

Abstract
Background
Congenital insensitivity to pain (CIP) conditions are a group of Mendelian disorders with clinical and genetic heterogeneity. CIP with anhidrosis (CIPA) is a distinct subtype caused by biallelic variants in the NTRK1 gene.
Methods
In this study, six families with CIPA were recruited and submitted to a series of clinical and genetic examinations. Whole‐exome sequencing and whole‐genome sequencing were applied to perform a comprehensive genetic analysis. Sanger sequencing was used as a validation method.
Results
These patients exhibited phenotypic variability. All probands in the six families were positive for biallelic pathogenic variants in NTRK1. Five individual variants, namely NTRK1: (NM_002529.3) c.851‐33T>A, c.717+2T>C, c.1806‐2A>G, c.1251+1G>A, and c.851‐794C>G, including three novel ones, were identified, which were carried by the six patients in a homozygous or compound heterozygous way. The validation results indicated that all the parents of the six probands, except for one father and one mother, were monoallelic carriers of a single variant.
Conclusions
The findings in our study extended the variation spectrum of the NTRK1 gene and highlighted the advantage of the integrated application of multiplatform genetic technologies.
Keywords: congenital insensitivity to pain with anhidrosis, NTRK1 gene, whole‐exome sequencing, whole‐genome sequencing
In this study, we recruited six families with probands displaying congenital insensitivity to pain with anhidrosis. A comprehensive genetic detection was performed, and the diagnostic variants were identified.

1. INTRODUCTION
Congenital Insensitivity to Pain is a category of rare congenital conditions mainly characterized by the inability to perceive pain (absence of nociception) (Schon et al., 2018). Patients with these conditions do not feel pain from any noxious stimuli, including inflammation and heat (Goldberg et al., 2007), so they often develop finger‐tip and oral cavity wounds due to self‐mutilating injuries in childhood and bony deformities due to recurrent fractures while getting older (Schon et al., 2018). Additionally, they have an increased susceptibility to the infection of Staphylococcus aureus (Drissi et al., 2020). Assessment of the remainder of the peripheral and central nervous system is typically normal (touch, vibration and position sense, motor functions, and deep tendon creflexes).
The prevalence of CIP phenotype is currently unknown, while the genes associated with it have been identified continuously (Nahorski et al., 2015). Up to our knowledge, at least 16 genes have been indicated to be CIP related, among which the NTRK1 gene is the most common one (Drissi et al., 2020; Indo, 2008; Schon et al., 2018). NTRK1‐CIP, also known as CIPA or hereditary sensory and autonomic neuropathy IV (HSAN IV, MIM #256800), is caused by the biallelic pathogenic variants in NTRK1 and is distinctive from other CIP subtypes by the anhidrosis symptom and susceptibility to recurrent febrile episodes (Indo, 2001). Besides, the patients of CIPA often exhibit a variable degree of intellectual disability, which enhances the difficulty in clinical diagnosis and often causes misdiagnosis (Caparros‐Martin et al., 2017).
The NTRK1 (MIM *191315) gene encodes the neurotrophic tyrosine kinase‐1 receptor and belongs to a family of nerve growth factor receptors whose ligands include neurotrophins (Indo, 2012), which play an important role in regulating the development of both the central and peripheral nervous systems (Bibel & Barde, 2000). NTRK1 contains 17 exons spanning 25 kb of DNA, of which exon 9 is alternatively spliced (https://www.omim.org/). Since Indo et al. initially identified NTRK1 as the causative gene for CIPA (Indo et al., 1996), more than 130 pathogenic variants including nonsense, splicing, and missense have been detected (The Human Gene Mutation Database, HGMD professional v2019.4, http://www.hgmd.cf.ac.uk/ac/index.php). There are different “hot‐spot” variants of NTRK1 in various descents; for instance, the p.Pro621Serfs*12 variant accounts for 89% in Israeli Bedouins (Shatzky et al., 2000), whereas the p.Arg554Glyfs*104, c.851‐33T>A, and p.Asp674Tyr variants together account for more than 70% in Japanese (Indo, 2001).
