

Abstract
Background and Objectives
To predict when cognitively normal individuals with brain amyloidosis will develop symptoms of Alzheimer disease (AD).
Methods
Brain amyloid burden was measured by amyloid PET with Pittsburgh compound B. The mean cortical standardized uptake value ratio (SUVR) was transformed into a timescale with the use of longitudinal data.
Results
Amyloid accumulation was evaluated in 236 individuals who underwent >1 amyloid PET scan. The average age was 66.5 ± 9.2 years, and 12 individuals (5%) had cognitive impairment at their baseline amyloid PET scan. A tipping point in amyloid accumulation was identified at a low level of amyloid burden (SUVR 1.2), after which nearly all individuals accumulated amyloid at a relatively consistent rate until reaching a high level of amyloid burden (SUVR 3.0). The average time between levels of amyloid burden was used to estimate the age at which an individual reached SUVR 1.2. Longitudinal clinical diagnoses for 180 individuals were aligned by the estimated age at SUVR 1.2. In the 22 individuals who progressed from cognitively normal to a typical AD dementia syndrome, the estimated age at which an individual reached SUVR 1.2 predicted the age at symptom onset (R2 = 0.54, p < 0.0001, root mean square error [RMSE] 4.5 years); the model was more accurate after exclusion of 3 likely misdiagnoses (R2 = 0.84, p < 0.0001, RMSE 2.8 years).
Conclusion
The age at symptom onset in sporadic AD is strongly correlated with the age at which an individual reaches a tipping point in amyloid accumulation.
Alzheimer disease (AD) is characterized neuropathologically by amyloid plaques and neurofibrillary tangles, which are thought to start accumulating approximately 20 years before the onset of dementia symptoms.1,2 Brain amyloid accumulation does not occur linearly over time; instead, the pattern is reminiscent of the multiphased nucleation-dependent aggregation of β-amyloid (Aβ) peptide in vitro.3-5 In a test tube, monomeric Aβ stochastically aggregates and disaggregates during an initial lag phase. Once a critical concentration of Aβ aggregates is reached (the nucleation threshold or tipping point), the aggregates serve as nuclei for the rapid and relatively predictable growth phase of Aβ aggregation. Similarly, in the human brain, studies suggest that the rate of amyloid accumulation is slow and highly variable at very low levels of amyloid burden.2,6 Once a threshold level of amyloid burden is crossed, the rate of amyloid accumulation increases and becomes relatively consistent across individuals, which allows the time of amyloid accumulation to be reliably estimated through a variety of mathematical approaches.2,6-8
Although the pattern of brain amyloid accumulation may or may not be directly related to the aggregation kinetics of Aβ in vitro and empirically investigating this potential connection was not the goal of this study, we used nucleation-dependent aggregation as a conceptual framework for modeling amyloid accumulation. We speculated that the level of amyloid burden that marks the transition from slow to rapid amyloid accumulation might represent the nucleation threshold for brain amyloid aggregation. Furthermore, we hypothesized that this tipping point in amyloid aggregation could be used to align longitudinal clinical data across individuals and enable prediction of when cognitively normal individuals with brain amyloidosis will develop the onset of AD symptoms.
Methods
Standard Protocol Approvals, Registrations, and Participant Consents
Community-dwelling participants enrolled in longitudinal studies of memory and aging at the Knight Alzheimer Disease Research Center (ADRC) who underwent an amyloid PET scan with Pittsburgh compound B (PiB) within 1 year of a comprehensive clinical assessment and had available APOE genotype data were included in the present study. Race and sex were self-reported. APOE genotyping was performed by the Knight ADRC Genetics Core.9 All procedures were approved by the Washington University Human Research Protection Office, and written informed consent in accordance with the Declaration of Helsinki was obtained from each participant or their legally authorized representative when appropriate. All data were used for research purposes only.
Clinical Assessment and Categorization of Dementia
Individuals who were cognitively normal or had cognitive impairment caused by an uncertain etiology or suspected AD etiology were enrolled. Comprehensive clinical assessments were performed every 3 years for participants <65 years old and yearly for participants ≥65 years of age. The clinical assessment included a detailed interview of a collateral source, a neurologic examination of the participant, the Clinical Dementia Rating (CDR),10 CDR Sum of Boxes,11 and the Mini-Mental State Examination.12 Individuals with a CDR score of ≥0.5 were considered to have a dementia syndrome, and the probable etiology of the dementia syndrome was formulated by clinicians on the basis of clinical features in accordance with standard criteria and methods.13
For this study, individuals with a CDR score of 0 were categorized as cognitively normal. Previous studies in this cohort established that individuals with a CDR score of 0.5 and an etiologic diagnosis of AD, who would have been described by some other studies as having mild cognitive impairment, had early AD dementia.14,15 Symptomatic AD is a continuum that includes mild cognitive impairment due to AD and AD dementia.14-16 Therefore, individuals with a CDR score of ≥0.5 were categorized as having a typical AD dementia syndrome if they met the McKhann et al.17 criteria, which include the insidious onset and slow progression of dementia with an amnestic presentation and without substantial evidence of confounding factors.17 Individuals with a typical AD dementia syndrome, who also had secondary disorders that could impair cognition (e.g., depression) that were not thought to be contributing significantly to the dementia syndrome, were also categorized as having a typical AD dementia syndrome. Individuals with a CDR score of ≥0.5 with any other etiologic dementia diagnoses were categorized as having other dementia syndrome. The most common diagnosis in the other dementia category was uncertain etiology, meaning that the clinical features were not clearly concordant with 1 dementia diagnosis. Possible AD dementia was included in the other dementia syndrome category when other disorders were present and thought to be contributing significantly to the dementia or when atypical clinical features were present that could potentially be caused by other neurologic disorders such as parkinsonism or early changes in language, visuospatial function, or social appropriateness. The other dementia syndrome category also included diagnoses of non-AD dementias, including dementia with Lewy bodies, frontotemporal dementia, and vascular dementia. Diagnostic categories were defined before the analyses described herein were performed.
