

Abstract
On the basis of potent and selective A3 adenosine receptor (AR) antagonist, 2-chloro-N6-(3-iodobenzyl)-4′-thioadeno-sine-5′-N,N-dimethyluronamide, structure–activity relationships were studied for a series of 5′-N,N-dialkyluronamide derivatives, synthesized from d-gulonic γ-lactone. From this study, it was revealed that removal of the hydrogen bond-donating ability of the 5′-uronamide was essential for the pure A3AR antagonism. 5′-N,N-Dimethyluronamide derivatives exhibited higher binding affinity than larger 5′-N,N-dialkyl or 5′-N,N-cycloalkylamide derivatives, indicating that steric factors are crucial in binding to the human A3AR. A N6-(3-bromobenzyl) derivative 6c (Ki = 9.32 nM) exhibited the highest binding affinity at the human A3AR with very low binding affinities to other AR subtypes.
Keywords: 4′-Thionucleoside, A3 adenosine receptor antagonist, Conformational change, Radioligand binding
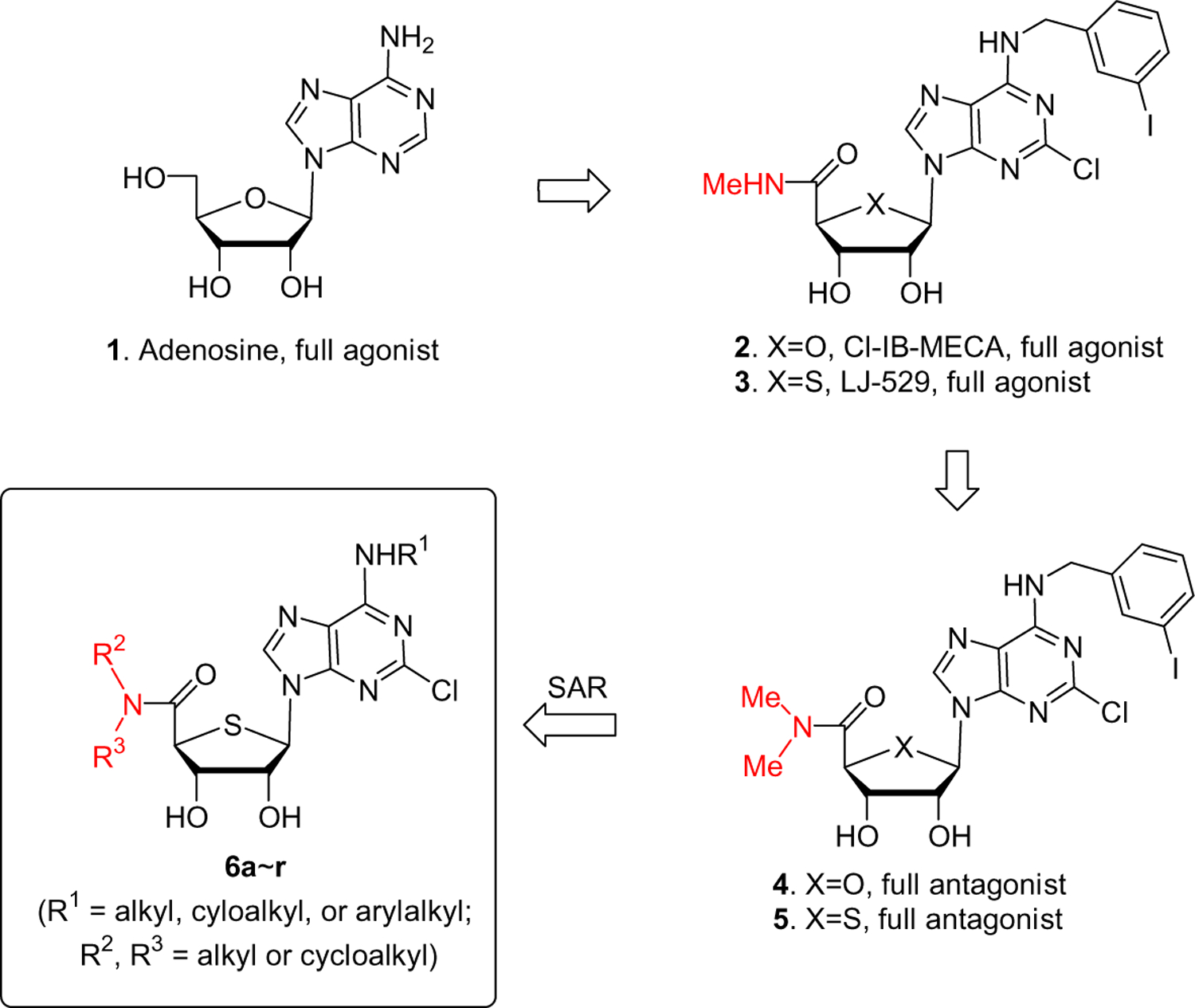
Adenosine (1) regulates many physiological functions through four subtypes (A1, A2a, A2b, and A3) of adenosine receptors (AR).1 Among these subtypes, activation of the A3AR inhibits adenylate cyclase to lower the level of cAMP.1 Also, activation of the A3AR stimulates phospholipase C (PLC) through the β, γ subunit of the G protein, to increase levels of inositol 1,4,5-tris-phosphate (IP3). Control of A3AR is thought to be relevant to experimental treatment modalities for a variety of disorders related to these signal transduction pathways, such as cancer, cerebral or cardiac ischemia, glaucoma, allergic conditions, and inflammation.1,2
