown in the plane of the membrane, depicting scaffolding and transport domains in red and green, respectively, substrate and sodium binding sites (orthosteric site or OS, in yellow), a proposed allosteric site (AS in pink, from reference [188]), and positions of the EA6 and epileptic encephalopathies mutations. Structure was modelled using the crystal structure of EAAT1 in complex with L-aspartate (PDB 5LLM, ref [147]) as template, using PyMol Molecular Graphic Systems, version 2.4.1, Schrodinger, LLC.
Note: Residues G82, L85 (EAAT2 numbering), M128, C186, P290 and V393 (EAAT1 numbering) are conserved between EAAT1 and EAAT2 transporter subtypes. However, T318 in EAAT1 is M317 in EAAT2, A329 in EAAT1 is G328 in EAAT2 and R499 in EAAT1 is K498 in EAAT2.
Knock out/down studies
One of the first studies examined loss of GLAST and GLT-1 in organotypic spinal cord cultures using antisense oligonucleotides, which demonstrated an essential role of these astroglial transporters, as their loss was toxic to motor neurons. Further, this study examined GLAST and GLT-1 knockdown in vivo in rats, and observed increased extracellular glutamate concentrations, neuronal degeneration, and progressive paralysis [124]. One year later, remarkable studies by Tanaka’s group show that GLT-1 knockout mice experience spontaneous lethal seizures at 6 weeks of age and increased susceptibility to acute cortical injury, directly proving the involvement of astrocytic glutamate uptake in the prevention epileptic seizures and neuronal death [46]. These mutant mice also showed an increase in synaptic concentrations of glutamate and histological analysis of brains taken from these mice revealed neurodegeneration in the CA1 region of the hippocampus, suggesting that GLT-1 plays an important role in the maintenance of low extracellular glutamate concentrations and prevention of excitotoxicity and seizure activity.
A study analyzed conditional deletion of astrocytic or neuronal GLT-1 in mice [125]. Elimination of astrocytic GLT-1 resulted in 80% loss of GLT-1 protein and of glutamate uptake activity, excess mortality, lower body weight, and seizures, suggesting that astrocytic GLT-1 is of major importance. On the other hand, neuronal GLT-1 deletion resulted in normal survival, weight gain, and no seizures. However, the glutamate uptake capacity into synaptosomes was significantly reduced, suggesting a greater contribution to synaptosomal glutamate uptake of neuronal GLT-1 than expected [125].
Tanaka’s group recently expanded their studies to examine the consequences of GLT-1 dysfunction in different encephalic brain regions. Region-specific GLT-1 knockout mice were generated by crossing floxed-GLT-1 mice with mice expressing the Cre recombinase in a particular domain of the ventricular zone. Selective deletion of GLT-1 in the diencephalon, brainstem and spinal cord was sufficient to reproduce the phenotypes of the global GLT-1 null mice, including excess mortality, decreased body weight, and lethal spontaneous seizure. On the other hand, dorsal forebrain-specific GLT-1 knockout mice had nonlethal complex seizures including myoclonic jerks, hyperkinetic running, spasm, and clonic convulsions. This suggests that GLT-1 dysfunction in the dorsal forebrain is involved in the pathogenesis of infantile epilepsy and GLT-1 in the diencephalon, brainstem and spinal cord might be very important in the prevention of seizure-induced sudden death [126].
Although this review is focused on glial transporter EAAT2, it is noteworthy to discuss a study on the system xc−, a glial transporter that mediates the release of glutamate in the synaptic cleft in exchange for cystine and represents the key source of hippocampal extracellular glutamate. A genetic knockout of xCT (the specific subunit of system xc−) in a Theiler’s murine encephalomyelitis model of viral-induced epilepsy observed a small decrease in neuronal injury, as well as increases in astrogliosis in the CA1 hippocampal region and in cortical EAAT2 expression, adding to our knowledge for future development of novel anti-seizure therapies [127].
No changes in expression
An early study in a rodent kindling model reported that GLT-1 levels remain unchanged [128]. Two other studies in surgically removed tissue from temporal lobe of patients with epilepsy found no change in EAAT2 expression [80,129].
Further studies (below) provided abundant evidence of decreased transporter expression in epilepsy, and some report transient increased expression of transporters, followed by decreased expression. Explanations for the different observations among studies may include differences in tissue processing, stage of epileptogenesis development or severity of the epileptic disease.
Increased expression
Some studies reported that glial EAATs were increased shortly after seizures, possibly indicating acute activation of astrocytes but decreased in the later chronic phase. In an in vivo model of kainic acid-induced seizures and in hippocampal organotypic slice cultures, decreased immunoreactivity of EAAC1 neuronal transporter was observed in hippocampal regions, whereas GLT-1 expression levels were increased, indicating a differential regulation of neuronal and glial glutamate transporters that could be playing a role in seizure induction, neurotoxicity and neuronal plasticity [130]. In a chronic model of epilepsy using PTZ, GLAST and GLT-1 expression in the hippocampus increased within 24 h of kindling, however, their levels returned to baseline 30 days after the last seizure [131]. In a SE model triggered by intrahippocampal application of kainic acid, the levels of GLT-1significantly increased within 24h post-SE; but decreased to lower levels than in control brains at later timepoints [35].
Collectively, these studies show a temporal upregulation followed by downregulation in these models, providing more evidence that astrocytic glutamate transporter dysregulation contributes to the development of epilepsy.
A recent study implicated GLT-1 dysfunction and increased extracellular glutamate levels in the hippocampus in a mouse model of Lafora disease, a fatal rare disease marked the presence of insoluble polyglucosan accumulation in brain, epilepsy and neurodegeneration.. In this study, the researchers found that the minor isoform of the GLT-1 gene, GLT-1b, was upregulated in the hippocampus, and they suggested this to be associated with compensatory mechanisms triggered by neuronal disturbance. Additionally, glutamate clearance dysfunction was observed in Lafora mice, observed by a challenge with a blockade of GLT-1 that resulted in these animals to be unable to clear glutamate [132].
Decreased expression
Human patients
Several studies have reported impaired glutamate transport function in human epilepsy. Decreased levels of both EAAT1 and EAAT2, increased extracellular glutamate levels and neuron loss were observed in the epileptogenic hippocampus of patients with TLE both during and after seizures [67,133,134]. One study found that decreased levels of EAAT2 protein and alternatively spliced forms of EAAT2 were present in sclerotic, but not the intact parts of the hippocampus in TLE patients [135]. One study reported reduced expression of EAAT1 and EAAT2 in the hippocampus CA1, which the authors suggest may either be an adaptive response to neuronal death or a causative event contributing to neuronal death [67]. Another study demonstrated down regulation of EAAT1 and EAAT2 expression in sclerotic hippocampi, and pronounced neuronal loss in the CA1, whereas in the CA2 there was no decrease or even an increase in areas with less neuronal loss. This suggests compensatory mechanisms of the transporters in response to the pathology [136]. Intriguingly, significant reductions in mRNAs and protein levels of EAAT3 and EAAT4 in human focal epileptic brain regions were observed, with no changes in EAAT2 mRNA, but decreases in EAAT2 protein expression at epileptic foci, suggesting regional reductions in EAAT expression associated with increased local glutamate levels and likely contribution to both hyperexcitability and spontaneous generation of epileptic discharges [137].
Human mutations
In this section we will discuss human mutations identified in EAAT1 and EAAT2 and their relationship to epileptic phenotypes. A study looked for mutations in SLC1A3 (the gene encoding EAAT1, see table 1) in episodic ataxia 6 (EA6), a neurological disease characterized by long attack duration, absent myokymia, nystagmus, tinnitus, and epilepsy. Mutant P290R in EAAT1 was identified, and in vitro studies showed decreased EAAT1 protein expression and functionality, which could contribute to neuronal hyperexcitability [138]. Later, the same group identified mutation C186S in a patient with severe episodic and progressive ataxia, migraine headache and seizures. This mutant had a significant reduction in glutamate uptake [139]. Another study confirmed that P290R mutation resulted in reduced EAAT1 cell surface expression and decreased glutamate uptake, and add that it resulted in increased anion currents, even though the transporter expression were decreased, suggesting a gain-of-function in anion conduction as a pathological process in EA6 [140]. A later study also determined this mutation to cause glial apoptosis [141]. In a Drosophila model of ataxia, this mutation disrupted EAAT1-mediated chloride channel, and caused astrocyte malformation and paralysis episodes, suggesting that the chloride channel properties of EAAT1 is correlated to the pathophysiological mechanisms causing dysfunction of neural circuit, which could be an important and novel mechanism for epilepsy [142]. A meticulous study on several mutations (M128R, C186S, P290R, T3128A, A329T, V393I, R499Q) expressed in heterologous cells observed impairments of multiple EAAT1 properties ranging from changes in transport function, impaired trafficking to increased protein expression [143]. Collectively, these genetic and functional assays studies depict a crucial role of cerebellar transporters (as EAAT1 is mainly expressed in the cerebellum), and therefore cerebellar function, in epileptogenesis, although much still needs to be unraveled.
Regarding EAAT2 mutants, a study reported de novo mutations in SLC1A2 (gene that encodes EAAT2) as an important cause of epileptic encephalopathies, a group of severe early-onset epilepsies. These mutations include G82R and L85P, and individuals with such mutations had a very severe phenotype, marked by seizures beginning in the first week of life and profound developmental impairment, which could be associated with dysregulation of EAAT2 and glutamate excitotoxicity [144,145]. A recent study further examined recurrent de novo L85P EAAT2 variant, which was associated with early onset developmental and severe epileptic encephalopathy, through a dominant negative mechanism that decreases the expression of EAAT2 [146]. Very intriguingly, this study suggests that there is a critical amount of functional EAAT2 protein (somewhere between 0 and 50% of wild type) that correlated to seizures development. These considerations could be crucial in the development of therapeutics.
The mutation sites described here are also shown in the figure 2, in which the glutamate transporter structure was drawn based on the structure of human EAAT1 [147]. Figure 2A shows the 2-D structure, and 2B the three-dimensional protein structure. Transmembrane domains 3 and 6-8 and two hairpin loops (HP1 and HP2) form the transport domain, shown in green, which contains the substrate binding sites (or orthosteric site). Transmembrane domains 1, 2, 4 and 5 and parts of the N-terminal surrounding the central core transport domain form a peripheral rigid scaffold, shown in red. This is designated the scaffolding domain, which provides the interaction interface between three subunits of the trimer and is thought to facilitate “the elevator-like movement” of the transport domain. Figure 2B also shows an allosteric site, which was essential for the identification of allosteric modulators, discussed below.