In formulating the CDR and etiologic diagnosis based on clinical features, clinicians were blinded to the participant's performance on neuropsychological tests, the results of prior assessments, and biomarker results. This blinding was critical to the current study: if the amyloid PET standardized uptake value ratio (SUVR) were considered in the diagnostic formulation, the relationship between amyloid PET SUVR and syndromic diagnosis could be confounded.
Amyloid PET and Structural Brain MRI
More detailed imaging methods are presented in eAppendix 1 (links.lww.com/WNL/B535). Briefly, participants underwent a dynamic scan with PiB18 in coordination with a structural MRI scan. Regional data from the 40- to 60 minute postinjection window were converted to SUVRs with cerebellar gray as a reference and partial volume corrected with a geometric transfer matrix approach based on the FreeSurfer parcellation.19,20 Values from regions where amyloid deposition occurs early in AD were averaged together to represent mean cortical SUVR: the left and right lateral orbitofrontal, medial orbitofrontal, precuneus, rostral middle frontal, superior frontal, superior temporal, and middle temporal cortices. Amyloid PET positivity for PiB has previously been defined as a mean cortical SUVR >1.42.21,22
Transformation of Mean Cortical SUVR Into Amyloid Time
Mean cortical SUVR was transformed into a timescale, as has been performed by other groups who evaluated the relationship between amyloid burden and time.6,7 This transformation was performed with the computer algorithm provided in eAppendix 2 (links.lww.com/WNL/B536) using data from individuals with ≥2 amyloid PET scans. The rate of amyloid accumulation for each individual was determined by linear regression and anchored at the estimated SUVR halfway through the follow-up period (SUVRmidpoint). Phases of amyloid accumulation, similar to those described by other studies,2,23 were delineated by binning individuals by SUVRmidpoint and evaluating the correlation of SUVRmidpoint with the rate of amyloid accumulation (eFigure 1, links.lww.com/WNL/B537). The lowest/first bin with a significant correlation between the rate of amyloid accumulation and SUVRmidpoint was SUVRmidpoint 1.1 to 1.2, and the upper boundary of this bin (SUVR 1.2) was chosen as the tipping point because nearly all individuals accumulated amyloid after SUVRmidpoint >1.2.
Because there was a significant but nonlinear correlation between the rate of amyloid accumulation and SUVRmidpoint for individuals with an SUVRmidpoint between 1.2 and 3.0, the relationship between the rate of amyloid accumulation and SUVRmidpoint was fitted with a cubic spline with knots at 1.2 and 3.0. The average time in years required to pass from one SUVR to another was then estimated by integrating the reciprocal of the modeled rate of amyloid accumulation, similar to previously reported methods.6,7 Amyloid time was defined as the estimated years from SUVR 1.2; at SUVR 1.2, amyloid time was equal to 0.
A cross-validation procedure was performed to evaluate the correspondence of amyloid time and actual time (by dates) using 80% of the cohort to generate the model and 20% of the cohort to test the model. PROC SURVEYSELECT was used to randomly select training and tests groups. This approach was applied to all individuals with at least 2 amyloid PET scans within the range of SUVR 1.2 to 3.0. The actual time interval between the first and last amyloid PET scan was calculated and compared to the amyloid time interval.
Alignment of Longitudinal Clinical Diagnoses by the Estimated Age at SUVR 1.2
The amyloid time transformation of SUVR derived from individuals with longitudinal amyloid PET was applied to all PiB scans between SUVR 1.2 and 3.0 in the Knight ADRC cohort. The age at SUVR 1.2 was estimated by subtracting the amyloid time value for the scan from the participant's age at the scan. For individuals with >1 amyloid PET scan between SUVR 1.2 and 3.0, the estimated age at SUVR 1.2 was averaged across all scans from the individual. The time from SUVR 1.2 for a clinical assessment was calculated as the age at a clinical assessment minus the estimated age at SUVR 1.2.
Estimating Years From Symptom Onset
Progressors to symptomatic AD were defined as individuals who were cognitively normal at their baseline clinical assessment and diagnosed with typical AD dementia syndrome at their last clinical assessment. The age at symptom onset was defined as the age when progressors were first diagnosed with a typical AD dementia syndrome. These definitions were formulated before data analysis. Logistical regression models were used to evaluate dementia syndrome category as a function of the estimated age at SUVR 1.2 and the estimated time from SUVR 1.2 or age and SUVR. Linear regression models were used to estimate the age at symptom onset as a function of the estimated age at SUVR 1.2.
Spearman correlations were used to evaluate potentially nonlinear relationships, while Pearson correlations were used to evaluate whether estimated values corresponded linearly with actual values. All statistical analyses and programming were conducted with SAS 9.4 (SAS Institute Inc, Cary, NC). Plots were created with GraphPad Prism version 9.1 (GraphPad Software, La Jolla, CA).
Data Availability
All data used for this study are from the Knight ADRC and are available on request by qualified investigators.