Advances in massive parallel sequencing have accelerated the discovery of novel NTRK1 variants (Geng et al., 2018; Nam et al., 2017; Zhao et al., 2020). However, the genotype–phenotype correlation of CIPA is still not well established due to the heterogeneity and variability of its clinical features, and the shortage of in‐depth functional study of specific variations (Li et al., 2019; Shaikh et al., 2017).
In the present study, a cohort of six families with CIPA was recruited and subjected to genetic analysis. All causative variants in these cases were identified and verified using a combination of various techniques, including WES, WGS, and Sanger sequencing. Our findings provided solid evidence to counseling for the affected families and expanded the mutation spectrum of NTRK1‐CIPA.
2. MATERIALS AND METHODS
2.1. Patients
Six unrelated cases with suspected CIPA were recruited from three medical institutions (institution 2/4/5 on the title page) between January 2017 and November 2020. These families were all from northern China and belonged to the Han population. Written informed consent was obtained from all patients or their guardians. A comprehensive physical examination was conducted on the patients. Imaging examination with X‐ray was also performed.
2.2. Genomic DNA extraction
Three milliliters of peripheral blood was collected from participants by means of BD Vacutainer™ tubes (BD Biosciences, USA). Genomic DNA was extracted using the QIAamp DNA Blood mini‐kits (Qiagen Sciences, USA) according to the manufacturer's protocol, and the DNA quality was verified by 1% agarose gels and Qubit® DNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA).
2.3. Whole‐exome sequencing (WES)
Briefly, the enrichment of the exon‐region sequence was conducted by the Sure Select Human Exon Sequence Capture Kit (Agilent, USA). The sequencing libraries were quantified using the Illumina DNA Standards and Primer Premix Kit (Kapa Biosystems, USA), and were massively parallel‐sequenced using the Illumina Novaseq6000 platform. After sequencing and filtering out the low‐quality readings, the high‐quality reads (%Q30 > 89%) were compared with the human genome reference sequence [hg19]. The GATK software was used to identify suspected pathogenic variants (https://software.broadinstitute.org/gatk/). The variations were identified by sequence alignment with the NCBI Reference Sequence (NG 011537.1) using Chromas v2.33. The pathogenicity of the identified variants was then assessed according to the common guidelines issued by the American Association of Medical Genetics and Genomics (ACMG) (Richards et al., 2015) referring to multiple databases (1000g2015aug_eas, https://www.internationalgenome.org/; ExAC_EAS, http://exac.broadinstitute.org; gnomAD_exome_EAS, http://gnomad.broadinstitute.org/); HGMD®: Human Gene Mutation Database (Professional Version 2019.4) with the Enliven® Variants Annotation Interpretation (Berry Genomics, China) system.
2.4. Whole‐genome sequencing (WGS)
To further detect the other diagnostic variant in one case with only one heterozygous allele detected by WES, WGS was introduced. Briefly, the sequencing library was generated using the CLEAN‐NGS DNA kit following the manufacturer's recommendations, and index codes were added to each sample. Afterward, the DNA sample was taken and enzymatically disrupted to a size of 350 bp. Then DNA fragments were end‐polished, A‐tailed, and ligated with the full‐length adapter for Illumina sequencing, followed by further PCR amplification. After PCR products were purified (AMPure XP system), libraries were analyzed for size distribution by Agilent 2100 Bioanalyzer and quantified by real‐time PCR (3 nM). The clustering of the index‐coded samples was performed on a cBot Cluster Generation System using Novaseq6000 S4 Reagent Kit (Illumina, USA) according to the manufacturer's instructions. After cluster generation, the DNA libraries were sequenced on the Illumina NovaSeq platform and 150 bp paired‐end reads were generated. Clean reads were compared with reference human genome (UCSC hg19), and the results were converted into bam format and sorted by SAMtools software. Finally, basic information statistics and map comparison statistics were conducted. Subsequently, the annotation and interpretation progress was fulfilled as in Section 2.4.
2.5. Sanger sequencing
The suspected diagnostic variant was validated by Sanger sequencing using ABI 3730 Automated Sequencer according to the manufacturer's protocol (Applied Biosystems, USA). Primers and methods for Sanger sequencing can be found in Supporting Information.