Animal studies
Animal studies have also been pivotal in examining changes in EAAT expression in several epilepsy models. A study on chronic seizures induced by injection of FeCl3 into the amygdala found that glial transporters GLAST and GLT-1 were down-regulated at 60 days in the hippocampal tissue, indicating a disturbance of glutamate regulation caused by the downregulation of these glutamate transporters [148]. In a model of amygdala-kindled rats, levels of GLAST protein were initially downregulated in the piriform cortex/amygdala region, and later increased in this region and in the hippocampus, whereas GLT-1 levels remained unchanged, suggesting differential regulation in kindling [149]. Downregulation of GLT-1 was also shown in pilocarpine models [104] and correlated with thalamic neuronal death in kainic acid-induced models of SE as well [150]. In an intrahippocampal kainic acid model of SE, it was observed an initial increase in GLT-1 immunoreactivity in the hippocampus, , followed by a downregulation of GLT-1 at 4- and 7-days post-insult, suggesting a differential transporter regulation in the pathophysiology of epileptogenesis [65,81]. Recently, a microarray analysis on differentially expressed genes in the pilocarpine induced mTLE model determined 3,232 potential genes highly correlated with epilepsy. The SLC1A2 gene, that encodes the human EAAT2 protein, was shown to be related to brain development and its expression is significantly decreased in the hippocampus of epilepsy patients, suggesting EAAT2 as potential biomarker of epilepsy [151].
Additionally, in agreement with findings from pharmacological epilepsy models, decreased expression and function of GLAST and GLT-1, associated with increased levels of extracellular glutamate and excitotoxic neuronal death were observed in a mouse model of tuberous sclerosis, a genetic disorder associated with epilepsy [152,153]. Rats subjected to traumatic brain injury (TBI), a condition that can also lead to the development of epileptic seizures, also had decreased brain expression of EAAT2 [154]. Furthermore, the spontaneously epileptic rat (SER, a double mutant zi/zi, tm/tm), presents both tonic convulsions and absence-like seizures from 8 weeks of age. Decreased expression of EAAT1 and mGluR1 were shown in the SERs hippocampus, whereas EAAT2 is increased, suggesting that epileptogenesis in SER is associated with regulation of EAAT1, EAAT2 and mGluR1 [155]. Another study found that a conditional astrocytic knock-out of mGluR5 during epileptogenesis impairs high-frequency glutamate uptake mediated by EAAT1 and EAAT2, suggesting a compensatory response to glutamate dysregulation [156].
The role of astrocytes in epileptogenesis has been investigated in a study in two models: blood-brain barrier (BBB)-breakdown with treatment with deoxycholic acid, and direct exposure of the neocortex to serum-derived albumin. These insult models result in fast upregulation GFAP (glial fibrillary acidic protein, a marker of astrocytes), followed by the development of an epileptic focus within 4-7 days, and decreased clearance capacity for both extracellular glutamate and potassium. Frequency-dependent synaptic facilitation leading to seizure-like activity were confirmed by in vitro electrophysiological recordings during epileptogenesis. These findings suggested that during early epileptogenesis there was a transcription-mediated astrocytic transformation, which was accompanied by downregulation of EAAT2 [157]. Recently, another study reinforced the notion that BBB dysfunction plays an important role in epilepsy. In an endothelial Cdk5-deficient mouse model, the authors observed spontaneous seizures, decreased glutamate reuptake through GLT-1, and increased glutamate synaptic function. Ceftriaxone, a β-lactam antibiotic that was previously shown to increase GTL-1/ EAAT2 protein expression levels [158], was shown to restore GLT-1 function and to inhibit seizures, revealing a previously unknown link between cerebrovascular factors and epileptogenesis [159].
Recently, a study explored sex differences in a model of pilocarpine-induced-SE in mice. It was found that males had shorter latency to seizures and higher prevalence of SE, which was accompanied by more hippocampal neuronal death, glial activation, higher levels of inflammatory markers, and higher levels of GABA transporter 3 (GAT-3). On the other hand, mRNA levels of GLAST and GLT-1 were higher in female mice. These findings suggest that male mice are more vulnerable to SE than female mice, and importantly, these observed sex differences correspond to the sex differences seen in GLAST and GLT-1 mRNA expression. These findings can contribute to development of tailored therapies for both sexes [160].
Collectively, these studies suggest that the involvement of downregulated or dysfunctional GLT-1/EAAT2 in seizure onset and epileptogenesis seems to be dependent on the specific epileptic syndrome or animal model. Taken together, it appears that the expression and functional activity of glial EAATs are decreased in the chronic phase of epilepsy in experimental rodent models and in patients, possibly contributing to brain damage and epileptogenesis. However, further studies are needed to accurately determine whether transporters regulation correspond to causative or compensatory changes during epileptogenesis.
Upregulation of EAAT2 expression as potential therapeutics
Many studies discussed above demonstrated the involvement of downregulated or dysfunctional GLT-1/EAAT2 in seizure onset and epileptogenesis. In the converse situation, several studies show that upregulation of GLT-1/EAAT2 seems to be neuroprotective in epilepsy models. Table 3 summarizes the studies demonstrating potential therapeutic strategies involving modulation of GLT-1/EAAT2 transporter.
Table 3.
Potential therapeutic strategies for epilepsy involving modulation of astrocytic glutamate transporters. Administration routes for compounds are included.
| Compound/ strategy | Model/study/administration route | Effects | References |
|---|---|---|---|
| Upregulation of GLT-1/EAAT2 expression as potential therapeutics | |||
| Genetic overexpression of EAAT2 | EAAT2 transgenic
mice Pilocarpine-induced model of SE |
1.5-2-fold overexpression of EAAT2 protein
levels in the hippocampus after SE: - decreased chronic seizure frequency - decreased mortality - reduced neuronal degeneration - decreased SE-induced neurogenesis and mossy fiber sprouting |
[161] |
| LDN/OSU-0212320, pyridazine
derivative ↑ GLT-1/EAAT2 expression through translational activation |
Pilocarpine-induced TLE mouse
model Administration route: i.p. injection 3 h before pilocarpine insult |
Treatment protects cultured neurons from
glutamate excitotoxic injury and death via GLT-1
activation Reduces mice mortality, neuronal death, and spontaneous recurrent seizures |
[162] |
|
Ceftriaxone, β-lactam antibiotic ↑ GLT-1/EAAT2 expression in the transcriptional level |
In vitro studies | Restores GLT-1 function | [158,163] |
| Mouse model of tuberous
sclerosis Administration route: i.p. injections either at postnatal day 21 or at six weeks of age until death or other pre-defined endpoint of experiment |
Inhibits seizures associated with decreased
extracellular glutamate levels in the hippocampus Reduces neuronal death |
[164] | |
| TBI-induced epilepsy in
rats Administration route: i.p. injections for 7 days after TBI |
Restores GLT-1 expression in the injured
cortex Reduces seizure activity |
[154] | |
| Endothelial Cdk5-deficient
mouse model Administration route: i.p. injections for 5 days before PTZ-induced convulsions in 4-wk-old Cdh5-Cre;Cdk5f/f mice |
Restores GLT-1 function Inhibits seizures |
[159] | |
| Rat model of PTZ-induced
convulsions Administration route: i.p. injections for 4 or 7 days before PTZ-insults [165] i.p. injections for 7 days before PTZ-insults [167] i.p. injections of ceftriaxone, valproic acid or the combination daily for 7 days after PTZ- insults [168] |
Reduces acute mortality In combination with valproic acid reduces seizures and enhances motor and cognitive functions |
[165,167] [168] |
|
| Pilocarpine-induced TLE rat
model Administration route: i.p. injections for 5 days 48 hours before pilocarpine administration |
In the acute phase of epileptogenesis (first
72 hours after SE): - reduces glutamate levels - reduces GS activity - reduces GLT-1 expression In the chronic phase of epileptogenesis (4 weeks after SE): - increases GLT-1 expression - attenuates impaired learning and memory ability |
[169] | |
|
Riluzole, FDA- approved drug for ALS |
In vitro studies | ↑ EAAT2 levels, stimulates glutamate uptake, prevents glutamate release from presynaptic terminals, acts as non-specific ion channel blocker | [170–177] |
| Bipolar electrode stimulation in the right
amygdala of rats to induce kindling Administration route: i.p. injection 30 min before kindling stimulation. |
Inhibits kindled seizures and the development of behavioral seizures in kindling acquisition | [178] | |
| Mice model of PTZ-induced
convulsions Administration route: i.p. injection 30 min before PTZ insults |
Enhances the anti-seizure actions of valproate, phenobarbital and ethosuximide | [179] | |
| 4-AP infusion in the hippocampus in
rats Administration route: i.p. 30 min before 4-AP perfusion |
Showed toxicity when combined with neuronal hyperexcitation | [180] | |
| Pilocarpine-induced limbic seizure model and
in GBL-induced absence seizure model in rats Administration route: i.p. after pilocarpine-insult, or 2 h before GBL-induced absence seizure |
Inhibits pre-ictal spikes and spike-wave discharges | [181] | |
| Hsp90 inhibition | Mouse model of TLE Administration route: i.p. injection of Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17AAG) 35-45 days after KA-induced SE [164] oral gavage of Hsp90 inhibitor HSP990 4-5 weeks KA-induced SE [165] |
↑ GLT-1 levels Prevents GLT-1 degradation Suppresses spontaneous recurrent seizures |
[182,183] |
| p38 MAPK signaling inhibition | Pilocarpine-induced limbic seizure model in
rats Administration route: i.p. injection of specific inhibitor of p38 MAPK, SB203580, 30 min before pilocarpine insults |
↑ GLT-1 levels Reduces time to first epileptic seizure Attenuates seizure severity |
[184] |
| Positive Allosteric Modulation of GLT-1/EAAT2 as potential therapeutics | |||
| GT949 and GT951 PAMs of EAAT2 | In vitro studies | ↑ glutamate uptake through
EAAT2 Do not change EAAT2 expression Offer neuroprotection in vitro |
[188,189] |
|
Parawixin10 Acylpolyamine PAM of EAAT1 and EAAT2 |
Seizures induced by intrahippocampal
injection of kainic acid, NMDA and PTZ in rats Administration route: right lateral ventricle injections for 1 week, 1 h or 24 h after NMDA injection [185] Intracerebroventricular injections 10 min before insults [186] |
Offers neuroprotection and anticonvulsant properties and prevents onset of seizures | [190–192]] |
| Pilocarpine-induced limbic seizure model in
rats Administration route: right lateral ventricle injections 60 min after induction of SE, and then daily for four days |
Chronic administration of Parawixin10 results in neuroprotection, increased latency of recurrent seizures, and decreased duration and severity of seizures | [193] | |
A study successfully demonstrated that EAAT2 transgenic mice with a 1.5-2-fold overexpression of EAAT2 protein levels, had decreased chronic seizure frequency, mortality, reduced neuronal degeneration in the hippocampus after SE, and decreased SE-induced neurogenesis and mossy fiber sprouting in a pilocarpine-induced model of SE. This suggests that this strategy can be neuroprotective against SE-induced neuropathological changes, chronic seizure development and SE-induced death [161]. This group later showed that treatment with compound LDN/OSU-0212320, a pyridazine derivative they previously discovered that increases EAAT2 expression through translational activation, had neuroprotective properties in primary cultured neurons, as well in an in vivo pilocarpine-induced TLE model [162].