Results
Participants
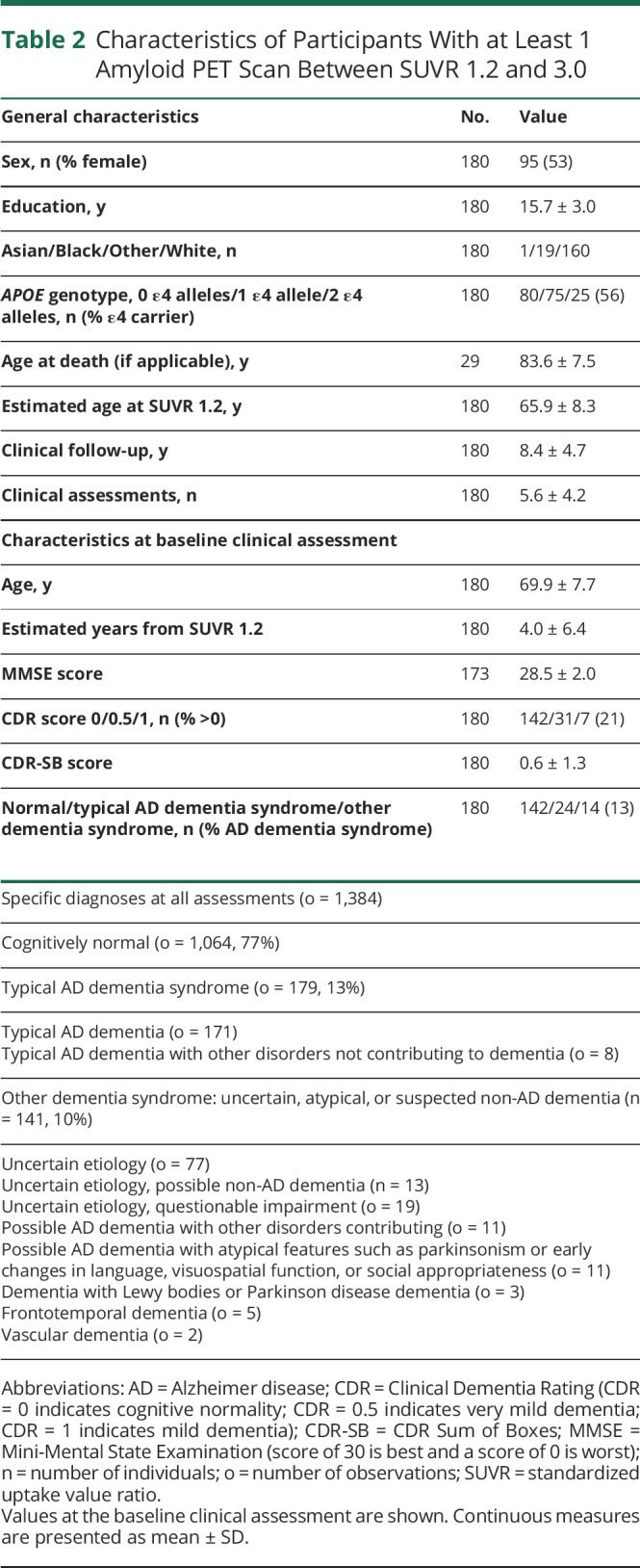
The longitudinal cohort was used to create of model of time as a function of amyloid PET SUVR and included 236 individuals with >1 amyloid PET scan. The majority of individuals had 2 scans (n = 139, 59%), and the average follow-up period between the last and first scan was 4.8 ± 2.2 years (Table 1). The average age was 66.5 ± 9.2 years, and 12 (5%) individuals had cognitive impairment (CDR score ≥0.5) at their baseline amyloid PET scan. The alignment cohort was used to visualize clinical diagnosis as a function of estimated years from SUVR 1.2 and included 180 individuals with at least 1 amyloid PET scan between SUVR 1.2 and 3.0 (centiloid values of 7 and 88, respectively; Table 2).
Table 1.
Characteristics of Participants With Longitudinal Amyloid PET Data

Table 2.
Characteristics of Participants With at Least 1 Amyloid PET Scan Between SUVR 1.2 and 3.0
Rate of Amyloid Accumulation
The average rate of amyloid accumulation as a function of amyloid burden, which was represented by the estimated SUVR halfway through the follow-up period (SUVRmidpoint), was plotted for each individual with longitudinal amyloid PET scans (Figure 1A). The rate of amyloid accumulation became significantly correlated with amyloid burden between SUVR 1.1 and 1.2 (Spearman ρ = 0.58, p = 0.02, eFigure 1, links.lww.com/WNL/B537), and SUVR 1.2 was identified as the tipping point after which nearly all individuals accumulated amyloid. For individuals with an SUVR ≤1.2, the rate of amyloid accumulation was positively but weakly correlated with amyloid burden (phase 1, Spearman ρ = 0.17, p = 0.03). Interestingly, there was a significant correlation in APOE ε4 carriers (Spearman ρ = 0.48, p = 0.003) but no correlation in APOE ε4 noncarriers (Spearman ρ = 0.06, p = 0.48, Figure 1B). After SUVR 1.2, amyloid accumulation was positive in nearly all individuals and did not vary significantly by APOE ε4 carrier status. This suggests that once an individual crossed the SUVR 1.2 threshold, he or she was almost always destined to develop significant amyloid accumulation regardless of APOE ε4 carrier status. There was a strong correlation between the rate of amyloid accumulation and amyloid burden between SUVR 1.2 and 3.0, but this relationship was nonlinear, with a positive correlation between SUVR 1.2 and 1.8 (phase 2a, Spearman ρ = 0.76, p < 0.0001) and a negative correlation between SUVR 1.8 and 3.0 (phase 2b, Spearman ρ = −0.47, p = 0.006, eTable 1, links.lww.com/WNL/B539). After individuals reached an amyloid burden of SUVR 3.0, the rate of amyloid accumulation was highly variable and no longer correlated with amyloid burden (phase 3, Spearman ρ = −0.11, p = 0.75).
Figure 1. Transformation of Amyloid PET Mean Cortical SUVR Into Amyloid Time.

Rate of amyloid accumulation for each individual was determined by linear regression and anchored at the estimated standardized uptake value ratio (SUVR) halfway through the follow-up period (SUVRmidpoint) (A). Note that the y-axis represents rate of change, not total amyloid burden. Solid red squares represent APOE ε4 homozygotes (ε4/ε4); red open squares represent ε4 heterozygotes (ε4/ε2 or ε4/ε3); and blue circles represent APOE ε4 noncarriers. Rate of change for each individual was correlated (Spearman) with SUVRmidpoint over phases denoted by vertical dotted lines. Solid lines indicate the linear regression of amyloid accumulation as a function of amyloid burden for APOE ε4 carriers (red) and noncarriers (blue). At SUVR ≤1.2, the rate of amyloid accumulation was correlated (Spearman) with amyloid burden in APOE ε4 carriers but not noncarriers (B). One point with a low rate was omitted for improved data visualization. Change in amyloid burden (last SUVR minus first SUVR) was linearly correlated (Pearson) with the time between scans (last scan date minus first scan date) (C). For individuals with an SUVRmidpoint between SUVR 1.2 and 3.0, the estimated years between 2 SUVR values was calculated by integrating the reciprocal of the modeled rate of amyloid accumulation (D). Amyloid time was defined as the estimated years from SUVR 1.2. A cross-validation approach showed that for individuals with at least 2 scans between SUVR 1.2 and 3.0, the amyloid time interval (based on the last SUVR minus the first SUVR) was highly correlated (Pearson) with actual time interval by dates (E). Solid line represents the linear regression between the amyloid time interval and actual time interval. Dashed black line represents a perfect correlation.