3. RESULTS
3.1. Clinical findings
The representative images of patients were summarized in Figure 1. And the clinical manifestations of the six probands were shown in Table 1.
FIGURE 1.
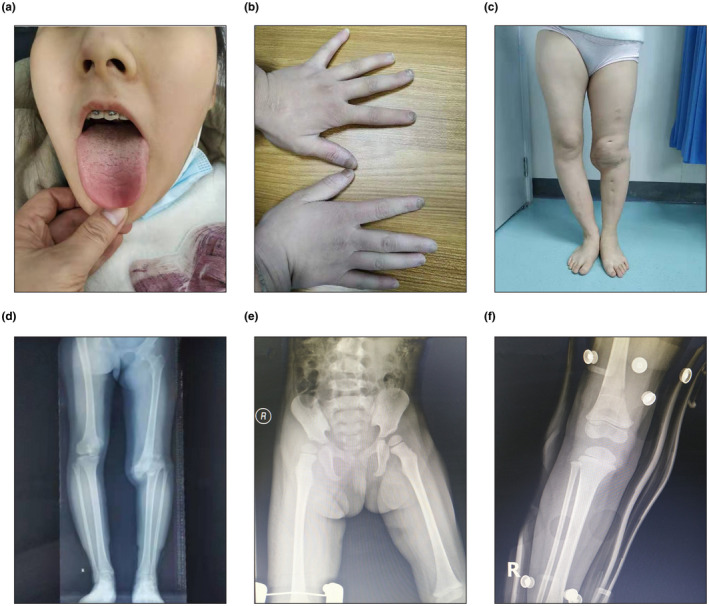
Representative clinical indications of patients in this study. (a) Oral image of patient 1.1 showing mild healing wounds to the lip and tongue. (b) Patient 1.1’s hands showing nail bite marks. (c and d) Patient 1.1’s appearance and X‐ray photographs of the lower extremity showing typical Charcot's joint formation. (e and f) Patient 3.1’s X‐ray photographs of lower extremity showing treatment for dislocation of the right hip
TABLE 1.
Clinical features of the six probands
| Patients | Gendera | Age at recruitment | Clinical manifestation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CIPA a | Bone fractures | Recurrent fever | Intellectual disability | Self‐mutilation | Irascibility | Othersa | |||
| 1.1 | F | 13 years | Y | Y | Y | Y | Y | Y | Charcot's osteoarthropathy in left knee; leukodystrophy |
| 2.1 | F | 3.5 years | Y | Y | Y | Y | Y | Y | N/A |
| 3.1 | F | 2.5 years | Y | N | Y | N | Y | N | Dislocation of the right hip |
| 4.1 | F | 3.5 years | Y | N | Y | Y | Y | Y | N/A |
| 5.1 | M | 10 years | Y | Y | Y | Y | Y | Y | N/A |
| 6.1 | M | 3 years | Y | Y | Y | Y | Y | Y | N/A |
Abbreviations: F, female; M, male; N, no; N/A, not available; Y, yes.
congenital insensitivity to pain with anhidrosis.
All patients exhibited insensitivity to pain with anhidrosis, recurrent unexplained fever, absence of reaction to noxious stimuli, and self‐mutilating behaviors. Apart from patient 3.1, all of the other patients displayed intellectual disability and irascibility. Older patients, such as 1.1, have formed “Charcot's joint” in the knee due to past fractures, resulting in limited movement and having to undergo surgery. Patient 1.1 exhibited leukodystrophy; and 3.1 exhibited dislocation of the right hip.
3.2. Genetic variations
Biallelic variants were identified in all of the six patients (see details in Table 2). Validation by Sanger sequencing indicated that their parents were all carriers of one single pathogenic allele, except for the father of 3.1 and mother of patient 4.1 (see patients 3.2 and 4.3 in Figure 2). The parental origin details of the variants can be seen in Table 2. Five various variants including three novel ones were detected (see representative sequence peak images in Figure 3).
TABLE 2.