Moreover, several studies examined the effects of ceftriaxone, an GLT-1/EAAT2 protein enhancer that acts in the transcriptional level [158,163], in epilepsy models. Ceftriaxone treatment restored the levels of GLT-1 in a mouse model of tuberous sclerosis, which was correlated with decreased hippocampal extracellular glutamate levels and reduced neurodegeneration [153,164]. Similarly, ceftriaxone had protective effects by reducing seizure activity in a traumatic brain injury-induced epilepsy [154], in a Cdk5-knockout model that leads to spontaneous seizures in mice [159] and in an acute mortality in PTZ models of epilepsy [165–167]. A combination of treatment with ceftriaxone and valproic acid in the PTZ rat model dose-dependently reduced seizures and enhanced motor and cognitive functions [168]. A recent study reported that ceftriaxone administration in a pilocarpine model of mTLE led to a reduction of glutamate levels, and elevation of GS activity and GLT-1 expression in the acute phase. However, 4 weeks after SE glutamate levels and GLT-1 expression were decreased, and impairment of learning and memory ability were attenuated of in the chronic phase of epileptogenesis [169]. It remains to be determined whether ceftriaxone treatment for human epilepsy is clinically translatable.
Additionally, riluzole, an FDA- approved drug for amyotrophic lateral sclerosis (ALS) [170], was also investigated as potentially antiepileptic. Previously, riluzole has been reported to decrease presynaptic glutamate release [171,172], to increase EAAT2 levels [173–176], and also acts as non-specific ion channel blocker [177]. A study demonstrated that riluzole inhibited the development of behavioral seizures in kindling acquisition [178]. Riluzole was also shown to enhance the anti-seizure action of conventional ASDs, namely valproate, phenobarbital and ethosuximide, in an mice model of PTZ-induced convulsions [179]. Conversely, one study raised concerns about the repurposing of riluzole for epilepsy, as it was shown to be toxic when combined with neuronal hyperexcitation, in a model of infusion of the K+ channel blocker 4-aminopyridine (4-AP) in the hippocampus [180]. Also, riluzole treatment inhibited pre-ictal spikes and spike-wave discharges in the models of gamma-hydroxybutyrate lactone (GBL)-induced absence seizure model and in the pilocarpine-induced limbic seizure model , These findings suggest that riluzole could be developed as antiepileptic treatment against limbic seizure and absence seizure [181]. Collectively, these studies suggest that, even though riluzole has promising neuroprotective actions in some studies, and this action seems to be accomplished primarily by inhibiting excitatory neurotransmission by modulation of presynaptic release, uptake and postsynaptic activities of glutamate, the mechanisms behind these actions are not well understood. Therefore, further studies are needed before riluzole can be proposed as candidate supplementary therapy for epilepsy.
Another strategy to increase GLT-1 levels in vivo is through inhibition of Hsp90, which was shown to prevent the degradation of this transporter and to suppress spontaneous recurrent seizures in a rodent TLE model [182,183]. These studies suggest the possibility of developing therapeutics with Hsp90 inhibitors that would prevent GLT-1/EAAT2 degradation. Such a strategy, both for TLE and other excitotoxicity-related disorders, warrants future investigations.
Lastly, a study reported that inhibition of p38 MAPK signaling results in increased GLT-1 expression in the brains of rats subjected to a lithium chloride-pilocarpine epilepsy model, which resulted in decreased latency of the first epileptic seizure and attenuated severity of seizures [184]. This study suggests a potential mechanism by which GLT-1/EAAT2 expression is upregulated, adding to our current understanding of the regulation of this transporter in epilepsy.
The findings described in experimental models suggest that inducing expression of EAAT2 could be developed as a therapeutic approach to treat epilepsy and be a disease modifying course of treatment. However, this approach raises a concern about unknown off target effects in other regions of the brain due to the prevalence of EAAT2 in the brain and spinal cord, as well as possible regulation of various other proteins. Thus, enhancement of the function of EAAT2 through a direct activation of EAAT2 emerges as an alternative approach. In the next section, we will discuss positive allosteric modulation of EAAT2 to enhance transport activity, a mechanism that may serve as a novel therapeutic to provide neuroprotection and prevent spontaneous recurrent seizures and epileptogenesis.
Positive Allosteric Modulation of EAAT2 as potential therapeutics
While NMDA and AMPA receptor antagonists and calcium channel blockers may reduce glutamate excitotoxicity in epilepsy, neither approach has been sufficient in addressing the unmet medical need in treating epilepsy without causing serious adverse events [5,107,119].
As many studies propose that glutamate excitotoxicity is a key event in both the initial insult and epileptogenic process, reducing extracellular glutamate concentrations by increasing glutamate uptake via glutamate transporters offers promise in preventing neuronal death after initial insult and preventing epileptogenesis with treatment during the latent period. EAAT2, as the predominant glutamate transporter in the brain [48], appears as a promising therapeutic target. Our lab has pursued the development of compounds that selectively target EAAT2. Previous studies have demonstrated that the venom P. bistriata spider stimulates glutamate uptake in rat brain synaptosomes [185] and the purified compound Parawixin 1 selectively and directly enhances the activity of EAAT2 [186]. Further, mutagenesis studies were performed to identify the region in EAAT2 involved in transport stimulation, these investigations identified an allosteric site [187]. Using this knowledge, an in-silico approach for screening was performed using a model based on the structure of a glutamate transporter homologue from Pyrococcus horikoshii. This study identified molecules that were characterized as positive allosteric modulators (PAMs) of EAAT2 upon evaluation in glutamate uptake in transfected COS-7 cells [188]. We propose that the molecules interact with the interface between transport and scaffolding domains (shown in figure 2B), to alter the equilibrium between different conformations of the transporter and result in altered transition dynamics, ultimately resulting in increased glutamate transport. Current studies using functional, structural, and biophysical approaches are aimed at understanding the specific mechanisms that are involved in allosteric transporter modulation. Importantly, these compounds were subsequently shown to have neuroprotective properties in vitro [189]. Medicinal chemistry efforts continue to develop further lead series based on these compounds to optimize their drug-like properties.
Further studies with this class of compounds in in vitro and in vivo models of epilepsy are needed to validate their therapeutic potential in epilepsy. Nonetheless, preliminary experiments suggest that this class of compounds are neuroprotective and decrease calcium influx in primary hippocampal cultures subjected to low magnesium insults, an in vitro epilepsy model.
Recently, our group elucidated the chemical structure of another purified compound isolated from P. bistriata venom, Parawixin10. This compound is an acylpolyamine that was determined to be a non-selective positive allosteric modulator of glial transporters EAAT1 and EAAT2, with in vitro neuroprotective properties [190]. In previous studies, Parawixin10 was demonstrated to be neuroprotective and to have anticonvulsant properties in an in vivo model of intrahippocampal injection of NMDA [191]. In another study, pretreatment with Parawixin10 prevented the onset of seizures induced by kainic acid, NMDA, and PTZ [192]]. Another study demonstrated that chronic administration of Parawixin10 in the pilocarpine-induced rat model of epilepsy resulted in neuroprotection, increased the latency of recurrent seizures, and decreased duration and severity of seizures, with no behavioral deficits analyzed by the Morris water maze approach [193]. Even though it is predicted that Parawixin10 does not yield well to drug development, these studies served as proof of concept that allosteric modulation of glial transporters EAAT1 and EAAT2 may offer neuroprotection and anticonvulsant properties.
Further studies with selective EAAT2 PAM in several epilepsy models, and other models that involve glutamatergic dysfunction, will further determine whether this class of compounds can be developed as therapy to provide neuroprotection, to prevent spontaneous recurrent seizures, and to halt epileptogenesis.
Acknowledgments:
We would like to thank Katelyn L. Reeb (Drexel University) with assistance with the figures, and for support of NIH grant NS111767 to A.C.F.K.
Abbreviations:
- ALS
amyotrophic lateral sclerosis
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR
AMPA receptor
- APV
(((2R)-amino-5-phosphonovaleric acid; (2R)-amino-5-phosphonopentanoate)
- ASDs
anti-seizure drugs
- BBB
blood-brain barrier
- CDC
Centers for Disease Control and Prevention
- Cdk5
Cell division protein kinase 5
- CNS
central nervous system
- EAAC1
excitatory amino acid carrier 1
- EAATs
excitatory amino acid transporters
- EAAT1-3
human excitatory amino acid transporter subtypes 1-3
- EAAT2
human glutamate transporter 2
- EA6
episodic ataxia 6
- EEG
electroencephalogram
- 4-AP
4-aminopyridine
- GAT-3
GABA transporter 3
- GFAP
glial fibrillary acidic protein
- GAD
glutamate decarboxylase
- GLAST
glutamate and aspartate transporters
- GLT-1
rat glutamate transporter 1
- GluN1
subunit of NMDA receptor
- HP
hairpin loop
- Hsp90
heat shock protein 90
- IL-1β
Interleukin 1 beta
- IL-1R
Interleukin-1 receptor
- i.p.
intraperitoneal
- LDN/OSU-0212320
3-[[(2-Methylphenyl)methyl]thio]-6-(2-pyridinyl)-pyridazine
- NMDA
N-methyl-D-aspartic acid
- NMDAR
N-methyl-D-aspartic acid receptor
- mTLE
mesial temporal lobe epilepsy
- MRI
Magnetic Resonance Imaging
- PAMs
positive allosteric modulators
- PTZ
pentylenetetrazole
- p38 MAPK
p38 mitogen-activated protein kinases
- SE
status epilepticus
- SER
spontaneously epileptic rat
- TNFαR
tumor necrosis factor α receptor
- VEGF-3
vascular endothelial growth factor-3
- vGluT
vesicular glutamate transporter
- xCT
glutamate-cystine exchangers
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Interests
Authors have no conflicts of interest.