Transforming SUVR Into a Proxy for Time
The relationship between amyloid burden and time was evaluated. For all individuals, the change in SUVR (SUVR at last scan minus SUVR at first scan) was significantly but relatively weakly correlated with the actual time interval (last scan date minus first scan date) on a linear scale (Pearson r = 0.31, p < 0.0001, Figure 1C). When only scans with an SUVR between 1.2 and 3.0 were evaluated, this linear relationship was much stronger (Pearson r = 0.75, p < 0.0001). To account for the nonlinear rate of amyloid accumulation between SUVR 1.2 and 3.0, a cubic spline was fitted to the rate of amyloid accumulation as a function of amyloid burden. The average time between levels of amyloid burden between SUVR 1.2 and 3.0 was estimated by integrating the reciprocal of the modeled rate of amyloid accumulation between 2 SUVR levels (eFigure 2, links.lww.com/WNL/B538, and eAppendix 2, links.lww.com/WNL/B536). Given that SUVR 1.2 was the threshold after which significant amyloid accumulation was likely to occur, SUVR 1.2 was set as the origin (0) for this scale, and the estimated years from SUVR 1.2 was called amyloid time.
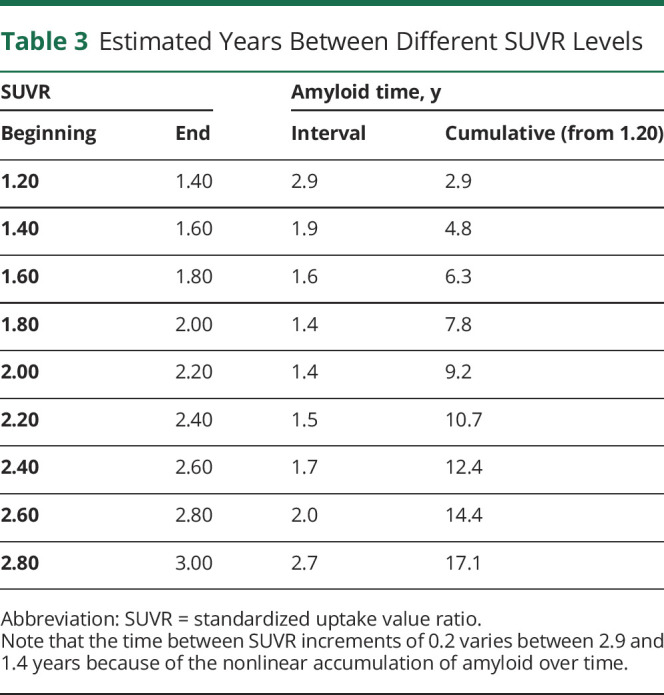
Between SUVR 1.2 and 3.0, the estimated years between SUVR increments of 0.2 ranged between 1.4 and 2.9 years, demonstrating the nonlinear relationship between SUVR and time over small intervals despite an appearance of relative linearity (Table 3 and Figure 1D). The amyloid time interval (amyloid time at the last scan minus amyloid time at the first scan) had a strong linear relationship with the actual time interval (last scan date minus first scan date) with a Pearson r = 0.83. To more rigorously examine the accuracy of the amyloid time estimates, a cross-validation approach was used such that each amyloid PET scan was assigned an amyloid time value estimated with a model in which that scan was not included. The actual time interval between the first and last scan (by dates) and the amyloid time interval based on the SUVR of the 2 scans was highly correlated (Pearson r = 0.83, p < 0.0001, Figure 1E).
Table 3.
Estimated Years Between Different SUVR Levels
Examining the Age at SUVR 1.2 as a Function of APOE Genotype
The estimated age at which individuals reached SUVR 1.2 was examined as a function of APOE ε4 genotype. Using the model created in participants with longitudinal amyloid PET, we estimated amyloid time for all amyloid PET scans with an SUVR between 1.2 and 3.0 in the entire Knight ADRC cohort (Table 2 gives participant characteristics). Amyloid time at the baseline amyloid PET scan was plotted as a function of age for APOE ε4 noncarriers (Figure 2A) and carriers (Figure 2B). For individuals with an SUVR between 1.2 and 3.0, the age at which an individual reached SUVR 1.2 could be estimated by subtracting amyloid time from the age at the scan. The estimated age at SUVR 1.2 across multiple amyloid PET scans in the same individual was highly consistent: the within-individual SD was only 0.8 years (Figure 2C). For example, if an individual's estimated age at SUVR 1.2 was 75 years at the baseline scan (age 80 years at the scan minus amyloid time of 5 years [SUVR 1.63]), it was almost exactly 75 years at a scan 5 years later (age 85 years at the scan minus amyloid time of 10 years [SUVR 2.31]). The estimated age at SUVR 1.2 was plotted by the number of APOE ε4 alleles: no alleles (n = 80), 69.3 ± 7.6 years; 1 allele (n = 75), 64.3 ± 7.0 years; and 2 alleles (n = 25), 59.9 ± 9.2 years (mean ± SD, Figure 2D).
Figure 2. Estimating the Age at Which an Individual Reached SUVR 1.2.