NTRK1 gene variation characteristics of the six probands
| Patients | Variant | Zygote | Pathogenicity (ACMG a evidence level) | Parental origin |
|---|---|---|---|---|
| 1.1 | c.851‐33T>A | Homozygote | Pathogenic (PS1 + PS3 + PM2 + PP4) | Both father (1.2) and mother (1.3) are heterozygous carriers |
| 2.1 | c.851‐33T>A | Homozygote | Pathogenic (PS1 + PS3 + PM2 + PP4) | Both father (2.2) and mother (2.3) are heterozygous carriers |
| 3.1 | c.851‐33T>A | Compound heterozygote | Pathogenic (PS1 + PS3 + PM2 + PP4) | Mother (3.3) is heterozygous carrier |
| c.717+2T>C | Likely pathogenic (PVS1 + PM2) | |||
| 4.1 | c.1806‐2A>G | Compound heterozygote | Pathogenic (PVS1 + PM3_Strong + PM2) | |
| c.1251+1G>A | Likely pathogenic (PVS1 + PM2) | Father (4.2) is heterozygous carrier | ||
| 5.1 | c.851‐33T>A | Homozygote | Pathogenic (PS1 + PS3 + PM2 + PP4) | Both father (2.2) and mother (2.3) are heterozygous carriers |
| 6.1 | c.851‐33T>A | Compound heterozygote | Pathogenic (PS1 + PS3 + PM2 + PP4) | Father (6.2) is heterozygous carrier |
| c.851‐794C>G | Likely pathogenic (PM2 + PM3 + PP3 + PP4) | Mother (6.3) is heterozygous carrier |
NTRK1 gene transcript: NM_002529.3.
ACMG, American Committee on Medical Genetics and Genomics (Richards et al., 2015; PMID 25741868).
FIGURE 2.
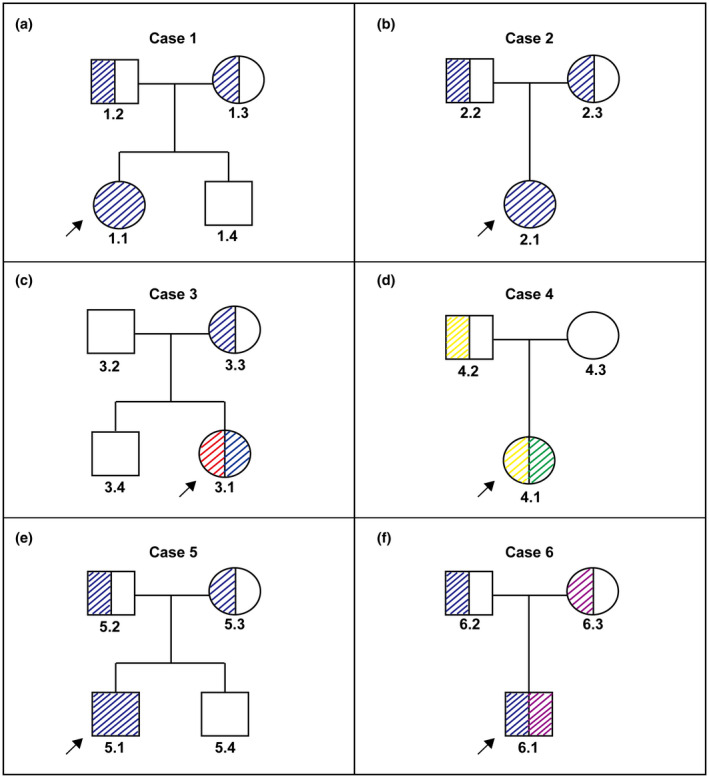
Pedigree schematics of the six families in this study. The color slashes represent the carrying status of each variant, and their colors are consistent with the block referents in Figure 3a,b. Black arrows represent probands
FIGURE 3.