On the basis of the structure of adenosine (1), worldwide efforts have been made to discover potent and selective human A3AR agonists and antagonists.2,3 Modification on the N6- and/or 4′-hydroxymethyl group of 1 resulted in 2 (Cl-IB-MECA) as a potent and selective human A3AR full agonist4 (Chart 1). Bioisosteric replacement of the furanose oxygen of 2 with a sulfur atom produced another potent and selective human A3AR full agonist 3 (LJ-529).5 However, until recently it has been difficult to discover potent and selective human A3AR full antagonists with a nucleoside skeleton because of the tendency of derivatives of adenosine (1) to act as full agonists. Thus, most of the potent and selective antagonists for the human A3AR belong to the category of nonpurine heterocyclic compounds,6–10 which act competitively with adenosine derivatives at the orthosteric binding site of the A3AR. However, these nonpurine heterocyclic compounds showed weak binding affinity at the rat A3AR11,12 and were unsuitable for efficacy evaluation in small animal models and thus not optimal for further drug development. Therefore, it has been highly desirable to discover potent and selective A3AR full antagonists of which the affinity and selectivity are independent of species. Since it was reported that antagonists with a nucleoside skeleton13 could be species-independent, potent, and selective A3AR full antagonists, we searched for novel nucleoside analogues, among which appending an additional methyl group to the 5′-uronamide nitrogen of compounds 2 and 3 converted these A3AR full agonists into the potent and selective A3AR full antagonists 4 and 5.14 4′-Thionucleoside analogue 5 was found to be a more potent and selective antagonist than the corresponding 4′-oxonucleoside analogue 4.14 Thus, on the basis of these findings, we modified the N6- and 4′-hydroxymethyl groups of 4′-thionucleoside analogue 5 in order to search for potent and selective A3AR full antagonists. Herein, we report the structure–activity relationship (SAR) study of 2-chloro-N6-substituted-4′-thioadenosine-5′-N,N-dialkylamides at the human A3AR.
Chart 1.