References
- [1].Ali R, Connolly ID, Feroze AH, Awad AJ, Choudhri OA, Grant GA, Epilepsy: A Disruptive Force in History, World neurosurgery 90 (2016) 685–690. 10.1016/j.wneu.2015.11.060. [DOI] [PubMed] [Google Scholar]
- [2].C.f.D. Control, Epilepsy Data and Statistics, https://www.cdc.gov/epilepsy/data/index.html. 2015. 2015).
- [3].Takahashi KK, Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease, Cellular and molecular life sciences : CMLS 72 3489–3506. 10.1007/s00018-015-1937-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Patel DC, Tewari BP, Chaunsali L, Sontheimer H, Neuron-glia interactions in the pathophysiology of epilepsy, Nature reviews. Neuroscience 20 (2019) 282–297. 10.1038/s41583-019-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chapman AG, Glutamate and epilepsy, The Journal of nutrition 130 (2000) 1043s–1045s. [DOI] [PubMed] [Google Scholar]
- [6].Werner FM, Covenas R, Review: Classical neurotransmitters and neuropeptides involved in generalized epilepsy in a multi-neurotransmitter system: How to improve the antiepileptic effect?, Epilepsy & behavior : E&B (2015). 10.1016/j.yebeh.2015.01.038. [DOI] [PubMed] [Google Scholar]
- [7].Walker MC, Pathophysiology of status epilepticus, Neuroscience letters 667 (2018) 84–91. 10.1016/j.neulet.2016.12.044. [DOI] [PubMed] [Google Scholar]
- [8].Albrecht J, Zielinska M, Mechanisms of Excessive Extracellular Glutamate Accumulation in Temporal Lobe Epilepsy, Neurochemical research 42 (2017) 1724–1734. 10.1007/s11064-016-2105-8. [DOI] [PubMed] [Google Scholar]
- [9].Riviello JJ, Classification of seizures and epilepsy, Current neurology and neuroscience reports 3 (2003) 325–331. [DOI] [PubMed] [Google Scholar]
- [10].Wiebe S, Epidemiology of temporal lobe epilepsy, The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiques 27 Suppl 1 (2000) S6–10; discussion S20-11. 10.1017/s0317167100000561. [DOI] [PubMed] [Google Scholar]
- [11].Aronica E, Mühlebner A, Neuropathology of epilepsy, Handbook of clinical neurology 145 (2017) 193–216. 10.1016/b978-0-12-802395-2.00015-8. [DOI] [PubMed] [Google Scholar]
- [12].Engelborghs S, D’Hooge R, De Deyn PP, Pathophysiology of epilepsy, Acta neurologica Belgica 100 (2000) 201–213. [PubMed] [Google Scholar]
- [13].Knake S, Hamer HM, Rosenow F, Status epilepticus: a critical review, Epilepsy & behavior : E&B 15 (2009) 10–14. 10.1016/j.yebeh.2009.02.027. [DOI] [PubMed] [Google Scholar]
- [14].Lothman E, The biochemical basis and pathophysiology of status epilepticus, Neurology 40 (1990) 13–23. [PubMed] [Google Scholar]
- [15].Delgado-Escueta AVA, Status epilepticus: summary, Advances in neurology 34 (1983) 537–541. [PubMed] [Google Scholar]
- [16].Dong X.-x., Wang Y, Qin Z.-h., Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases, Acta pharmacologica Sinica 30 (2009) 379–387. 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McNamara JO, Huang YZ, Leonard AS, Molecular signaling mechanisms underlying epileptogenesis, Science’s STKE : signal transduction knowledge environment 2006 (2006) re12. 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- [18].Pitkanen A, Lukasiuk K, Mechanisms of epileptogenesis and potential treatment targets, Lancet neurology 10 (2011) 173–186. 10.1016/s1474-4422(10)70310-0. [DOI] [PubMed] [Google Scholar]
- [19].Lin CL, Kong Q, Cuny GD, Glicksman MA, Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases, Future medicinal chemistry 4 (2012) 1689–1700. 10.4155/fmc.12.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jarero-Basulto JJ, Gasca-Martínez Y, Rivera-Cervantes MC, Ureña-Guerrero ME, Feria-Velasco AI, Beas-Zarate C, Interactions Between Epilepsy and Plasticity, Pharmaceuticals (Basel) 11 (2018) 17. 10.3390/ph11010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vasilev DS, Tumanova NL, Kim KK, Lavrentyeva VV, Lukomskaya NY, Zhuravin IA, Magazanik LG, Zaitsev AV, Transient Morphological Alterations in the Hippocampus After Pentylenetetrazole-Induced Seizures in Rats, Neurochemical research 43 (2018) 1671–1682. 10.1007/s11064-018-2583-y. [DOI] [PubMed] [Google Scholar]
- [22].Dhaher R, Gruenbaum SE, Sandhu MRS, Ottestad-Hansen S, Tu N, Wang Y, Lee TW, Deshpande K, Spencer DD, Danbolt NC, Zaveri HP, Eid T, Network-Related Changes in Neurotransmitters and Seizure Propagation During Rodent Epileptogenesis, Neurology (2021). 10.1212/wnl.0000000000011846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Barker-Haliski M, White HS, Glutamatergic Mechanisms Associated with Seizures and Epilepsy, Cold Spring Harbor perspectives in medicine 5 (2015) a022863. 10.1101/cshperspect.a022863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rakhade SN, Jensen FE, Epileptogenesis in the immature brain: emerging mechanisms, Nature reviews. Neurology 5 (2009) 380–391. 10.1038/nrneurol.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bialer M, White HS, Key factors in the discovery and development of new antiepileptic drugs, Nature reviews. Drug discovery 9 (2010) 68–82. 10.1038/nrd2997. [DOI] [PubMed] [Google Scholar]
- [26].Bromfield E, Cavazos J, Sirven J, Neuropharmacology of Antiepileptic Drugs, An Introduction to Epilepsy, American Epilepsy Society, West Hartford, CT, 2006. [PubMed] [Google Scholar]
- [27].Rogawski MA, Cavazos JE, Mechanisms of action of antiepileptic drugs, Wyllie’s Treatment of Epilepsy: Principles and Practice: Sixth Edition2015. [Google Scholar]
- [28].Löscher W, Potschka H, Sisodiya SM, Vezzani A, Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options, Pharmacological reviews 72 (2020) 606–638. 10.1124/pr.120.019539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen Z, Brodie MJ, Liew D, Kwan P, Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study, JAMA neurology 75 (2018) 279–286. 10.1001/jamaneurol.2017.3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sankar R, Holmes GL, Mechanisms of action for the commonly used antiepileptic drugs: relevance to antiepileptic drug-associated neurobehavioral adverse effects, Journal of child neurology 19 Suppl 1 (2004) S6–14. 10.1177/088307380401900102. [DOI] [PubMed] [Google Scholar]
- [31].de Kinderen RJ, Evers SM, Rinkens R, Postulart D, Vader CI, Majoie MH, Aldenkamp AP, Side-effects of antiepileptic drugs: The economic burden, Seizure : the journal of the British Epilepsy Association 23 (2014) 184–190. 10.1016/j.seizure.2013.11.009. [DOI] [PubMed] [Google Scholar]
- [32].Cramer JA, Mintzer S, Wheless J, Mattson RH, Adverse effects of antiepileptic drugs: a brief overview of important issues, Expert review of neurotherapeutics 10 (2010) 885–891. 10.1586/ern.10.71. [DOI] [PubMed] [Google Scholar]
- [33].Zwart R, Sher E, Ping X, Jin X, Sims JR Jr., Chappell AS, Gleason SD, Hahn PJ, Gardinier K, Gernert DL, Hobbs J, Smith JL, Valli SN, Witkin JM, Perampanel, an antagonist of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, for the treatment of epilepsy: studies in human epileptic brain and nonepileptic brain and in rodent models, The Journal of pharmacology and experimental therapeutics 351 (2014) 124–133. 10.1124/jpet.114.212779. [DOI] [PubMed] [Google Scholar]
- [34].Klein P, Tyrlikova I, No prevention or cure of epilepsy as yet, Neuropharmacology 168 (2020) 107762. 10.1016/j.neuropharm.2019.107762. [DOI] [PubMed] [Google Scholar]
- [35].Peterson AR, Binder DK, Regulation of Synaptosomal GLT-1 and GLAST during Epileptogenesis, Neuroscience 411 (2019) 185–201. 10.1016/j.neuroscience.2019.05.048. [DOI] [PubMed] [Google Scholar]
- [36].Fontana AC, Current approaches to enhance glutamate transporter function and expression, Journal of neurochemistry 134 (2015) 982–1007. 10.1111/jnc.13200. [DOI] [PubMed] [Google Scholar]
- [37].Sheldon AL, Robinson MB, The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention, Neurochemistry international 51 (2007) 333–355. 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Willard SS, Koochekpour S, Glutamate, glutamate receptors, and downstream signaling pathways, Int J Biol Sci 9 (2013) 948–959. 10.7150/ijbs.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Danbolt NC, Glutamate uptake, Progress in neurobiology 65 (2001) 1–105. [DOI] [PubMed] [Google Scholar]
- [40].Vandenberg RJ, Ryan RM, Mechanisms of glutamate transport, Physiological reviews 93 (2013) 1621–1657. 10.1152/physrev.00007.2013. [DOI] [PubMed] [Google Scholar]
- [41].van den Berg CJ, Garfinkel D, A simulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain, The Biochemical journal 123 (1971) 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Westergaard N, Sonnewald U, Schousboe A, Metabolic trafficking between neurons and astrocytes: the glutamate/glutamine cycle revisited, Developmental neuroscience 17 (1995) 203–211. 10.1159/000111288. [DOI] [PubMed] [Google Scholar]
- [43].Shigeri Y, Seal RP, Shimamoto K, Molecular pharmacology of glutamate transporters, EAATs and VGLUTs, Brain research. Brain research reviews 45 (2004) 250–265. 10.1016/j.brainresrev.2004.04.004. [DOI] [PubMed] [Google Scholar]
- [44].Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI, Cloning and expression of a rat brain L-glutamate transporter, Nature 360 (1992) 464–467. 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- [45].Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danbolt NC, Brain glutamate transporter proteins form homomultimers, The Journal of biological chemistry 271 (1996) 27715–27722. [DOI] [PubMed] [Google Scholar]
- [46].Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K, Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1, Science 276 (1997) 1699–1702. [DOI] [PubMed] [Google Scholar]
- [47].Lehre KP, Danbolt NC, The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain, The Journal of neuroscience : the official journal of the Society for Neuroscience 18 (1998) 8751–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Suchak SK, Baloyianni NV, Perkinton MS, Williams RJ, Meldrum BS, Rattray M, The ‘glial’ glutamate transporter, EAAT2 (Glt-1) accounts for high affinity glutamate uptake into adult rodent nerve endings, Journal of neurochemistry 84 (2003) 522–532. [DOI] [PubMed] [Google Scholar]