Amyloid time at the baseline amyloid PET scan was plotted as a function of an individual's age at the time of the scan in APOE ε4 noncarriers (A) and carriers (B). Solid red squares represent APOE ε4 homozygotes (ε4/ε4);, red open squares represent ε4 heterozygotes (ε4/ε2 or ε4/ε3); and blue circles represent APOE ε4 noncarriers. Individuals with a standardized uptake value ratio (SUVR) ≤1.2 were assigned an amyloid time of 0 for visualization. Dashed vertical lines represent age 65 years. Age at which an individual reached SUVR 1.2 was estimated by subtracting amyloid time (if > 0) from age and was consistent across multiple scans (C). Estimated age at which an individual reached SUVR 1.2 was averaged across all scans from the individual and was plotted as a function of APOE ε4 genotype. Horizontal lines represent the mean and SD for each group (D). Trajectories of SUVR as a function of age were plotted for individuals with longitudinal amyloid PET (E). Red lines represent APOE ε4 carriers; blue lines represent noncarriers. Subtracting the estimated age at SUVR 1.2 from the age at each scan aligned SUVR trajectories across the cohort (F). Horizontal dotted lines represent SUVR 1.2 and 3.0 (E and F).
The estimated age at SUVR 1.2 was used to align amyloid PET trajectories. Plotting SUVR as a function of age depicted accurate slopes of change in amyloid burden over time (Figure 2E), but these trajectories were not aligned because individuals started accumulating amyloid at different ages. For the participants with at least 1 amyloid PET scan between SUVR 1.2 and 3.0 (Table 2), the estimated age at SUVR 1.2 was subtracted from the age at each scan, which aligned amyloid PET trajectories (Figure 2F). Some individuals with an estimated age at SUVR 1.2 had undergone earlier amyloid PET scans with an SUVR ≤1.2 or later amyloid PET scans with an SUVR ≥3.0; therefore, the trajectories aligned by age at SUVR 1.2 extended outside the SUVR 1.2 to 3.0 range.
Estimation of Years Until Symptom Onset Using Age at SUVR 1.2
An estimate of the age at SUVR 1.2 allowed alignment of longitudinal clinical diagnoses, enabling visualization of 1,384 clinical assessments from 180 individuals with an average total follow-up time of 8.4 ± 4.7 years (Figure 3A). Each point represented the dementia syndrome category from a single clinical assessment; data from each individual were arranged in a single row and aligned on the x-axis by the estimated time from SUVR 1.2 (the age at each clinical assessment minus the estimated age at SUVR 1.2). Points with a negative value on the x-axis denoted clinical assessments that occurred before the individual was estimated to have reached SUVR 1.2. Because clinicians were blinded to biomarker status, some individuals were diagnosed with typical AD dementia syndrome at a low amyloid PET burden. Clinicians were also blinded to diagnoses at previous assessments; some individuals fluctuated from year to year between a typical AD dementia syndrome and other dementia syndrome (usually with an uncertain dementia diagnosis), potentially reflecting changing conditions that affected diagnostic certainty. The 180 individuals in Figure 3A were further arranged vertically from oldest (top) to youngest (bottom) estimated age at SUVR 1.2. With the dementia syndrome category at the baseline assessment for all 180 individuals in the alignment cohort, a logistic regression was used to estimate the years from SUVR 1.2 at which 50% of individuals would have a typical AD dementia syndrome (eTable 2, links.lww.com/WNL/B540); this estimate is denoted by a black line in Figure 3A and represents the expected age at symptom onset based on the estimated age at SUVR 1.2 and estimated time from SUVR 1.2.
Figure 3. Visualizing Dementia Syndromes Over Time and Estimating Age at Symptom Onset.

At each clinical assessment, clinicians who were blinded to biomarker status and the results of prior clinical assessments formulated the Clinical Dementia Rating (CDR) score; individuals with a CDR of ≥0.5 were considered to have a dementia syndrome, and the probable etiology of the dementia syndrome was diagnosed from clinical features. Diagnoses from 1,384 assessments on 180 individuals were plotted by the estimated years from standardized uptake value ratio (SUVR) 1.2 (A). Each row of points corresponds to 1 individual. Each point represents 1 clinical assessment, and the color of the point denotes the dementia syndrome category: green, cognitively normal; yellow, other dementia syndrome (uncertain, atypical, or suspected non–Alzheimer disease [AD] dementia); and red, typical AD dementia syndrome. Black points represent the date of death if applicable. Individuals were arranged vertically in order of estimated age at SUVR 1.2. Dotted vertical line represents SUVR 1.2; dashed vertical line represents SUVR 1.42, the established cutoff for amyloid PET positivity. Solid black line represents the estimated time from SUVR 1.2 when 50% of individuals would have a typical AD dementia syndrome based on a logistic regression of the dementia syndrome category at the baseline clinical assessment (eTable 2, links.lww.com/WNL/B540). Solid red line represents the estimated time of symptom onset as predicted by the model in (D). Twenty-two individuals who were cognitively normal at their baseline clinical assessment and had a typical AD dementia syndrome at their last clinical assessment were defined as progressors to symptomatic AD (B). Purple line represents the predicted age at which the progressors were first diagnosed with typical AD dementia syndrome on the basis of the estimated age at which individuals reached SUVR 1.2 (C). Omitting 3 individuals (purple arrowheads in B, purple points in C) who were amyloid PET negative at the onset of cognitive decline improved prediction of the age at symptom onset (D).
Of the 180 individuals with an estimated age at SUVR 1.2 available, 22 were cognitively normal at their baseline clinical assessment and had a typical AD syndrome at their last clinical assessment; these individuals were defined as progressors to symptomatic AD (Figure 3B and eTable 3, links.lww.com/WNL/B541). The age at which the progressors first developed a typical AD dementia syndrome was predicted by the estimated age at SUVR 1.2 (R2 = 0.54, p < 0.0001, root mean square error [RMSE] 4.5 years, Figure 3C). Three progressors who were amyloid PET negative (SUVR ≤1.42) at the onset of cognitive decline (CDR score >0) could have experienced a typical AD dementia syndrome caused by a non-AD etiology; omission of these individuals improved prediction of the age at symptom onset (R2 = 0.84, RMSE 2.8 years, p < 0.0001, Figure 3D). Sex, years of education, APOE ε4 status, and race were not significant predictors when included in the models. The model for age at symptom onset derived in the progressors (n = 19, Figure 3D) was plotted with a red line in Figure 3A. It was largely overlapping with the model derived in the entire alignment cohort (n = 180) that included individuals who were cognitively stable throughout follow-up and individuals who with diagnosed with dementia at study entry.