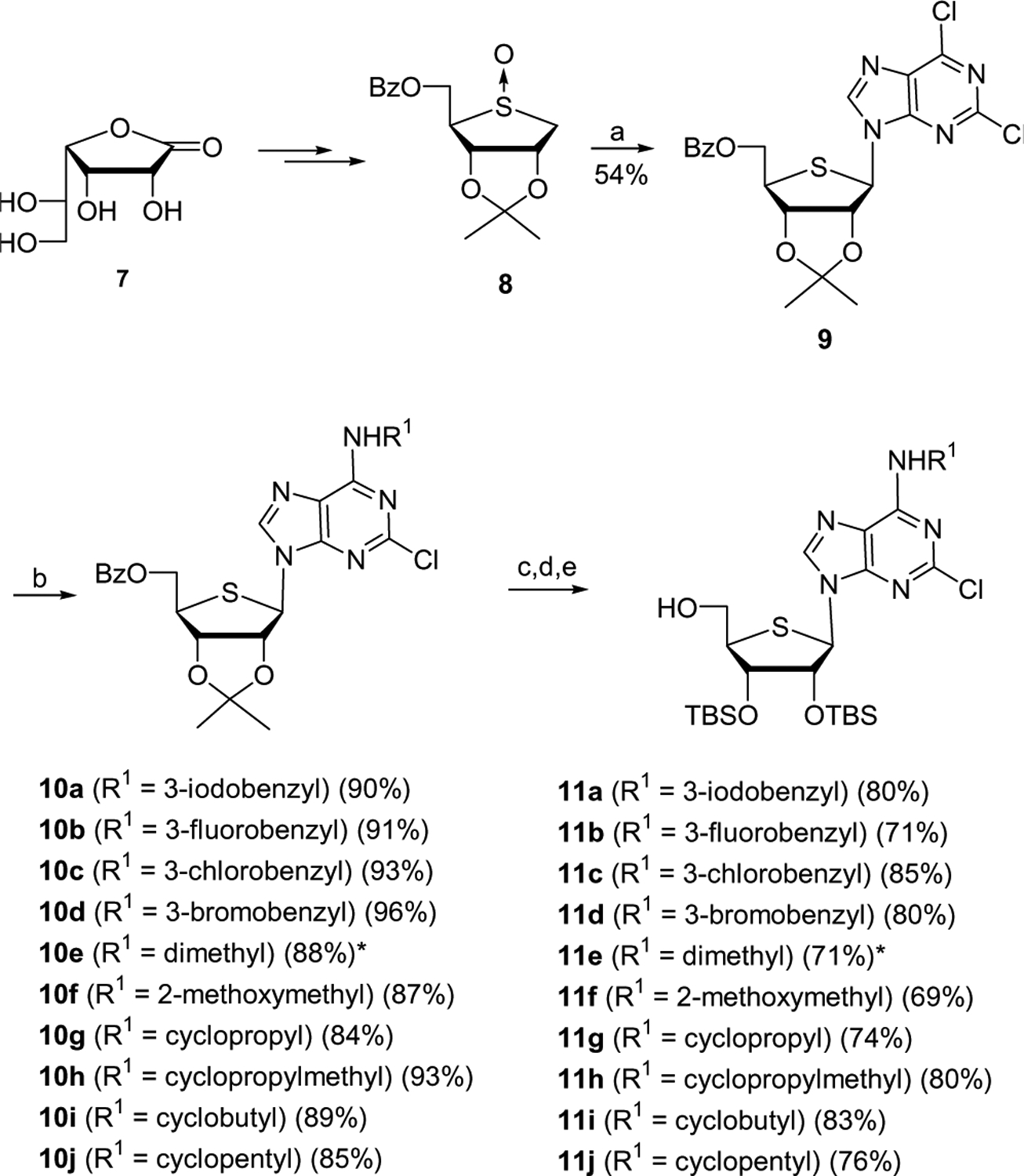
Synthesis of the key intermediates 11a–j started from d-gulonic γ-lactone (7), as shown in Scheme 1.
Scheme 1.
Reagents and conditions: (a) 2,6-dichloropurine, TMSOTf, Et3N, CH3CN–ClCH2CH2Cl, rt to 83 °C; (b) R1NH2, Et3N, EtOH, rt; (c) 80% AcOH, 70 °C; (d) TBSOTf, pyridine, 50 °C; (e) NaOMe, MeOH. *N6 (CH3)2.
d-Gulonic γ-lactone (7) was converted to the glycosyl donor 8, using our previously published procedure.5,15 Pummerer-type condensation of 8 with 2,6-dichloropurine in the presence of TMSOTf and Et3N afforded the desired nucleoside 95,15 as a single diastereomer.
Treatment of 2,6-dichloropurine derivative 9 with various alkyl or arylalkyl amines gave N6-substituted analogues 10a–j. Because of the difficulty in removing the isopropylidene group at the final step, the isopropylidene intermediates 10a–j were converted to the TBS derivatives by treatment with 80% acetic acid followed by reprotection with TBSOTf. The resulting compounds were treated with sodium methoxide to give the key intermediates 11a–j.