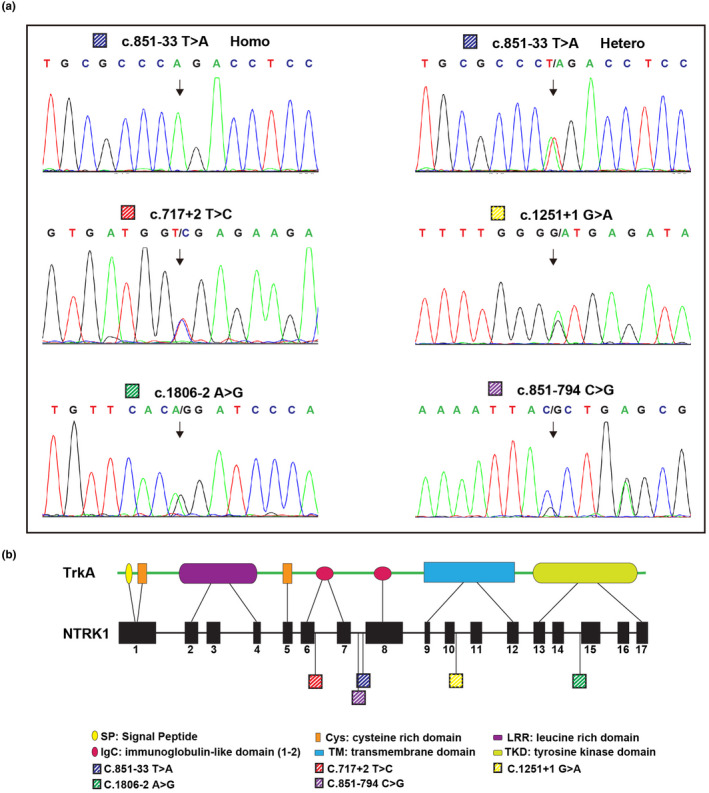
(a) The sequences of five detected variants are illustrated by Sanger sequencing. Variant c.851‐33T>A was exhibited in both homozygous (Homo; an example from patient 1.1) and heterozygous (Hetero; an example from patient 3.1) patterns. (b) Schematics of the NTRK1 gene and its product, TrkA peptide, showing the locations of these five variants
The most commonly carried variant, c.851‐33T>A, was present in five patients of the six, namely 1.1, 2.1, 3.1, 5.1, and 6.1, in a homozygous or compound heterozygous way (Figure 2 and Table 2). The other four NTRK1 variants, namely (NM_002529.3) c.717+2T>C, c.1806‐2A>G, c.1251+1G>A, and c.851‐794C>G, were detected in 3.1, 4.1, and 6.1, respectively, in a compound heterozygous way (Figure 2 and Table 2). Three variants, c.717+2T>C, c.1251+1G>A, and c.851‐794C>G, were novelly identified in this study. The variant c.851‐794C>G harbored by patient 6.1 was identified by WGS, as WES failed in detecting it.
4. DISCUSSION
Pain is essential in teaching us how to use our bodies optimally and avoid or respond to injuries, being permanently painless results in significant morbidity and mortality (Drissi et al., 2020). To date, up to nine genes have been identified to be associated with the human CIP phenotype (Drissi et al., 2020). Conditions caused by these genetic defects have a commonality in pain insensitivity, yet each has its own characteristics (Drissi et al., 2020). NTRK1 is the first identified and most common pathogenesis among these genes. To the best of our knowledge, however, the HSAN IV it leads to has a high phenotypic overlap with at least two rare autosomal recessive CIP subtypes, namely, HSAN V (MIM #608654) caused by NGF and HSAN6 (MIM #616488) caused by PRDM12, mainly in the symptoms of anhidrosis and increased S. aureus infection risk (Chen et al., 2015; Einarsdottir et al., 2004). Therefore, in order to clarify the inheritance pattern of specific CIP/CIPA cases and to evaluate the recurrence risk in affected families, comprehensive and systematic genetic analysis is pivotal (Li et al., 2019; Zhao et al., 2020).
In this study, all six affected children not only showed typical CIPA phenotypes but also showed some degree of phenotypic heterogeneity, mainly in the presence of mental abnormalities or not. This may be related to the degree of damage caused by specific variations to protein function or may be due to genetic background differences among different individuals. But whatever the reason is, further functional studies are needed. Besides, as there is an ongoing therapeutic clinical trial for NTRK1‐HSAN IV (No. NCT02424942, https://www.clinicaltrials.gov/), early detection of the causative variation is valuable for possible treatments.