A cross-validation approach in which the amyloid time model was generated without data from the progressors resulted in nearly identical models and model fits for the estimated age at symptom onset in progressors (R2 = 0.54, RMSE 4.5 years for all progressors; and R2 = 0.85, RMSE 2.7 years after 3 potential misdiagnoses were omitted). A logistic regression of the dementia syndrome category at the baseline amyloid PET scan found that older individuals were more likely to have a typical AD dementia syndrome at a given SUVR (eTable 4, links.lww.com/WNL/B542).
Discussion
Amyloid accumulated at a nonlinear but relatively consistent rate across individuals from SUVR 1.2 until SUVR 3.0, an approximately17-year period that included much of the preclinical phase of AD for most individuals. Amyloid time, the estimated years from SUVR 1.2, was highly correlated with actual time by dates (Pearson r = 0.83, p < 0.0001) and was used to estimate the age when an individual reached SUVR 1.2. A variety of approaches have been used to calculate the time course of amyloid accumulation,2,6-8,24,25 and lowest level of amyloid burden that predicts cognitive decline has been evaluated.26 In the work most conceptually similar to the current study, a completely different mathematical approach was used to align amyloid PET trajectories, which were remarkably consistent across individuals, and to estimate the age at amyloid positivity.24 A longer time of amyloid positivity was associated with a higher risk of cognitive decline and clinical progression.24 Compared to other studies, the major innovation of the current study was the use of estimated age at SUVR 1.2 to align longitudinal clinical diagnostic data collected many years distant from the amyloid PET scan and to predict the age at AD symptom onset. The agreement and convergence of our findings with other studies suggest that the amyloid time concept is robust across different cohorts and mathematical approaches.
At low levels of amyloid burden (SUVR ≤1.2) analogous to the lag phase of Aβ aggregation in vitro, the rate of amyloid accumulation was correlated with amyloid burden in APOE ε4 carriers but not noncarriers. Biochemical studies demonstrate that adding preformed nuclei or seeds shortens or eliminates the lag phase of Aβ aggregation in vitro.3,5 The abbreviated lag phase in APOE ε4 carriers, as demonstrated by the correlation between the rate of amyloid accumulation and amyloid burden at SUVR ≤1.2, may indicate that APOE ε4 has a seeding effect on brain amyloid aggregation. The earlier age at amyloid deposition in APOE ε4 carriers seen in this study and many others25,27-32 also suggests that APOE ε4 has a seeding effect on amyloid aggregation in the human brain. Previous studies in our cohort28,33 and other cohorts6,30,32,34-36 have reported inconsistent results on how and whether APOE ε4 affects the rate of amyloid accumulation, likely because the studies were examining different phases of amyloid accumulation depending on their analytic approach. A study that stratified by amyloid burden found results similar to this study: faster amyloid accumulation in APOE ε4 carriers at lower levels of amyloid burden but no difference in amyloid accumulation by APOE ε4 status at higher levels of amyloid burden.36 The lack of effect of APOE ε4 status on amyloid accumulation between SUVR 1.2 and 3.0, which may be analogous to the growth phase of amyloid aggregation, suggests that APOE ε4 modulates primarily initiation of amyloid accumulation (seeding) and not the rate of amyloid accumulation later in the disease course.
The most important finding of this study was that the age at AD symptom onset for cognitively normal individuals with brain amyloidosis could be estimated with a single amyloid PET scan. Previous studies have shown that parental age at symptom onset in autosomal dominant AD predicted individual age at onset with an R2 = 0.38 to 0.56.1,37 In this study of sporadic AD, the model including all progressors predicted individual age at onset at a similar level (R2 = 0.54, RMSE 4.5 years). Exclusion of 3 of the 22 progressors (14%) with cognitive decline (CDR score >0) before amyloid PET positivity (SUVR 1.42) improved the model fit (R2 = 0.84, RMSE 2.8 years). The 3 progressors who developed cognitive decline before SUVR 1.42 may have had a non-AD cause of their symptoms.15,38 When the model for symptom onset that was created in 19 progressors was applied to 180 individuals across the cognitive spectrum, it was closely aligned to the cross-sectional logistic regression estimate for when 50% of the 180 individuals had a typical AD dementia syndrome. In studies of autosomal dominant AD, approximately half of individuals are expected to be cognitively impaired at the estimated age at symptom onset.1,37
Our results suggest that older individuals develop symptomatic AD after a shorter period of amyloid accumulation, which translates to a lower level of amyloid burden. According to our model using the age at SUVR 1.2 to align cognitive trajectories, a 45-year-old who reached SUVR 1.2 would be expected to develop symptoms 21 years later at SUVR >3.0, whereas an 85-year-old would develop symptoms in only 9 years at SUVR ≈2.2. Furthermore, using a simple logistic regression, we found that older individuals were more likely to manifest symptoms of AD dementia at the same level of amyloid burden (SUVR). The increased expression of AD symptoms in older individuals could be related to decreased cognitive reserve from normal aging processes or comorbid conditions.39,40 Brain amyloid deposition is necessary but not sufficient for symptomatic AD, and many other genetic, biological, and social factors are likely involved in expression of symptoms.41,42 Analyses of larger numbers of progressors will likely improve modeling of factors that are known to modify the risk of symptomatic AD associated with a particular level of amyloid burden21,39,43,44 and may further improve estimation of the age at symptom onset. In addition, analyses of cohorts that include assessment of prodromal AD (e.g., subjective cognitive decline) may enable study of the evolution of AD symptom onset.