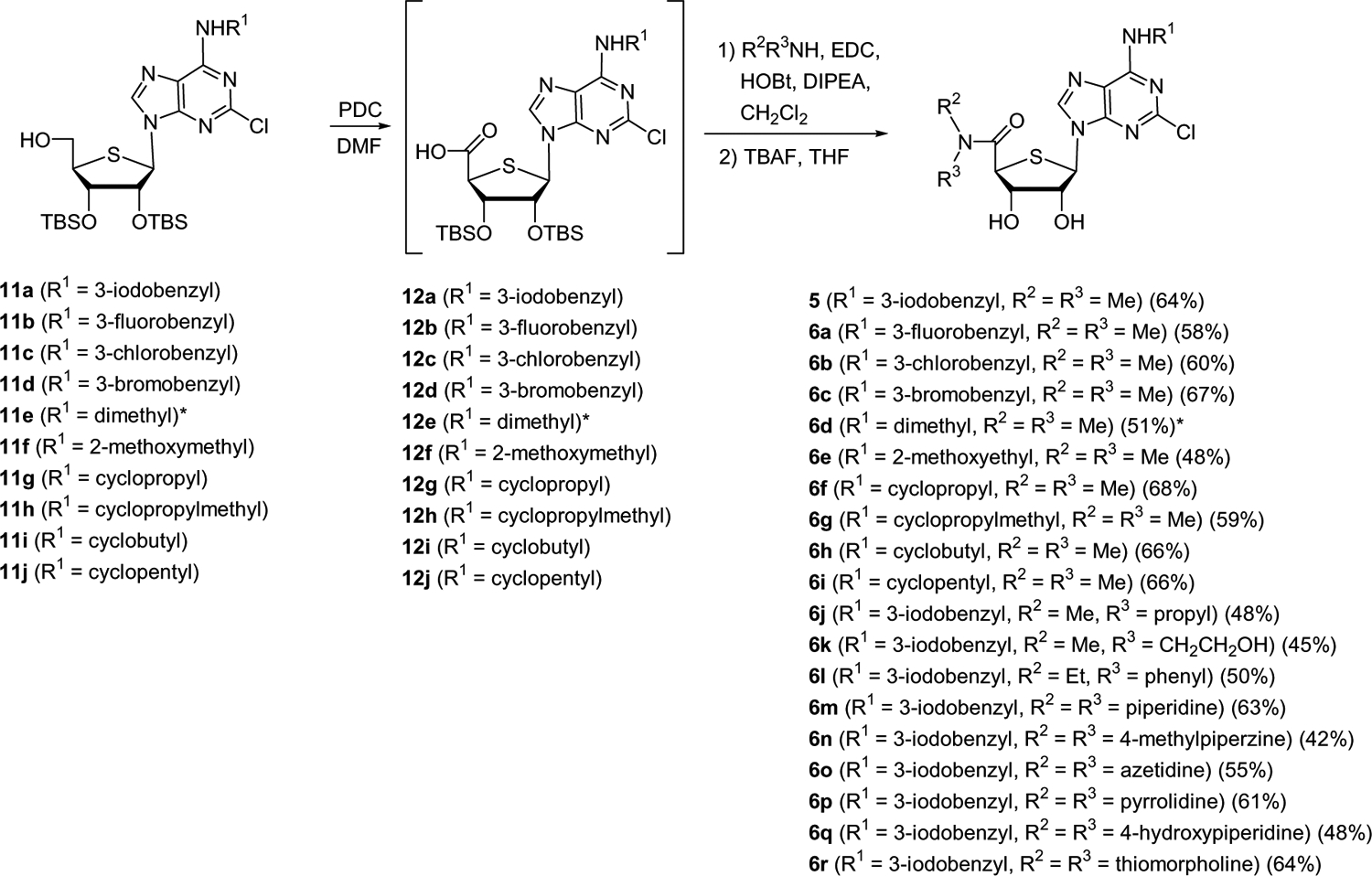
Conversion of 11a–j to the target nucleosides 6a–r is illustrated in Scheme 2. The primary hydroxyl groups of 11a–j were oxidized by treatment with PDC in DMF to the carboxylic acids 12a–j, which were converted to various tertiary amides 6a–r16 by coupling the acids 12a–j with various dialkyl or cycloalkyl amines in the presence of EDC and HOBt.
Scheme 2.
*N6 (CH3)2.