The genetic etiology of six probands in this study was all clear, which are biallelic variants in NTRK1. Intriguingly, all the variants we detected were in noncoding regions, either were splicing variation or in deep‐introns, which is inconsistent with the fact that missense mutations account for most of the previously reported mutations (http://www.hgmd.cf.ac.uk/) (Drissi et al., 2020; Indo, 2008). This may be because the sample size of our study is relatively small and does not have universal significance. We identified three novel variants, which were two splicings, c.717+2T>C and c.1251+1G>A, and one deep‐intronic variant, c.851‐794C>G. The last one was not detected by WES and was successfully revealed by WGS, indicating the importance of setting a more comprehensive genetic detection strategy. Combined with the results of this and previous studies, the c.851‐33T>A variant has a high prevalence in East‐Asian people (Geng et al., 2018; Li et al., 2019; Nam et al., 2017; Zhao et al., 2020), so it is worth to design a screening kit that targeting it as a carrier screening product. In addition, two separate parents of two probands were wild‐type, which is rare. We inferred from the results that the two probands (3.1 and 4.1) may have a de novo variant each, but we also could not rule out the possibility that their parents had gonadal chimerism (van der Martin et al., 1995). If so, care should be taken and appropriate advice, such as prenatal diagnosis, should be provided when assessing the family's risk of recurrence of future pregnancy.
According to the research by Lee et al. (2009) and Altassan et al. (2017), NTRK1 gene's specific types of variants are related to the severity of the phenotype. The symptoms of mental development and sweat gland development level caused by specific types of missense variants and splicing variants are “mild.” However, for the variants detected in our research, there is still insufficient evidence on the association between genotype and phenotype. Therefore, a more in‐depth research is required on these functional genes in order to clarify in future research (Franco et al., 2016; Shaikh et al., 2017).
The limitation of this study is that the sample size is relatively small, and we have not carried out the functional verification of the novel variations for the time being, which is pivotal to understand the impact of specific NTRK1 variants on gene expression and protein function and will be arranged in the subsequent study (Shaikh et al., 2017).
In conclusion, the current study has genetically diagnosed six CIPA cases and detected bi‐allelic variants in all patients in the NTRK1 gene, including three novel unreported variants. The findings of this study expanded the mutation spectrum of NTRK1 gene and provide a solid evidential basis for genetic counseling and reproductive guidance to affected families.
ETHICAL COMPLIANCE
This study was approved by the Ethics Committee of Peking University People's Hospital. All legal guardians of the participants signed informed consent allowing the publication of relevant information.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Shang Li and Hua‐ying Hu: project conceptualization, methodology. Kai Yang: writing‐ original draft preparation. Jun‐jun Xu and Zhan‐ke Feng: validation, investigation. Yong‐qing Sun: data curation, visualization. Xu Chen: resources. Dong‐liang Zhang: project administration and supervision; draft reviewing.
Supporting information
Table S1‐S2
ACKNOWLEDGMENTS
We thank all the patients who participated in this study.
Li, S. , Hu, H.‐Y. , Xu, J.‐J. , Feng, Z.‐K. , Sun, Y.‐Q. , Chen, X. , Yang, K. , Li, Y.‐Z. , & Zhang, D.‐L. (2021). Identification of novel variations in the NTRK1 gene causing congenital insensitivity to pain with anhidrosis. Molecular Genetics & Genomic Medicine, 9, e1839. 10.1002/mgg3.1839
Shang Li and Hua‐ying Hu contributed equally to this work.
Funding information
This study was supported by the Hebei Natural Science Foundation Precision Medicine Joint Fund Cultivation Project (No. H2021206242).
Contributor Information
Ya‐zhou Li, Email: 176304578@qq.com.
Dong‐liang Zhang, Email: zhangdongliang@mail.ccmu.edu.cn.
DATA AVAILABILITY STATEMENT
All available clinical data are shared in this article.