- [49].Scofield MD, Kalivas PW, Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis, The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 20 (2014) 610–622. 10.1177/1073858413520347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Maragakis NJ, Dykes-Hoberg M, Rothstein JD, Altered expression of the glutamate transporter EAAT2b in neurological disease, Annals of neurology 55 (2004) 469–477. 10.1002/ana.20003. [DOI] [PubMed] [Google Scholar]
- [51].Lauriat TL, McInnes LA, EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders, Molecular psychiatry 12 (2007) 1065. 10.1038/sj.mp.4002065. [DOI] [PubMed] [Google Scholar]
- [52].Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, Dasgupta S, Barral PM, Hedvat M, Diaz P, Reed JC, Stebbins JL, Pellecchia M, Sarkar D, Fisher PB, Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics, Journal of cellular physiology 226 (2011) 2484–2493. 10.1002/jcp.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nilsson P, Hillered L, Ponten U, Ungerstedt U, Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 10 (1990) 631–637. 10.1038/jcbfm.1990.115. [DOI] [PubMed] [Google Scholar]
- [54].Vespa P, Prins M, Ronne-Engstrom E, Caron M, Shalmon E, Hovda DA, Martin NA, Becker DP, Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: a microdialysis study, Journal of neurosurgery 89 (1998) 971–982. 10.3171/jns.1998.89.6.0971. [DOI] [PubMed] [Google Scholar]
- [55].Faden AI, Demediuk P, Panter SS, Vink R, The role of excitatory amino acids and NMDA receptors in traumatic brain injury, Science 244 (1989) 798–800. [DOI] [PubMed] [Google Scholar]
- [56].Raghupathi R, Cell death mechanisms following traumatic brain injury, Brain Pathol 14 (2004) 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lewerenz J, Klein M, Methner A, Cooperative action of glutamate transporters and cystine/glutamate antiporter system Xc- protects from oxidative glutamate toxicity, Journal of neurochemistry 98 (2006) 916–925. 10.1111/j.1471-4159.2006.03921.x. [DOI] [PubMed] [Google Scholar]
- [58].Aniksztejn L, Cattani AA, Represa A, Milh M, Villeneuve N, TRANSPORTERS | Function of Cell-Surface Glutamate Transporters in the Brain: An Important Role for Development and Preventing Seizures, in: Schwartzkroin PA (Ed.) Encyclopedia of Basic Epilepsy Research, Academic Press, Oxford, 2009, pp. 1373–1379. [Google Scholar]
- [59].Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, Bannai S, Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain, The Journal of neuroscience : the official journal of the Society for Neuroscience 22 (2002) 8028–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sorensen MF, Heimisdottir SB, Sorensen MD, Mellegaard CS, Wohlleben H, Kristensen BW, Beier CP, High expression of cystine-glutamate antiporter xCT (SLC7A11) is an independent biomarker for epileptic seizures at diagnosis in glioma, Journal of neuro-oncology 138 (2018) 49–53. 10.1007/s11060-018-2785-9. [DOI] [PubMed] [Google Scholar]
- [61].Zorec R, Araque A, Carmignoto G, Haydon PG, Verkhratsky A, Parpura V, Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route, ASN neuro 4 (2012). 10.1042/an20110061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Perea G, Navarrete M, Araque A, Tripartite synapses: astrocytes process and control synaptic information, Trends in neurosciences 32 (2009) 421–431. 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- [63].Peterson AR, Binder DK, Astrocyte Glutamate Uptake and Signaling as Novel Targets for Antiepileptogenic Therapy, Frontiers in neurology 11 (2020) 1006–1006. 10.3389/fneur.2020.01006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wetherington J, Serrano G, Dingledine R, Astrocytes in the Epileptic Brain, Neuron 58 (2008) 168–178. 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hubbard JA, Szu JI, Yonan JM, Binder DK, Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy, Experimental neurology 283 (2016) 85–96. 10.1016/j.expneurol.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sofroniew MV, Vinters HV, Astrocytes: biology and pathology, Acta neuropathologica 119 (2010) 7–35. 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sarac S, Afzal S, Broholm H, Madsen FF, Ploug T, Laursen H, Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy, APMIS : acta pathologica, microbiologica, et immunologica Scandinavica 117 (2009) 291–301. 10.1111/j.1600-0463.2009.02443.x. [DOI] [PubMed] [Google Scholar]
- [68].Tilleux S, Hermans E, Neuroinflammation and regulation of glial glutamate uptake in neurological disorders, Journal of neuroscience research 85 (2007) 2059–2070. 10.1002/jnr.21325. [DOI] [PubMed] [Google Scholar]
- [69].Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E, Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ, The Journal of neuroscience : the official journal of the Society for Neuroscience 22 (2002) 9430–9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Haroon E, Miller AH, Sanacora G, Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders, Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 42 (2017) 193–215. 10.1038/npp.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Vargas-Sanchez K, Mogilevskaya M, Rodriguez-Perez J, Rubiano MG, Javela JJ, Gonzalez-Reyes RE, Astroglial role in the pathophysiology of status epilepticus: an overview, Oncotarget 9 (2018) 26954–26976. 10.18632/oncotarget.25485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Vezzani A, Baram TZ, New roles for interleukin-1 Beta in the mechanisms of epilepsy, Epilepsy currents / American Epilepsy Society 7 (2007) 45–50. 10.1111/j.1535-7511.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Crino PB, Mechanistic target of rapamycin (mTOR) signaling in status epilepticus, Epilepsy & behavior : E&B 101 (2019) 106550. 10.1016/j.yebeh.2019.106550. [DOI] [PubMed] [Google Scholar]
- [74].Jeong KH, Cho KO, Lee MY, Kim SY, Kim WJ, Vascular endothelial growth factor receptor-3 regulates astroglial glutamate transporter-1 expression via mTOR activation in reactive astrocytes following pilocarpine-induced status epilepticus, Glia 69 (2021) 296–309. 10.1002/glia.23897. [DOI] [PubMed] [Google Scholar]
- [75].Robinson MB, Jackson JG, Astroglial glutamate transporters coordinate excitatory signaling and brain energetics, Neurochemistry international (2020). 10.1016/j.neuint.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Boison D, Steinhauser C, Epilepsy and astrocyte energy metabolism, Glia 66 (2018) 1235–1243. 10.1002/glia.23247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Robinson MB, Lee ML, DaSilva S, Glutamate Transporters and Mitochondria: Signaling, Co-compartmentalization, Functional Coupling, and Future Directions, Neurochemical research 45 (2020) 526–540. 10.1007/s11064-020-02974-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Onodera M, Meyer J, Furukawa K, Hiraoka Y, Aida T, Tanaka K, Tanaka KF, Rose CR, Matsui K, Exacerbation of epilepsy by astrocyte alkalization and gap junction uncoupling, The Journal of Neuroscience (2021) JN-RM-2365-2320. 10.1523/jneurosci.2365-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Rogawski MA, Loscher W, Rho JM, Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet, Cold Spring Harbor perspectives in medicine 6 (2016). 10.1101/cshperspect.a022780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, de Lanerolle NC, Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy, Lancet 363 (2004) 28–37. [DOI] [PubMed] [Google Scholar]
- [81].Eid T, Lee TW, Patrylo P, Zaveri HP, Astrocytes and Glutamine Synthetase in Epileptogenesis, Journal of neuroscience research 97 (2019) 1345–1362. 10.1002/jnr.24267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zhou Y, Dhaher R, Parent M, Hu QX, Hassel B, Yee SP, Hyder F, Gruenbaum SE, Eid T, Danbolt NC, Selective deletion of glutamine synthetase in the mouse cerebral cortex induces glial dysfunction and vascular impairment that precede epilepsy and neurodegeneration, Neurochemistry international 123 (2019) 22–33. 10.1016/j.neuint.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA, Glia and epilepsy: excitability and inflammation, Trends in neurosciences 36 174–184. 10.1016/j.tins.2012.11.008. [DOI] [PubMed] [Google Scholar]
- [84].Bedner P, Steinhäuser C, Neuron-glia interaction in epilepsy, Journal of neuroscience research 94 (2016) 779–780. 10.1002/jnr.23800. [DOI] [PubMed] [Google Scholar]
- [85].Olney JW, Collins RC, Sloviter RS, Excitotoxic mechanisms of epileptic brain damage, Advances in neurology 44 (1986) 857–877. [PubMed] [Google Scholar]
- [86].Smolders I, Khan GM, Manil J, Ebinger G, Michotte Y, NMDA receptor-mediated pilocarpine-induced seizures: characterization in freely moving rats by microdialysis, British journal of pharmacology 121 (1997) 1171–1179. 10.1038/sj.bjp.0701231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].During MJ, Spencer DD, Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain, Lancet 341 (1993) 1607–1610. [DOI] [PubMed] [Google Scholar]
- [88].Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, Krystal JH, Spencer DD, Abi-Saab WM, Extracellular metabolites in the cortex and hippocampus of epileptic patients, Annals of neurology 57 (2005) 226–235. 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- [89].McDonough JH Jr., Shih TM, Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology, Neuroscience and biobehavioral reviews 21 (1997) 559–579. 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- [90].Thomas PM, Phillips JP, O’Connor WT, Hippocampal microdialysis during spontaneous intraoperative epileptiform activity, Acta neurochirurgica 146 (2004) 143–151. 10.1007/s00701-003-0189-9. [DOI] [PubMed] [Google Scholar]
- [91].Coulter DA, Eid T, Astrocytic regulation of glutamate homeostasis in epilepsy, Glia 60 (2012) 1215–1226. 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jabs R, Seifert G, Steinhauser C, Astrocytic function and its alteration in the epileptic brain, Epilepsia 49 Suppl 2 (2008) 3–12. 10.1111/j.1528-1167.2008.01488.x. [DOI] [PubMed] [Google Scholar]