Binding assays were carried out using standard radioligands in Chinese hamster ovary (CHO) cells stably expressing a human AR subtype,14,17 and binding affinities of the final nucleosides 6a–r at the three subtypes of human ARs are shown in Table 1. In general, 5′-N,N-dimethylamide derivatives exhibited higher binding affinity than larger 5′-N,N-dialkyl or 5′-N,N-cycloalkylamide derivatives indicating that steric factors are crucial in binding to human A3AR. The effects on the binding affinity of various N6-substituted analogues also having a 5′-N,N-dimethyl group at the 5′-uronamide position were examined. The N6-(3-halobenzyl) series generally showed better binding affinity at the human A3AR than the N6-dialkyl or N6-cycloalkyl series. Within the N6-(3-halobenzyl) series, the rank order of binding affinity at the human A3AR was 3-bromobenzyl > 3-iodobenzyl > 3-chlorobenzyl > 3-fluorobenzyl. This result indicated that the human A3AR prefers bromo as an optimal size of 3-halo substitution, and larger (I) or smaller (F) substitution reduced binding affinity. Among compounds tested, compound 6c (Ki = 9.32 nM) exhibited the highest binding affinity at the human A3AR with very low binding affinities to other AR subtypes.
Table 1.
Potency of 2-chloro-4′-thioadenosine-5′-dialkylamide derivatives in binding at human A1, A2A, and A3ARs expressed in CHO cells
|
|||
|---|---|---|---|
| Compound No. (R1,R2,R3) | Ki (nM ± SEM) or % inhibition at 10 μMa | ||
| hA1ARb | hA2AARb | hA3ARb | |
| 2 c | 222 ± 22 | 5360 ± 2470 | 1.4 ± 0.3 |
| 3 c | 193 ± 46 | 223 ± 36 | 0.38 ± 0.07 |
| 4 d | 5870 ± 930 | >10,000 | 29.0 ± 4.9 |
| 5d (3-iodobenzyl, Me, Me) | 6220 ± 640 | >10,000 | 15.5 ± 3.1 |
| 6a (3-fluorobenzyl, Me, Me) | (16 ± 4%) | (1%) | 121 ± 11 |
| 6b (3-chlorobenzyl, Me, Me) | (10 ± 8%) | (−1%) | 21.3 ± 2.1 |
| 6c (3-bromobenzyl, Me, Me) | (37 ± 2%) | (7%) | 9.32 ± 3.20 |
| 6d (Me2, Me, Me)e | (8 ± 2%) | (−2%) | 136 |
| 6e (2-methoxyethyl, Me, Me) | (−7 ± 7%) | (1%) | 425 |
| 6f (cyclopropyl, Me, Me) | (7 ± 16%) | (−8 ± 4%) | 109 ± 16 |
| 6g (cyclopropylmethyl, Me, Me) | 404 ± 120 | (0 ± 6%) | 30.7 ± 9.4 |
| 6h (cyclobutyl, Me, Me) | 345 ± 152 | (10%) | 115 ± 50 |
| 6i (cyclopentyl, Me, Me) | (18 ± 21%) | (4 ± 27%) | 837 |
| 6j (3-iodobenzyl, Me, Propyl) | (50 ± 1%) | (5%) | 727 |
| 6k (3-iodobenzyl, Me, CH2CH2OH) | 237 ± 6 | (13%) | 126 ± 17 |
| 6l (3-iodobenzyl, Et, phenyl) | (18 ± 5%) | (8%) | 398 |
| 6m (3-iodobenzyl, piperidine) | (27 ± 5%) | (7%) | 565 |
| 6n (3-iodobenzyl, 4-methylpiperazine) | (19 ± 5%) | (−7%) | 667 |
| 6o (3-iodobenzyl, azetidine) | (8 ± 6%) | (12%) | 43.4 ± 2.6 |
| 6p (3-iodobenzyl, pyrrolidine) | (20 ± 3%) | (14%) | 117 ± 31 |
| 6q (3-iodobenzyl, 4-hydroxypiperidine) | (16 ± 7%) | (10%) | 1530 |
| 6r (3-iodobenzyl, thiomorpholine) | (19 ± 9%) | (15%) | 867 |
All experiments were done on CHO cells stably expressing one of three subtypes of human ARs. The binding affinity for A1, A2A, and A3ARs was expressed as Ki values and was determined by using agonist radioligands ([3H]CCPA), ([3H]CGS21680), [125I]I-AB-MECA, respectively. Values in parentheses are for weak binding, corresponding to an IC50 ⩾ 10 μM. Data are expressed as means ± standard error.
Ki in binding, unless noted.
Ki values from Ref. 5.
Ki values from Ref. 14.
N6 (CH3)2.