REFERENCES
- Altassan, R. , Saud, H. A. , Masoodi, T. A. , Dosssari, H. A. , Khalifa, O. , Al‐Zaidan, H. , Sakati, N. , Rhabeeni, Z. , Al‐Hassnan, Z. , Binamer, Y. , Alhashemi, N. , Wade, W. , Al‐Zayed, Z. , Al‐Sayed, M. , Al‐Muhaizea, M. A. , Meyer, B. , Al‐Owain, M. , & Wakil, S. M. (2017). Exome sequencing identifies novel NTRK1 mutations in patients with HSAN‐IV phenotype. American Journal of Medical Genetics: Part A, 173(4), 1009–1016. 10.1002/ajmg.a.38120 [DOI] [PubMed] [Google Scholar]
- Bibel, M. , & Barde, Y.‐A. (2000). Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes & Development, 14, 2919–2937. 10.1101/gad.841400 [DOI] [PubMed] [Google Scholar]
- Caparros‐Martin, J. A. , Aglan, M. S. , Temtamy, S. , Otaify, G. A. , Valencia, M. , Nevado, J. , Vallespin, E. , Del Pozo, A. , Prior de Castro, C. , Calatrava‐Ferreras, L. , Gutierrez, P. , Bueno, A. M. , Sagastizabal, B. , Guillen‐Navarro, E. , Ballesta‐Martinez, M. , Gonzalez, V. , Basaran, S. Y. , Buyukoglan, R. , Sarikepe, B. , … Ruiz‐Perez, V. L. (2017). Molecular spectrum and differential diagnosis in patients referred with sporadic or autosomal recessive osteogenesis imperfecta. Molecular Genetics & Genomic Medicine, 5(1), 28–39. 10.1002/mgg3.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y.‐C. , Auer‐Grumbach, M. , Matsukawa, S. , Zitzelsberger, M. , Themistocleous, A. C. , Strom, T. M. , Samara, C. , Moore, A. W. , Cho, L.‐Y. , Young, G. T. , Weiss, C. , Schabhüttl, M. , Stucka, R. , Schmid, A. B. , Parman, Y. , Graul‐Neumann, L. , Heinritz, W. , Passarge, E. , Watson, R. M. , … Senderek, J. (2015). Transcriptional regulator PRDM12 is essential for human pain perception. Nature Genetics, 47(7), 803–808. 10.1038/ng.3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drissi, I. , Woods, W. A. , & Woods, C. G. (2020). Understanding the genetic basis of congenital insensitivity to pain. British Medical Bulletin, 133(1), 65–78. 10.1093/bmb/ldaa003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einarsdottir, E. , Carlsson, A. , Minde, J. , Toolanen, G. , Svensson, O. , Solders, G. , Holmgren, G. , Holmberg, D. , & Holmberg, M. (2004). A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Human Molecular Genetics, 13(8), 799–805. 10.1093/hmg/ddh096 [DOI] [PubMed] [Google Scholar]
- Franco, M. L. , Melero, C. , Sarasola, E. , Acebo, P. , Luque, A. , Calatayud‐Baselga, I. , García‐Barcina, M. , & Vilar, M. (2016). Mutations in TrkA causing congenital insensitivity to pain with anhidrosis (CIPA) induce misfolding, aggregation, and mutation‐dependent neurodegeneration by dysfunction of the autophagic flux. Journal of Biological Chemistry, 291(41), 21363–21374. 10.1074/jbc.M116.722587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng, X. , Liu, Y. , Ren, X. , Guan, Y. , Wang, Y. , Mao, B. , & Zhang, X. (2018). Novel NTRK1 mutations in Chinese patients with congenital insensitivity to pain with anhidrosis. Molecular Pain, 14, 1744806918781140. 10.1177/1744806918781140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, Y. P. , MacFarlane, J. , MacDonald, M. L. , Thompson, J. , Dube, M.‐P. , Mattice, M. , Fraser, R. , Young, C. , Hossain, S. , Pape, T. , Payne, B. , Radomski, C. , Donaldson, G. , Ives, E. , Cox, J. , Younghusband, H. B. , Green, R. , Duff, A. , Boltshauser, E. , … Hayden, M. R. (2007). Loss‐of‐function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clinical Genetics, 71(4), 311–319. 10.1111/j.1399-0004.2007.00790.x [DOI] [PubMed] [Google Scholar]
- Indo, Y. (2001). Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): Mutations and polymorphisms in TRKA(NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Human Mutation, 18, 462–471. 10.1002/humu.1224 [DOI] [PubMed] [Google Scholar]