- [93].Matute C, Domercq M, Sanchez-Gomez MV, Glutamate-mediated glial injury: mechanisms and clinical importance, Glia 53 (2006) 212–224. 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- [94].Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG, Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus, The Journal of neuroscience : the official journal of the Society for Neuroscience 27 (2007) 10674–10684. 10.1523/jneurosci.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW, Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity, Proceedings of the National Academy of Sciences of the United States of America 90 (1993) 6591–6595. 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Rothstein JD, Kuncl RW, Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity, Journal of neurochemistry 65 (1995) 643–651. [DOI] [PubMed] [Google Scholar]
- [97].Bar-Peled O, O’Brien RJ, Morrison JH, Rothstein JD, Cultured motor neurons possess calcium-permeable AMPA/kainate receptors, Neuroreport 10 (1999) 855–859. 10.1097/00001756-199903170-00034. [DOI] [PubMed] [Google Scholar]
- [98].Maragakis NJ, Jackson M, Ganel R, Rothstein JD, Topiramate protects against motor neuron degeneration in organotypic spinal cord cultures but not in G93A SOD1 transgenic mice, Neuroscience letters 338 (2003) 107–110. 10.1016/s0304-3940(02)01386-1. [DOI] [PubMed] [Google Scholar]
- [99].Rogawski MA, AMPA receptors as a molecular target in epilepsy therapy, Acta Neurol Scand Suppl (2013) 9–18. 10.1111/ane.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M, An astrocytic basis of epilepsy, Nature medicine 11 (2005) 973–981. 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Benarroch EE, Astrocyte-neuron interactions: implications for epilepsy, Neurology 73 (2009) 1323–1327. 10.1212/WNL.0b013e3181bd432d. [DOI] [PubMed] [Google Scholar]
- [102].Wasterlain CG, Naylor DE, Liu H, Niquet J, Baldwin R, Trafficking of NMDA receptors during status epilepticus: therapeutic implications, Epilepsia 54 Suppl 6 (2013) 78–80. 10.1111/epi.12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Cunha AO, Mortari MR, Carolino RO, Coutinho-Netto J, Dos Santos WF, Glutamate binding is altered in hippocampus and cortex of Wistar rats after pilocarpine-induced Status Epilepticus, Neuroscience letters 424 (2007) 51–54. 10.1016/j.neulet.2007.07.010. [DOI] [PubMed] [Google Scholar]
- [104].Lopes MW, Soares FM, de Mello N, Nunes JC, Cajado AG, de Brito D, de Cordova FM, da Cunha RM, Walz R, Leal RB, Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy, Experimental brain research 226 (2013) 153–163. 10.1007/s00221-013-3421-8. [DOI] [PubMed] [Google Scholar]
- [105].Frasca A, Aalbers M, Frigerio F, Fiordaliso F, Salio M, Gobbi M, Cagnotto A, Gardoni F, Battaglia GS, Hoogland G, Di Luca M, Vezzani A, Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity, Neurobiology of disease 43 (2011) 507–515. 10.1016/j.nbd.2011.04.024. [DOI] [PubMed] [Google Scholar]
- [106].Das A, Wallace G.C.t., Holmes C, McDowell ML, Smith JA, Marshall JD, Bonilha L, Edwards JC, Glazier SS, Ray SK, Banik NL, Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors, Neuroscience 220 (2012) 237–246. 10.1016/j.neuroscience.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].McGinnity CJ, Koepp MJ, Hammers A, Riano Barros DA, Pressler RM, Luthra S, Jones PA, Trigg W, Micallef C, Symms MR, Brooks DJ, Duncan JS, NMDA receptor binding in focal epilepsies, Journal of neurology, neurosurgery, and psychiatry 86 (2015) 1150–1157. 10.1136/jnnp-2014-309897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, Hvalby O, Nagelhus EA, Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10), Glia 59 (2011) 1635–1642. 10.1002/glia.21205. [DOI] [PubMed] [Google Scholar]
- [109].Okamoto OK, Janjoppi L, Bonone FM, Pansani AP, da Silva AV, Scorza FA, Cavalheiro EA, Whole transcriptome analysis of the hippocampus: toward a molecular portrait of epileptogenesis, BMC genomics 11 (2010) 230. 10.1186/1471-2164-11-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Naylor DE, Liu H, Niquet J, Wasterlain CG, Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus, Neurobiology of disease 54 (2013) 225–238. 10.1016/j.nbd.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ryley Parrish R, Albertson AJ, Buckingham SC, Hablitz JJ, Mascia KL, Davis Haselden W, Lubin FD, Status epilepticus triggers early and late alterations in brain-derived neurotrophic factor and NMDA glutamate receptor Grin2b DNA methylation levels in the hippocampus, Neuroscience 248 (2013) 602–619. 10.1016/j.neuroscience.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].DeLorenzo RJ, Pal S, Sombati S, Prolonged activation of the N-methyl-D-aspartate receptor-Ca2+ transduction pathway causes spontaneous recurrent epileptiform discharges in hippocampal neurons in culture, Proceedings of the National Academy of Sciences of the United States of America 95 (1998) 14482–14487. 10.1073/pnas.95.24.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Sutula T, Koch J, Golarai G, Watanabe Y, McNamara JO, NMDA Receptor Dependence of Kindling and Mossy Fiber Sprouting: Evidence that the NMDA Receptor Regulates Patterning of Hippocampal Circuits in the Adult Brain, The Journal of Neuroscience 16 (1996) 7398–7406. 10.1523/jneurosci.16-22-07398.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].McNamara RK, Routtenberg A, NMDA receptor blockade prevents kainate induction of protein F1/GAP-43 mRNA in hippocampal granule cells and subsequent mossy fiber sprouting in the rat, Brain research. Molecular brain research 33 (1995) 22–28. 10.1016/0169-328x(95)00083-5. [DOI] [PubMed] [Google Scholar]
- [115].Dorandeu F, Barbier L, Dhote F, Testylier G, Carpentier P, Ketamine combinations for the field treatment of soman-induced self-sustaining status epilepticus. Review of current data and perspectives, Chemico-biological interactions 203 (2013) 154–159. 10.1016/j.cbi.2012.09.013. [DOI] [PubMed] [Google Scholar]
- [116].Dorandeu F, Dhote F, Barbier L, Baccus B, Testylier G, Treatment of status epilepticus with ketamine, are we there yet?, CNS Neurosci Ther 19 (2013) 411–427. 10.1111/cns.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME, A brain-specific microRNA regulates dendritic spine development, Nature 439 (2006) 283–289. 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- [118].Jimenez-Mateos EM, Engel T, Merino-Serrais P, McKiernan RC, Tanaka K, Mouri G, Sano T, O’Tuathaigh C, Waddington JL, Prenter S, Delanty N, Farrell MA, O’Brien DF, Conroy RM, Stallings RL, DeFelipe J, Henshall DC, Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects, Nature medicine 18 (2012) 1087–1094. 10.1038/nm.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wada Y, Hasegawa H, Nakamura M, Yamaguchi N, The NMDA receptor antagonist MK-801 has a dissociative effect on seizure activity of hippocampal-kindled cats, Pharmacology Biochemistry and Behavior 43 (1992) 1269–1272. 10.1016/0091-3057(92)90513-F. [DOI] [PubMed] [Google Scholar]
- [120].Lipton SA, Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults, NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics 1 (2004) 101–110. 10.1602/neurorx.1.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Lim DK, Ketamine associated psychedelic effects and dependence, Singapore medical journal 44 (2003) 31–34. [PubMed] [Google Scholar]
- [122].Hanada T, Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors, Biomolecules 10 (2020). 10.3390/biom10030464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zaitsev AV, Smolensky IV, Jorratt P, Ovsepian SV, Neurobiology, Functions, and Relevance of Excitatory Amino Acid Transporters (EAATs) to Treatment of Refractory Epilepsy, CNS drugs 34 (2020) 1089–1103. 10.1007/s40263-020-00764-y. [DOI] [PubMed] [Google Scholar]
- [124].Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF, Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate, Neuron 16 (1996) 675–686. [DOI] [PubMed] [Google Scholar]
- [125].Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA, Conditional Deletion of the Glutamate Transporter GLT-1 Reveals That Astrocytic GLT-1 Protects against Fatal Epilepsy While Neuronal GLT-1 Contributes Significantly to Glutamate Uptake into Synaptosomes, The Journal of neuroscience : the official journal of the Society for Neuroscience 35 (2015) 5187–5201. 10.1523/jneurosci.4255-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Sugimoto J, Tanaka M, Sugiyama K, Ito Y, Aizawa H, Soma M, Shimizu T, Mitani A, Tanaka K, Region-specific deletions of the glutamate transporter GLT1 differentially affect seizure activity and neurodegeneration in mice, Glia 66 (2018) 777–788. 10.1002/glia.23281. [DOI] [PubMed] [Google Scholar]
- [127].Loewen JL, Albertini G, Dahle EJ, Sato H, Smolders IJ, Massie A, Wilcox KS, Genetic and pharmacological manipulation of glial glutamate transporters does not alter infection-induced seizure activity, Experimental neurology 318 (2019) 50–60. 10.1016/j.expneurol.2019.04.010. [DOI] [PubMed] [Google Scholar]
- [128].Akbar MT, Torp R, Danbolt NC, Levy LM, Meldrum BS, Ottersen OP, Expression of glial glutamate transporters GLT-1 and GLAST is unchanged in the hippocampus in fully kindled rats, Neuroscience 78 (1997) 351–359. [DOI] [PubMed] [Google Scholar]
- [129].Tessler S, Danbolt NC, Faull RL, Storm-Mathisen J, Emson PC, Expression of the glutamate transporters in human temporal lobe epilepsy, Neuroscience 88 (1999) 1083–1091. [DOI] [PubMed] [Google Scholar]
- [130].Simantov R, Crispino M, Hoe W, Broutman G, Tocco G, Rothstein JD, Baudry M, Changes in expression of neuronal and glial glutamate transporters in rat hippocampus following kainate-induced seizure activity, Brain research. Molecular brain research 65 (1999) 112–123. [DOI] [PubMed] [Google Scholar]
- [131].Doi T, Ueda Y, Nagatomo K, Willmore LJ, Role of glutamate and GABA transporters in development of pentylenetetrazol-kindling, Neurochemical research 34 (2009) 1324–1331. 10.1007/s11064-009-9912-0. [DOI] [PubMed] [Google Scholar]