Many additional studies, including with both late-onset AD and autosomal dominant AD cohorts, are needed to refine these approaches and to validate these findings, especially before application to clinical settings. Innumerable approaches could be used to calculate amyloid time and to determine the tipping point in amyloid accumulation.2,6-8 We chose a simple approach that could be easily applied to other datasets, including datasets with relatively few scans from each individual. The specific values used in the amyloid time model (SUVR 1.2 and 3.0, which correspond to centiloid values of 7 and 8845) will vary depending on center-specific factors, including the amyloid PET tracer, brain regions evaluated, and PET processing. Improvements in MRI and PET scanners and sequences may provide greater sensitivity to pathology, and such differences should be systematically evaluated in future studies. PiB is a highly sensitive tracer,22 and models created with less sensitive tracers may not be as accurate. In addition, differences in the assessment of dementia diagnosis may affect models for prediction of symptom onset.
Accurate estimation of the age at symptom onset would improve our understanding of the AD time course and facilitate identification of cognitively normal participants at high risk of progression to symptomatic AD for prevention trials. As has been shown in drug trials for autosomal dominant AD, accurate prediction of symptom onset increases power and decreases costs.46 Furthermore, if specific therapies are most effective within a certain period of the disease course, precise staging is critical. Therefore, accurate prediction of symptom onset may accelerate efforts to develop effective preventative treatments for AD.
Acknowledgment
The authors express their gratitude to the research volunteers who participated in the studies from which these data were obtained and their supportive families. They thank the Clinical, Biomarker, and Imaging Cores at the Knight ADRC for sample and data collection. They also thank Andrianus Kardjaja for assistance in data visualization.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADRC
Alzheimer Disease Research Center
- CDR
Clinical Dementia Rating
- PiB
Pittsburgh compound B
- RMSE
root mean square error
- SUVR
standardized uptake value ratio
Appendix. Authors
Study Funding
This study was supported by National Institute on Aging grants R03AG050921 (S. Schindler), K23AG053426 (S. Schindler), P30AG066444 (J.C. Morris), P01AG003991 (J.C. Morris), P01AG026276 (J.C. Morris), and R01AG053550 (C. Xiong).
Disclosure
S. Schindler, Y. Li, V.D. Buckles, and B.A. Gordon report no disclosures. T.L.S. Benzinger has received research support from Avid Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly) and Biogen. She has or is currently participating in clinical trials sponsored by Janssen, Eli Lilly, Pfizer, Biogen, and Roche. She has received travel support from Biogen, the American Society for Neuroradiology, the Alzheimer's Association, and the People's Republic of China. G. Wang and D. Coble report no disclosures; W.E. Klunk is supported by NIH grants P50 AG005133, RF1 AG025516, and P01 AG025204. GE Healthcare holds a license agreement with the University of Pittsburgh based on the PiB PET technology described in this article. W.E. Klunk is a coinventor of PiB and thus has a financial interest in this license agreement and receives royalty payments. GE Healthcare provided no grant support for this study. A.M. Fagan has received research funding from the National Institute on Aging of the NIH, Biogen, Centene, Fujirebio, and Roche Diagnostics. She is a member of the Scientific Advisory boards for Roche Diagnostics, Genentech, and AbbVie and also consults for Araclon/Grifols, DiademRes, DiamiR, and Otsuka. D. Holtzman cofounded and is on the scientific advisory board of C2N Diagnostics. Washington University and D. Holtzman have equity ownership interest in C2N Diagnostics and receive royalty income based on technology (stable isotope labeling kinetics, blood plasma assay, anti-tau antibodies) licensed by Washington University to C2N Diagnostics. He receives income from C2N Diagnostics for serving on the Scientific Advisory Board. He is on the Scientific Advisory Board of Denali and Genentech. He consults for Merck, Cajal Neurosciences, and Takeda. His laboratory receives research support from C2N Diagnostics, NextCure, and Novartis. R.J. Bateman cofounded C2N Diagnostics. Washington University and Dr. Bateman have equity ownership interest in C2N Diagnostics and receive royalty income based on technology (stable isotope labeling kinetics and blood plasma assay) licensed by Washington University to C2N Diagnostics. He receives income from C2N Diagnostics for serving on the Scientific Advisory Board. Washington University, with R.J. Bateman as coinventor, has submitted the US provisional patent application “Plasma Based Methods for Detecting CNS Amyloid Deposition.” He consults for Roche, Genentech, AbbVie, Pfizer, Boehringer-Ingelheim, and Merck. J.C. Morris does not own stock or have equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. C. Xiong reports no disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Bateman RJ, Xiong C, Benzinger TL, et al. . Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):795-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villemagne VL, Burnham S, Bourgeat P, et al. . Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357-367. [DOI] [PubMed] [Google Scholar]
- 3.Harper JD, Lansbury PT Jr. Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385-407. [DOI] [PubMed] [Google Scholar]
- 4.Morel B, Conejero-Lara F. Early mechanisms of amyloid fibril nucleation in model and disease-related proteins. Biochim Biophys Acta Proteins Proteom. 2019;1867(11):140264. [DOI] [PubMed] [Google Scholar]
- 5.Srivastava AK, Pittman JM, Zerweck J, et al. . β-Amyloid aggregation and heterogeneous nucleation. Protein Sci. 2019;28(9):1567-1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jack CR Jr, Wiste HJ, Lesnick TG, et al. . Brain beta-amyloid load approaches a plateau. Neurology. 2013;80(10):890-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budgeon CA, Murray K, Turlach BA, et al. . Constructing longitudinal disease progression curves using sparse, short-term individual data with an application to Alzheimer's disease. Stat Med. 2017;36(17):2720-2734. [DOI] [PubMed] [Google Scholar]
- 8.Jagust WJ, Landau SM; Alzheimer's Disease Neuroimaging Initiative. Temporal dynamics of beta-amyloid accumulation in aging and Alzheimer disease. Neurology. 2021;96(9):e1347–e1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pastor P, Roe CM, Villegas A, et al. . Apolipoprotein Eepsilon4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol. 2003;54(2):163-169. [DOI] [PubMed] [Google Scholar]