- Indo, Y. (2008). NTRK1 congenital insensitivity to pain with anhidrosis. In Adam M. P., Ardinger H. H., Pagon R. A., & Wallace S. E. (Eds.)., GeneReviews® [Internet]. University of Washington, Seattle. [PubMed] [Google Scholar]
- Indo, Y. (2012). Nerve growth factor and the physiology of pain: Lessons from congenital insensitivity to pain with anhidrosis. Clinical Genetics, 82(4), 341–350. 10.1111/j.1399-0004.2012.01943.x [DOI] [PubMed] [Google Scholar]
- Indo, Y. , Tsuruta, M. , Hayashida, Y. , Karim, M. A. , Ohta, K. , Kawano, T. , Mitsubuchi, H. , Tonoki, H. , Awaya, Y. , & Matsuda, I. (1996). Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nature Genetics, 13(4), 485–488. 10.1038/ng0896-485 [DOI] [PubMed] [Google Scholar]
- Lee, S. T. , Lee, J. , Lee, M. , Kim, J. W. , & Ki, C. S. (2009). Clinical and genetic analysis of Korean patients with congenital insensitivity to pain with anhidrosis. Muscle and Nerve, 40(5), 855–859. 10.1002/mus.21340 [DOI] [PubMed] [Google Scholar]
- Li, N. , Guo, S. , Wang, Q. , Duan, G. , Sun, J. , Liu, Y. , Zhang, J. , Wang, C. , Zhu, C. , Liu, J. , & Zhang, X. (2019). Heterogeneity of clinical features and mutation analysis of NTRK1 in Han Chinese patients with congenital insensitivity to pain with anhidrosis. Journal of Pain Research, 12, 453–465. 10.2147/JPR.S188566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahorski, M. S. , Chen, Y. C. , & Woods, C. G. (2015). New Mendelian disorders of painlessness. Trends in Neurosciences, 38(11), 712–724. 10.1016/j.tins.2015.08.010 [DOI] [PubMed] [Google Scholar]
- Nam, T. S. , Li, W. , Yoon, S. , Eom, G. H. , Kim, M. K. , Jung, S. T. , & Choi, S. Y. (2017). Novel NTRK1 mutations associated with congenital insensitivity to pain with anhidrosis verified by functional studies. Journal of the Peripheral Nervous System, 22(2), 92–99. 10.1111/jns.12205 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon, K. R. , Parker, A. P. J. , & Woods, C. G. (2018). Congenital insensitivity to pain overview. In Adam M. P., Ardinger H. H., Pagon R. A., & Wallace S. E. (Eds.)., GeneReviews® [Internet]. University of Washington, Seattle. [PubMed] [Google Scholar]
- Shaikh, S. S. , Chen, Y. C. , Halsall, S. A. , Nahorski, M. S. , Omoto, K. , Young, G. T. , Phelan, A. , & Woods, C. G. (2017). A comprehensive functional analysis of NTRK1 missense mutations causing hereditary sensory and autonomic neuropathy type IV (HSAN IV). Human Mutation, 38(1), 55–63. 10.1002/humu.23123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatzky, S. , Moses, S. , Levy, J. , Pinsk, V. , Hershkovitz, E. , Herzog, L. , Shorer, Z. , Luder, A. , & Parvari, R. (2000). Congenital insensitivity to pain with anhidrosis (CIPA) in Israeli‐Bedouins: Genetic heterogeneity, novel mutations in the TRKA/NGF receptor gene, clinical findings, and results of nerve conduction studies. American Journal of Medical Genetics, 92, 353–360. [DOI] [PubMed] [Google Scholar]
- van der Meulen, M. A. , van der Meulen, M. J. , & te Meerman, G. J. (1995). Recurrence risk for germinal mosaics revisited. Journal of Medical Genetics, 32, 102–104. 10.1136/jmg.32.2.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, F. , Mao, B. , Geng, X. , Ren, X. , Wang, Y. , Guan, Y. , Li, S. , Li, L. , Zhang, S. , You, Y. , Cao, Y. , Yang, T. , & Zhao, X. (2020). Molecular genetic analysis in 21 Chinese families with congenital insensitivity to pain with or without anhidrosis. European Journal of Neurology, 27(8), 1697–1705. 10.1111/ene.14234 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2
Data Availability Statement
All available clinical data are shared in this article.