- [132].Munoz-Ballester C, Santana N, Perez-Jimenez E, Viana R, Artigas F, Sanz P, In vivo glutamate clearance defects in a mouse model of Lafora disease, Experimental neurology 320 (2019) 112959. 10.1016/j.expneurol.2019.112959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L, Born DE, Sakamoto AC, Assirati JA, Fried I, Peacock WJ, Ojemann GA, Adelson PD, Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy, Neurology 52 (1999) 453–472. [DOI] [PubMed] [Google Scholar]
- [134].Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, de Graan PN, Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy, Brain : a journal of neurology 125 (2002) 32–43. 10.1093/brain/awf001. [DOI] [PubMed] [Google Scholar]
- [135].Hoogland G, van Oort RJ, Proper EA, Jansen GH, van Rijen PC, van Veelen CW, van Nieuwenhuizen O, Troost D, de Graan PN, Alternative splicing of glutamate transporter EAAT2 RNA in neocortex and hippocampus of temporal lobe epilepsy patients, Epilepsy research 59 (2004) 75–82. 10.1016/j.eplepsyres.2004.03.003. [DOI] [PubMed] [Google Scholar]
- [136].Bjornsen LP, Eid T, Holmseth S, Danbolt NC, Spencer DD, de Lanerolle NC, Changes in glial glutamate transporters in human epileptogenic hippocampus: inadequate explanation for high extracellular glutamate during seizures, Neurobiology of disease 25 (2007) 319–330. 10.1016/j.nbd.2006.09.014. [DOI] [PubMed] [Google Scholar]
- [137].Rakhade SN, Loeb JA, Focal reduction of neuronal glutamate transporters in human neocortical epilepsy, Epilepsia 49 (2008) 226–236. 10.1111/j.1528-1167.2007.01310.x. [DOI] [PubMed] [Google Scholar]
- [138].Jen JC, Wan J, Palos TP, Howard BD, Baloh RW, Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures, Neurology 65 (2005) 529–534. 10.1212/01.wnl.0000172638.58172.5a. [DOI] [PubMed] [Google Scholar]
- [139].de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, Haan J, Terwindt GM, Boon EM, Howard BD, Frants RR, Baloh RW, Ferrari MD, Jen JC, van den Maagdenberg AM, Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake, Archives of neurology 66 (2009) 97–101. 10.1001/archneurol.2008.535. [DOI] [PubMed] [Google Scholar]
- [140].Winter N, Kovermann P, Fahlke C, A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents, Brain : a journal of neurology 135 (2012) 3416–3425. 10.1093/brain/aws255. [DOI] [PubMed] [Google Scholar]
- [141].Kovermann P, Untiet V, Kolobkova Y, Engels M, Baader S, Schilling K, Fahlke C, Increased glutamate transporter-associated anion currents cause glial apoptosis in episodic ataxia 6, Brain Commun 2 (2020) fcaa022. 10.1093/braincomms/fcaa022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Parinejad N, Peco E, Ferreira T, Stacey SM, van Meyel DJ, Disruption of an EAAT-Mediated Chloride Channel in a Drosophila Model of Ataxia, The Journal of neuroscience : the official journal of the Society for Neuroscience 36 (2016) 7640–7647. 10.1523/jneurosci.0197-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Chivukula AS, Suslova M, Kortzak D, Kovermann P, Fahlke C, Functional consequences of SLC1A3 mutations associated with episodic ataxia 6, Human mutation 41 (2020) 1892–1905. 10.1002/humu.24089. [DOI] [PubMed] [Google Scholar]
- [144].De Novo Mutations in SLC1A2 and CACNA1A Are Important Causes of Epileptic Encephalopathies, American journal of human genetics 99 (2016) 287–298. 10.1016/j.ajhg.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Guella I, McKenzie MB, Evans DM, Buerki SE, Toyota EB, Van Allen MI, Adam S, Boelman C, Bolbocean C, Candido T, Eydoux P, Horvath G, Huh L, Nelson TN, Sinclair G, van Karnebeek C, Vercauteren S, Suri M, Elmslie F, Simon MEH, van Gassen KLI, Héron D, Keren B, Nava C, Connolly MB, Demos M, Farrer MJ, De Novo Mutations in YWHAG Cause Early-Onset Epilepsy, The American Journal of Human Genetics 101 (2017) 300–310. 10.1016/j.ajhg.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Stergachis AB, Pujol-Gimenez J, Gyimesi G, Fuster D, Albano G, Troxler M, Picker J, Rosenberg PA, Bergin A, Peters J, El Achkar CM, Harini C, Manzi S, Rotenberg A, Hediger MA, Rodan LH, Recurrent SLC1A2 variants cause epilepsy via a dominant negative mechanism, Annals of neurology 85 (2019) 921–926. 10.1002/ana.25477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Canul-Tec JC, Assal R, Cirri E, Legrand P, Brier S, Chamot-Rooke J, Reyes N, Structure and allosteric inhibition of excitatory amino acid transporter 1, Nature 544 (2017) 446–451. 10.1038/nature22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Ueda Y, Doi T, Nagatomo K, Willmore LJ, Nakajima A, Functional role for redox in the epileptogenesis: molecular regulation of glutamate in the hippocampus of FeCl3-induced limbic epilepsy model, Experimental brain research 181 (2007) 571–577. 10.1007/s00221-007-0954-8. [DOI] [PubMed] [Google Scholar]
- [149].Miller HP, Levey AI, Rothstein JD, Tzingounis AV, Conn PJ, Alterations in glutamate transporter protein levels in kindling-induced epilepsy, Journal of neurochemistry 68 (1997) 1564–1570. [DOI] [PubMed] [Google Scholar]
- [150].Sakurai M, Kurokawa H, Shimada A, Nakamura K, Miyata H, Morita T, Excitatory amino acid transporter 2 downregulation correlates with thalamic neuronal death following kainic acid-induced status epilepticus in rat, Neuropathology : official journal of the Japanese Society of Neuropathology 35 (2015) 1–9. 10.1111/neup.12141. [DOI] [PubMed] [Google Scholar]
- [151].Zhang Y, Dong H, Duan L, Yuan G, Liang W, Li Q, Zhang X, Pan Y, SLC1A2 mediates refractory temporal lobe epilepsy with an initial precipitating injury by targeting the glutamatergic synapse pathway, IUBMB life 71 (2019) 213–222. 10.1002/iub.1956. [DOI] [PubMed] [Google Scholar]
- [152].Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, Mennerick S, Yamada KA, Gutmann DH, Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model, Annals of neurology 54 (2003) 251–256. 10.1002/ana.10648. [DOI] [PubMed] [Google Scholar]
- [153].Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC, Yamada KA, Wozniak DF, Holtzman DM, Gutmann DH, Wong M, Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex, Neurobiology of disease 28 (2007) 184–196. 10.1016/j.nbd.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A, Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat, Journal of neurotrauma 30 (2013) 1434–1441. 10.1089/neu.2012.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Guo F, Sun F, Yu JL, Wang QH, Tu DY, Mao XY, Liu R, Wu KC, Xie N, Hao LY, Cai JQ, Abnormal expressions of glutamate transporters and metabotropic glutamate receptor 1 in the spontaneously epileptic rat hippocampus, Brain research bulletin 81 (2010) 510–516. 10.1016/j.brainresbull.2009.10.008. [DOI] [PubMed] [Google Scholar]
- [156].Umpierre AD, West PJ, White JA, Wilcox KS, Conditional Knock-out of mGluR5 from Astrocytes during Epilepsy Development Impairs High-Frequency Glutamate Uptake, The Journal of Neuroscience 39 (2019) 727–742. 10.1523/jneurosci.1148-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A, Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis?, The Journal of neuroscience : the official journal of the Society for Neuroscience 29 (2009) 10588–10599. 10.1523/jneurosci.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB, Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression, Nature 433 (2005) 73–77. 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- [159].Liu XX, Yang L, Shao LX, He Y, Wu G, Bao YH, Lu NN, Gong DM, Lu YP, Cui TT, Sun NH, Chen DY, Shi WX, Fukunaga K, Chen HS, Chen Z, Han F, Lu YM, Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis, The Journal of experimental medicine (2019). 10.1084/jem.20180992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Hong GP, Kim MH, Kim HJ, Sex-related Differences in Glial Fibrillary Acidic Protein-positive GABA Regulate Neuropathology Following Pilocarpine-induced Status Epilepticus, Neuroscience 472 (2021) 157–166. 10.1016/j.neuroscience.2021.08.002. [DOI] [PubMed] [Google Scholar]
- [161].Kong Q, Takahashi K, Schulte D, Stouffer N, Lin Y, Lin CL, Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus, Neurobiology of disease 47 (2012) 145–154. 10.1016/j.nbd.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Kong Q, Chang LC, Takahashi K, Liu Q, Schulte DA, Lai L, Ibabao B, Lin Y, Stouffer N, Das Mukhopadhyay C, Xing X, Seyb KI, Cuny GD, Glicksman MA, Lin CL, Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection, The Journal of clinical investigation 124 (2014) 1255–1267. 10.1172/jci66163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB, Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes, The Journal of biological chemistry 283 (2008) 13116–13123. 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Zeng LH, Bero AW, Zhang B, Holtzman DM, Wong M, Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis Complex, Neurobiology of disease 37 (2010) 764–771. 10.1016/j.nbd.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Jelenkovic AV, Jovanovic MD, Stanimirovic DD, Bokonjic DD, Ocic GG, Boskovic BS, Beneficial effects of ceftriaxone against pentylenetetrazole-evoked convulsions, Experimental biology and medicine (Maywood, N.J.) 233 (2008) 1389–1394. 10.3181/0803-rm-83. [DOI] [PubMed] [Google Scholar]
- [166].Takahashi K, Foster JB, Lin CL, Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease, Cell Mol Life Sci 72 (2015) 3489–3506. 10.1007/s00018-015-1937-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Fachim HA, Guizzo R, Cunha AOS, Pereira AC, Dos Anjos LC, Mortari MR, Dos Santos WF, Ceftriaxone pretreatment confers neuroprotection in rats with acute glaucoma and reduces the score of seizures induced by pentylenotetrazole in mice, Journal of biochemical and molecular toxicology (2020) e22578. 10.1002/jbt.22578. [DOI] [PubMed] [Google Scholar]
- [168].Li H-H, Lin P-J, Wang W-H, Tseng L-H, Tung H, Liu W-Y, Lin C-L, Liu C-H, Liao W-C, Hung C-S, Ho Y-J, Treatment effects of the combination of ceftriaxone and valproic acid on neuronal and behavioral functions in an epilepsy rat model, Experimental physiology n/a (2021). 10.1113/EP089624. [DOI] [PubMed] [Google Scholar]