The functional efficacy of compounds 514 and 6c showed that high binding affinity at human A3AR was determined by inhibition of forskolin-stimulated cAMP production in AR-transfected CHO cells and measured at a concentration of 10 μM, in comparison to the maximal effect of a full agonist, N-ethyl-5′-N-ethylcarboxamidoadenosine (NECA), at 10 μM. In these functional assays, A3AR agonism was absent in these compounds, indicating that these analogues are pure A3AR antagonists, while the concentration–response curve for Cl-IB-MECA (2) indicated full agonism, as previously reported,4 with an EC50 of 1.2 nM at the human A3AR. To probe species differences, the affinity of N,N-5′-dimethylamide derivative 5 was also measured at the rat A3AR. Although the affinity decreased with respect to the affinity at the human A3AR, this compound showed moderate affinity (Ki = 321 ± 74 nM) at the rat A3AR.14
Molecular modeling of the A3AR indicated that the hydrogen bond-donating ability of the 5′-uronamide is responsible for the conformational change needed for receptor activation.18,19 Because the synthesized 5′-N,N-dialkyl derivatives which removed the hydrogen bond-donating ability of the 5′-uronamide could not lead to the conformational change, resulting in the loss of ability to activate the A3AR, the 5′-N,N-dialkyl series of compounds act as pure antagonists.
In summary, on the basis of potent and selective human A3AR antagonism of the dimethyl derivative 5, we carried out structure–activity relationship studies of 2-chloro-N6-substituted-4′-thioadenosine-5′-N,N-dial-kylamides. From this study, we identified compound 6c as a highly potent and selective human A3AR antagonist, in which removal of the hydrogen bond-donating ability of the 5′-uronamide was essential for the pure A3AR antagonism. 5′-N,N-dimethyluronamide derivatives generally exhibited higher binding affinity than larger 5′-N,N-dialkyl or 5′-N,N-dicycloalkylamide derivatives, indicating that steric factors in the 5′ region are important in binding of nucleosides to the human A3AR. The newly synthesized A3AR antagonists studied here could be evaluated in models of a number of disorders related to the A3AR, such as glaucoma, inflammation, and asthma.
Acknowledgments
This research was supported by the National Core Research Center (NCRC) program (No. R15-2006-020) of the Ministry of Science and Technology (MOST) and the Korea Science and Engineering Foundation (KOSEF) and by the National Institutes of Health, NIDDK Intramural Research Program.
References and notes
- 1.Fredholm BB; IJzerman AP; Jacobson KA; Klotz KN; Linden J Pharmacol. Rev 2001, 53, 527. [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson KA; Gao Z-G Nat. Rev. Drug Discov 2006, 5, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baraldi PG; Cacciari B; Romagnoli R; Merighi S; Varani K; Borea PA; Spalluto G Med. Res. Rev 2000, 20, 103. [DOI] [PubMed] [Google Scholar]
- 4.Kim HO; Ji X.-d.; Siddiqi SM; Olah ME; Stiles GL; Jacobson KA J. Med. Chem 1994, 37, 3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Jeong LS; Jin DZ; Kim HO; Shin DH; Moon HR; Gunaga P; Chun MW; Kim Y-C; Melman N; Gao Z-G; Jacobson KA J. Med. Chem 2003, 46, 3775; [DOI] [PubMed] [Google Scholar]; (b) Jeong LS; Lee HW; Jacobson KA; Kim HO; Shin DH; Lee JA; Gao ZG; Lu C; Duong HT; Gunaga P; Lee SK; Jin DZ; Chun MW; Moon HR J. Med. Chem 2006, 49, 273. [DOI] [PubMed] [Google Scholar]
- 6.Kim YC; Ji XD; Jacobson KA J. Med. Chem 1996, 39, 4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Muijlwijk-Koezen JE; Timmerman H; Link R; van der Goot H; IJzerman AP J. Med. Chem 1998, 41, 3994. [DOI] [PubMed] [Google Scholar]