- 10.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412-2414. [DOI] [PubMed] [Google Scholar]
- 11.Berg L, McKeel DW Jr, Miller JP, Baty J, Morris JC. Neuropathological indexes of Alzheimer's disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50(4):349-358. [DOI] [PubMed] [Google Scholar]
- 12.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198. [DOI] [PubMed] [Google Scholar]
- 13.Morris JC, Weintraub S, Chui HC, et al. . The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer disease centers. Alzheimer Dis Assoc Disord. 2006;20(4):210-216. [DOI] [PubMed] [Google Scholar]
- 14.Morris JC, Storandt M, Miller JP, et al. . Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58(3):397-405. [DOI] [PubMed] [Google Scholar]
- 15.Storandt M, Grant EA, Miller JP, Morris JC. Longitudinal course and neuropathologic outcomes in original vs revised MCI and in pre-MCI. Neurology. 2006;67(3):467-473. [DOI] [PubMed] [Google Scholar]
- 16.Morris JC, Blennow K, Froelich L, et al. . Harmonized diagnostic criteria for Alzheimer's disease: recommendations. J Intern Med. 2014;275(3):204-213. [DOI] [PubMed] [Google Scholar]
- 17.McKhann GM, Knopman DS, Chertkow H, et al. . The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klunk WE, Engler H, Nordberg A, et al. . Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound-B. Ann Neurol. 2004;55(3):306-319. [DOI] [PubMed] [Google Scholar]
- 19.Su Y, Blazey TM, Snyder AZ, et al. . Partial volume correction in quantitative amyloid imaging. NeuroImage. 2015;107:55-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 1998;39(5):904-911. [PubMed] [Google Scholar]
- 21.Vlassenko AG, McCue L, Jasielec MS, et al. . Imaging and cerebrospinal fluid biomarkers in early preclinical Alzheimer disease. Ann Neurol. 2016;80(3):379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su Y, Flores S, Wang G, et al. . Comparison of Pittsburgh compound B and florbetapir in cross-sectional and longitudinal studies. Alzheimers Dement (Amst). 2019;11:180-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo T, Dukart J, Brendel M, et al. . Rate of beta-amyloid accumulation varies with baseline amyloid burden: implications for anti-amyloid drug trials. Alzheimers Dement. 2018;14(11):1387-1396. [DOI] [PubMed] [Google Scholar]
- 24.Koscik RL, Betthauser TJ, Jonaitis EM, et al. . Amyloid duration is associated with preclinical cognitive decline and tau PET. Alzheimers Dement. 2020;12(1):e12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bilgel M, An Y, Zhou Y, et al. . Individual estimates of age at detectable amyloid onset for risk factor assessment. Alzheimers Dement. 2016;12(4):373-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farrell ME, Jiang S, Schultz AP, et al. . Defining the lowest threshold for amyloid-PET to predict future cognitive decline and amyloid accumulation. Neurology. 2021;96(4):e619-e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bussy A, Snider BJ, Coble D, et al. . Effect of apolipoprotein E4 on clinical, neuroimaging, and biomarker measures in noncarrier participants in the Dominantly Inherited Alzheimer Network. Neurobiol Aging. 2019;75:42-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishra S, Blazey TM, Holtzman DM, et al. . Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE epsilon4 genotype. Brain. 2018;141(6):1828-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villemagne VL, Pike KE, Chetelat G, et al. . Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleisher AS, Chen K, Liu X, et al. . Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging. 2013;34(1):1-12. [DOI] [PubMed] [Google Scholar]
- 31.Jack CR Jr, Wiste HJ, Weigand SD, et al. . Age, sex, and APOE epsilon4 effects on memory, brain structure, and beta-amyloid across the adult life span. JAMA Neurol. 2015;72(5):511-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopresti BJ, Campbell EM, Yu Z, et al. . Influence of apolipoprotein-E genotype on brain amyloid load and longitudinal trajectories. Neurobiol Aging. 2020;94:111-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vlassenko AG, Mintun MA, Xiong C, et al. . Amyloid-beta plaque growth in cognitively normal adults: longitudinal [11C]Pittsburgh compound B data. Ann Neurol. 2011;70(5):857-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grimmer T, Tholen S, Yousefi BH, et al. . Progression of cerebral amyloid load is associated with the apolipoprotein E epsilon4 genotype in Alzheimer's disease. Biol Psychiatry. 2010;68(10):879-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Resnick SM, Bilgel M, Moghekar A, et al. . Changes in Abeta biomarkers and associations with APOE genotype in 2 longitudinal cohorts. Neurobiol Aging. 2015;36(8):2333-2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim YY, Mormino EC, Alzheimer's Disease Neuroimaging Initiative. APOE genotype and early beta-amyloid accumulation in older adults without dementia. Neurology. 2017;89(10):1028-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryman DC, Acosta-Baena N, Aisen PS, et al. . Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83(3):253-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shim YS, Roe CM, Buckles VD, Morris JC. Clinicopathologic study of Alzheimer's disease: Alzheimer mimics. J Alzheimers Dis. 2013;35(4):799-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ossenkoppele R, Jansen WJ, Rabinovici GD, et al. . Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA. 2015;313(19):1939-1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol. 2012;11(11):1006-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and 'wingmen. Nat Neurosci. 2015;18(6):800-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arboleda-Velasquez JF, Lopera F, O'Hare M, et al. . Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25(11):1680-1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roe CM, Ances BM, Head D, et al. . Incident cognitive impairment: longitudinal changes in molecular, structural and cognitive biomarkers. Brain. 2018;141(11):3233-3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris JC, Roe CM, Grant EA, et al. . Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66(12):1469-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Su Y, Flores S, Hornbeck RC, et al. . Utilizing the centiloid scale in cross-sectional and longitudinal PiB PET studies. Neuroimage Clin. 2018;19:406-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bateman RJ, Benzinger TL, Berry S, et al. . The DIAN-TU Next Generation Alzheimer's Prevention Trial: adaptive design and disease progression model. Alzheimers Dement. 2017;13(1):8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used for this study are from the Knight ADRC and are available on request by qualified investigators.