- [169].Ramandi D, Elahdadi Salmani M, Moghimi A, Lashkarbolouki T, Fereidoni M, Pharmacological upregulation of GLT-1 alleviates the cognitive impairments in the animal model of temporal lobe epilepsy, PloS one 16 (2021) e0246068. 10.1371/journal.pone.0246068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Traynor BJ, Bruijn L, Conwit R, Beal F, O’Neill G, Fagan SC, Cudkowicz ME, Neuroprotective agents for clinical trials in ALS: a systematic assessment, Neurology 67 (2006) 20–27. 10.1212/01.wnl.0000223353.34006.54. [DOI] [PubMed] [Google Scholar]
- [171].Wang SJ, Wang KY, Wang WC, Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes), Neuroscience 125 (2004) 191–201. 10.1016/j.neuroscience.2004.01.019. [DOI] [PubMed] [Google Scholar]
- [172].Kretschmer BD, Kratzer U, Schmidt WJ, Riluzole, a glutamate release inhibitor, and motor behavior, Naunyn-Schmiedeberg’s archives of pharmacology 358 (1998) 181–190. 10.1007/pl00005241. [DOI] [PubMed] [Google Scholar]
- [173].Azbill RD, Mu X, Springer JE, Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes, Brain research 871 (2000) 175–180. [DOI] [PubMed] [Google Scholar]
- [174].Carbone M, Duty S, Rattray M, Riluzole elevates GLT-1 activity and levels in striatal astrocytes, Neurochemistry international 60 (2012) 31–38. 10.1016/j.neuint.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T, Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1, European journal of pharmacology 578 (2008) 171–176. 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- [176].Frizzo ME, Dall’Onder LP, Dalcin KB, Souza DO, Riluzole enhances glutamate uptake in rat astrocyte cultures, Cellular and molecular neurobiology 24 (2004) 123–128. 10.1023/b:cemn.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Farinato A, Altamura C, Desaphy JF, Effects of Benzothiazolamines on Voltage-Gated Sodium Channels, Handbook of experimental pharmacology 246 (2018) 233–250. 10.1007/164_2017_46. [DOI] [PubMed] [Google Scholar]
- [178].Yoshida M, Noguchi E, Tsuru N, Ohkoshi N, Effect of riluzole on the acquisition and expression of amygdala kindling, Epilepsy research 46 (2001) 101–109. 10.1016/s0920-1211(01)00251-0. [DOI] [PubMed] [Google Scholar]
- [179].Borowicz KK, Sekowski A, Drelewska E, Czuczwar SJ, Riluzole enhances the anti-seizure action of conventional antiepileptic drugs against pentetrazole-induced convulsions in mice, Polish journal of pharmacology 56 (2004) 187–193. [PubMed] [Google Scholar]
- [180].Peña F, Tapia R, Seizures and neurodegeneration induced by 4-aminopyridine in rat hippocampus in vivo: role of glutamate- and GABA-mediated neurotransmission and of ion channels, Neuroscience 101 (2000) 547–561. 10.1016/s0306-4522(00)00400-0. [DOI] [PubMed] [Google Scholar]
- [181].Kim JE, Kim DS, Kwak SE, Choi HC, Song HK, Choi SY, Kwon OS, Kim YI, Kang TC, Anti-glutamatergic effect of riluzole: comparison with valproic acid, Neuroscience 147 (2007) 136–145. 10.1016/j.neuroscience.2007.04.018. [DOI] [PubMed] [Google Scholar]
- [182].Sha L, Wang X, Li J, Shi X, Wu L, Shen Y, Xu Q, Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy, The Journal of experimental medicine 214 (2017) 547–563. 10.1084/jem.20160667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Sha L, Chen T, Deng Y, Du T, Ma K, Zhu W, Shen Y, Xu Q, Hsp90 inhibitor HSP990 in very low dose upregulates EAAT2 and exerts potent antiepileptic activity, Theranostics 10 (2020) 8415–8429. 10.7150/thno.44721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Yang Z, Wang J, Yu C, Xu P, Zhang J, Peng Y, Luo Z, Huang H, Zeng J, Xu Z, Inhibition of p38 MAPK Signaling Regulates the Expression of EAAT2 in the Brains of Epileptic Rats, Frontiers in neurology 9 (2018) 925–925. 10.3389/fneur.2018.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Fontana AC, Guizzo R, de Oliveira Beleboni R, Meirelles ESAR, Coimbra NC, Amara SG, dos Santos WF, Coutinho-Netto J, Purification of a neuroprotective component of Parawixia bistriata spider venom that enhances glutamate uptake, British journal of pharmacology 139 (2003) 1297–1309. 10.1038/sj.bjp.0705352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Fontana AC, de Oliveira Beleboni R, Wojewodzic MW, Ferreira Dos Santos W, Coutinho-Netto J, Grutle NJ, Watts SD, Danbolt NC, Amara SG, Enhancing glutamate transport: mechanism of action of Parawixin1, a neuroprotective compound from Parawixia bistriata spider venom, Molecular pharmacology 72 (2007) 1228–1237. 10.1124/mol.107.037127. [DOI] [PubMed] [Google Scholar]
- [187].Mortensen OV, Liberato JL, Coutinho-Netto J, Dos Santos WF, Fontana AC, Molecular determinants of transport stimulation of EAAT2 are located at interface between the trimerization and substrate transport domains, Journal of neurochemistry 133 (2015) 199–210. 10.1111/jnc.13047. [DOI] [PubMed] [Google Scholar]
- [188].Kortagere S, Mortensen OV, Xia J, Lester W, Fang Y, Srikanth Y, Salvino JM, Fontana ACK, Identification of Novel Allosteric Modulators of Glutamate Transporter EAAT2, ACS chemical neuroscience 9 (2018) 522–534. 10.1021/acschemneuro.7b00308. [DOI] [PubMed] [Google Scholar]
- [189].Falcucci RM, Wertz R, Green JL, Meucci O, Salvino J, Fontana ACK, Novel Positive Allosteric Modulators of Glutamate Transport Have Neuroprotective Properties in an in Vitro Excitotoxic Model, ACS chemical neuroscience 10 (2019) 3437–3453. 10.1021/acschemneuro.9b00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Forster YM, Green JL, Khatiwada A, Liberato JL, Reddy PAN, Salvino JM, Bienz S, Bigler L, Dos Santos WF, Fontana ACK, Elucidation of the structure and synthesis of neuroprotective low molecular mass components of the Parawixia bistriata spider venom, ACS chemical neuroscience (2020). 10.1021/acschemneuro.0c00007. [DOI] [PubMed] [Google Scholar]
- [191].Fachim HA, Mortari MR, Gobbo-Netto L, Dos Santos WF, Neuroprotective activity of parawixin 10, a compound isolated from Parawixia bistriata spider venom (Araneidae: Araneae) in rats undergoing intrahippocampal NMDA microinjection, Pharmacognosy magazine 11 (2015) 579–585. 10.4103/0973-1296.160450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Fachim HA, Cunha AO, Pereira AC, Beleboni RO, Gobbo-Neto L, Lopes NP, Coutinho-Netto J, dos Santos WF, Neurobiological activity of Parawixin 10, a novel anticonvulsant compound isolated from Parawixia bistriata spider venom (Araneidae: Araneae), Epilepsy & behavior : E&B 22 (2011) 158–164. 10.1016/j.yebeh.2011.05.008. [DOI] [PubMed] [Google Scholar]
- [193].Liberato JL, Godoy LD, Mortari MR, Gobbo-Neto L, Lopes NP, Santos WF, 034 — (LIB0083) Parawixin10: A new natural compound from <em>Parawixia bistriata</em> spider venom that presents neuroprotective, memory-saving, and disease-modifying effects in the pilocarpine model of TLE in Wistar rats, Epilepsy & Behavior 38 (2014) 196. 10.1016/j.yebeh.2014.08.067. [DOI] [Google Scholar]
- [194].Storck T, Schulte S, Hofmann K, Stoffel W, Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain, Proceedings of the National Academy of Sciences of the United States of America 89 (1992) 10955–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Tanaka K, Expression cloning of a rat glutamate transporter, Neuroscience research 16 (1993) 149–153. [DOI] [PubMed] [Google Scholar]
- [196].Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG, Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex, The Journal of neuroscience : the official journal of the Society for Neuroscience 14 (1994) 5559–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Kanai Y, Hediger MA, Primary structure and functional characterization of a high-affinity glutamate transporter, Nature 360 (1992) 467–471. 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- [198].Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG, An excitatory amino-acid transporter with properties of a ligand-gated chloride channel, Nature 375 (1995) 599–603. 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- [199].Arriza JL, Eliasof S, Kavanaugh MP, Amara SG, Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance, Proceedings of the National Academy of Sciences of the United States of America 94 (1997) 4155–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Vigneault É, Poirel O, Riad M, Prud’homme J, Dumas S, Turecki G, Fasano C, Mechawar N, El Mestikawy S, Distribution of vesicular glutamate transporters in the human brain, Front Neuroanat 9 (2015) 23–23. 10.3389/fnana.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Bellocchio EE, Reimer RJ, Fremeau RT Jr., Edwards RH, Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter, Science 289 (2000) 957–960. 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- [202].Takamori S, Rhee JS, Rosenmund C, Jahn R, Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons, Nature 407 (2000) 189–194. 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- [203].Fremeau RT Jr., Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH, The expression of vesicular glutamate transporters defines two classes of excitatory synapse, Neuron 31 (2001) 247–260. 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- [204].Fremeau RT, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH, The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate, Proceedings of the National Academy of Sciences 99 (2002) 14488–14493. 10.1073/pnas.222546799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Cho Y, Bannai S, Uptake of Glutamate and Cystine in C-6 Glioma Cells and in Cultured Astrocytes, Journal of neurochemistry 55 (1990) 2091–2097. 10.1111/j.1471-4159.1990.tb05800.x. [DOI] [PubMed] [Google Scholar]
- [206].Piani D, Fontana A, Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons, J Immunol 152 (1994) 3578–3585. [PubMed] [Google Scholar]
- [207].Bridges RJ, Natale NR, Patel SA, System xc− cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS, British journal of pharmacology 165 (2012) 20–34. 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]