- 8.Baraldi PG; Cacciari B; Moro S; Spalluto G; Pastorin G; Da Ros T; Klotz K-N; Varani K; Gessi S; Borea PA J. Med. Chem 2002, 45, 770. [DOI] [PubMed] [Google Scholar]
- 9.Maconi A; Pastorin G; Da Ros T; Spalluto G; Gao ZG; Jacobson KA; Baraldi PG; Cacciari B; Varani K; Moro S; Borea PA J. Med. Chem 2002, 45, 3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okamura T; Kurogi Y; Hashimoto K; Sato S; Nishikawa H; Kiryu K; Nagao Y Bioorg. Med. Chem. Lett 2004, 14, 3775. [DOI] [PubMed] [Google Scholar]
- 11.Moro S; Spalluto G; Gao ZG; Jacobson KA Med. Res. Rev 2006, 26, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H; Avila MY; Peterson-Yantorno K; Coca-Prados M; Stone RA; Jacobson KA; Civan MM Curr. Eye Res 2005, 30, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao ZG; Kim SK; Biadatti T; Chen W; Lee K; Barak D; Kim SG; Johnson CR; Jacobson KA J. Med. Chem 2002, 45, 4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao Z-G; Joshi BV; Klutz A; Kim S-K; Lee HW; Kim HO; Jeong LS; Jacobson KA Bioorg. Med. Chem. Lett 2006, 16, 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeong LS; Lee HW; Kim HO; Jung JY; Gao Z-G; Duong HT; Rao S; Jacobson KA; Shin DH; Lee JA; Gunaga P; Lee SK; Jin DZ; Chun MW Bioorg. Med. Chem 2006, 14, 4718. [DOI] [PubMed] [Google Scholar]
- 16. General procedure for the synthesis of 6a–r: To a stirred solution of per mmol of 4′-hydroxymethyl analogues (11a–j) in dry DMF (10 mL) was added pyridinium dichromate (10.0 equiv), and the reaction mixture was stirred at rt for 20 h. After being poured into water (50 mL/mmol), the reaction mixture was stirred at rt for 1 h. The precipitate was filtered, and the filter cake was washed with excess water and dried under high vacuum to give brownish solid, which was used in the next step without further purification. To a solution of per mmol of acid derivatives (12a–j), EDC (1.5 equiv), HOBt (1.5 equiv), and appropriate amine (1.5 equiv) in CH2Cl2 (20 mL) was added DIPEA (3.0 equiv), and the mixture was stirred at rt for 12 h. The reaction mixture was evaporated, and the residue was purified by a silica gel column chromatography (hexane/EtOAc = 10:1–5:1) to give silyl amides as a white foam. To a stirred solution of per mmol of silyl amides in THF (5 mL) was added TBAF (2.5 equiv) and the reaction mixture was stirred at rt for 1 h. The solvent was evaporated and the resulting residue was purified by silica gel column chromatography (CH2Cl2/EtOAc/MeOH = 20:20:1) to give 6a–r. Compound 6c: yield 67%; white solid; mp 180.4–182.0 °C; (c 0.10, MeOH); UV (MeOH) λmax 274 nm (pH 7); 1H NMR (CD3OD) δ 2.94 (s, 3 H, N–CH3), 3.03 (s, 3 H, N–CH3), 4.41 (d, 1H, J = 4.6 Hz, 4′-H), 4.57 (br dd, 1H, J = 4.6, 8.4 Hz, 3′-H), 4.64 (m, 1H, 2′-H), 4.73 (d, 2H, J = 5.7 Hz, N–CH2), 5.96 (d, 1H, J = 5.4 Hz, 1′-H), 7.22–7.56 (m, 4H, aromatic H), 8.52 (s, 1H, H-8); 13C NMR (CD3OD) δ 36.6, 38.2, 44.6, 54.3, 64.6, 77.2, 80.7, 127.4, 128.8, 130.0, 131.1, 134.9, 141.4, 142.8, 143.1, 151.7, 155.8, 156.4, 172.8; FAB-MS m/z 528 (M++H); Anal. (C19H20BrClN6O3S) C, H, N, S.
- 17.(a) Gao Z-G; Blaustein JB; Gross AS; Melman N; Jacobson KA Biochem. Pharmacol 2003, 65, 1675; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao Z-G; Jeong LS; Moon HR; Kim HO; Choi WJ; Shin DH; Elhalem E; Comin MJ; Melman N; Mamedova L; Gross AS; Rodriguez JB; Jacobson KA Biochem. Pharmacol 2004, 67, 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Tilburg EW; von Frijtag Drabbe Künzel J; de Groote M; IJzerman AP J. Med. Chem 2002, 45, 420. [DOI] [PubMed] [Google Scholar]
- 19.Kim SK; Jacobson KA J. Chem. Inf. Model 2007, 47, 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]