

RÉSUMÉ
Introduction.
Les myopathies des ceintures sont un groupe de maladies héréditaires se caractérisant par un déficit des muscles des ceintures et segments des membres. Ce sont des maladies rares qui ont été peu étudiées en Afrique subsaharienne. C’est ainsi que nous avons entrepris cette étude avec comme but de décrire les signes cliniques et paracliniques des myopathies des ceintures récessives au Service de Neurologie du CHU Point G.
Patients et méthodes.
Il s’agit d’une étude longitudinale, prospective ayant duré de Mars 2014 à Mai 2019, portant sur des patients atteints de myopathie des ceintures récessives et ayant donné leur consentement écrit. Les variables analysées étaient socio-démographiques, cliniques et paracliniques.
Résultats.
Nous avons enrôlé 46 familles (67 patients), soit une fréquence de 16,7% parmi les maladies neurodégénératives vues dans le service au sein desquels 45,6% était originaire de la région de Sikasso. Une transmission autosomique récessive a été retrouvée dans 67,4% des familles. Les symptômes apparaissaient généralement durant les 10 premières années de vie. Un déficit moteur proximal a été retrouvé chez la quasi-totalité des patients. L’échographie cardiaque a révélé une cardiomyopathie dilatée dans 4,5% des cas.
Conclusion:
Les myopathies des ceintures sont des maladies invalidantes présentes au Mali. Une exploration génétique pourrait étayer les différents gènes en cause.
Abstract
Introduction.
Limb-Girdle Muscular dystrophies (LGMD) is a group of inherited diseases characterized by predominantly proximal and limb muscle weakness. These are rare diseases that have not been well studied in sub-saharan Africa. The aim of our was the clinical and paraclinical characterization of patients with recessive LGMD at the Department of Neurology of the Teaching Hospital of Point G.
Patients and methods.
We conducted a longitudinal prospective study which took place from March 2014 to May 2019. Patients with recessive LGMD phenotype were enrolled. Sociodemographic, clinical and laboratory data were analyzed.
Results.
We enrolled 46 families (67 patients), i.e. a frequency of 16.7% among the neurodegenerative diseases seen in the service. Among them, 45.6% came from the Sikasso region. Autosomal recessive inheritance pattern was suspected in 67.4% of the families. Symptoms appeared mainly in the first decade of life. Proximal muscle weakness was found in almost all patients. Cardiac examination showed dilated cardiomyopathy in 4.5% of cases.
Conclusion.
Limb-Girdle muscular dystrophy is a disabling disease that is found in Mali. Further study of these cases could elucidate the underlying genetic defects.
Keywords: LGMD, Teaching Hospital of Point G, clinic, Mali
INTRODUCTION
Les myopathies des ceintures représentent une entité neurologique hétérogène subdivisée en plusieurs sous types cliniques et génétiques. Elles se caractérisent par une dégénérescence progressive des fibres musculaires aboutissant à un handicap moteur surtout des muscles proximaux qui va se manifester par une marche dandinante, des chutes fréquentes et des difficultés à courir ou à monter les escaliers. La transmission se fait de façon courante selon le mode autosomique dominant ou autosomique récessif même si quelques cas de transmission liée au chromosome X ou mitochondriale ont été rapportés. Cette maladie touche 5 à 6 personnes sur 1 million dans le monde d’après l’EuropeanNeuromuscular Center. Elles représentent 10 à 50% des dystrophies musculaires en Afrique du nord [1], et précisément 61,7% de celles-ci en Tunisie [2]. En Afrique au sud du Sahara les études sont rares voire inexistantes. Au Mali, une étude pilote menée en 2011 sur les maladies neurologiques héréditaires a rapporté deux cas de myopathie des ceintures confirmés par le test génétique [3], d’où l’intérêt d’une étude sur un échantillon plus large avec comme objectif de décrire les signes cliniques et paracliniques des myopathies des ceintures.
PATIENTS ET MÉTHODES
L’étude a été approuvée par les comités d’éthique de la FMOS de Bamako, Mali avec le financement de la National Institute of NeurologicalDisorders and Stroke du NIH (USA) sous le numéro U01HG007044. Cette étude a été menée au service de Neurologie du CHU Point G de Bamako de Mars 2014 à Mai 2019, soit une période d’environ cinq ans. Elle a concerné des patients présentant des signes cliniques évocateurs de myopathies des ceintures avec ou sans notion de cas familial similaire, ayant consenti verbalement et par écrit. Les données ont été recueillies à l’aide d’une fiche d’enquête préétablie et analysées à partir du logiciel SPSS. Après un recueil des variables socio-démographiques (âge à la première consultation, sexe, ethnie, origine géographique, consanguinité parentale), les patients ont subi un examen neurologique minutieux. Selon le besoin, une consultation cardiologique, ophtalmologique ou ORL a été faite. Un prélèvement sanguin a été fait chez les patients pour le dosage des enzymes musculaires telles la créatine phosphokinase (CPK), l’Aldolase et la LDH. D’autres examens biologiques tels que la glycémie, la numération formule sanguine (NFS), les transaminases ont été réalisés selon le cas. L’électroneuromyographie (ENMG) et l’échographie cardiaque ont été réalisées chez presque tous les patients. Par ailleurs, la confidentialité des données a été strictement respectée.
RÉSULTATS
Caractéristiques sociodémographiques
L’étude a porté sur un total de 46 familles totalisant 67 patients, soit 16,7% des maladies neurologiques héréditaires vues dans le service pendant la même période. L’enquête familiale a retrouvé une consanguinité dans 34,3% des familles. La figure 1 correspond au pedigree d’une famille avec une suspicion de transmission autosomique récessive.
Figure 1:
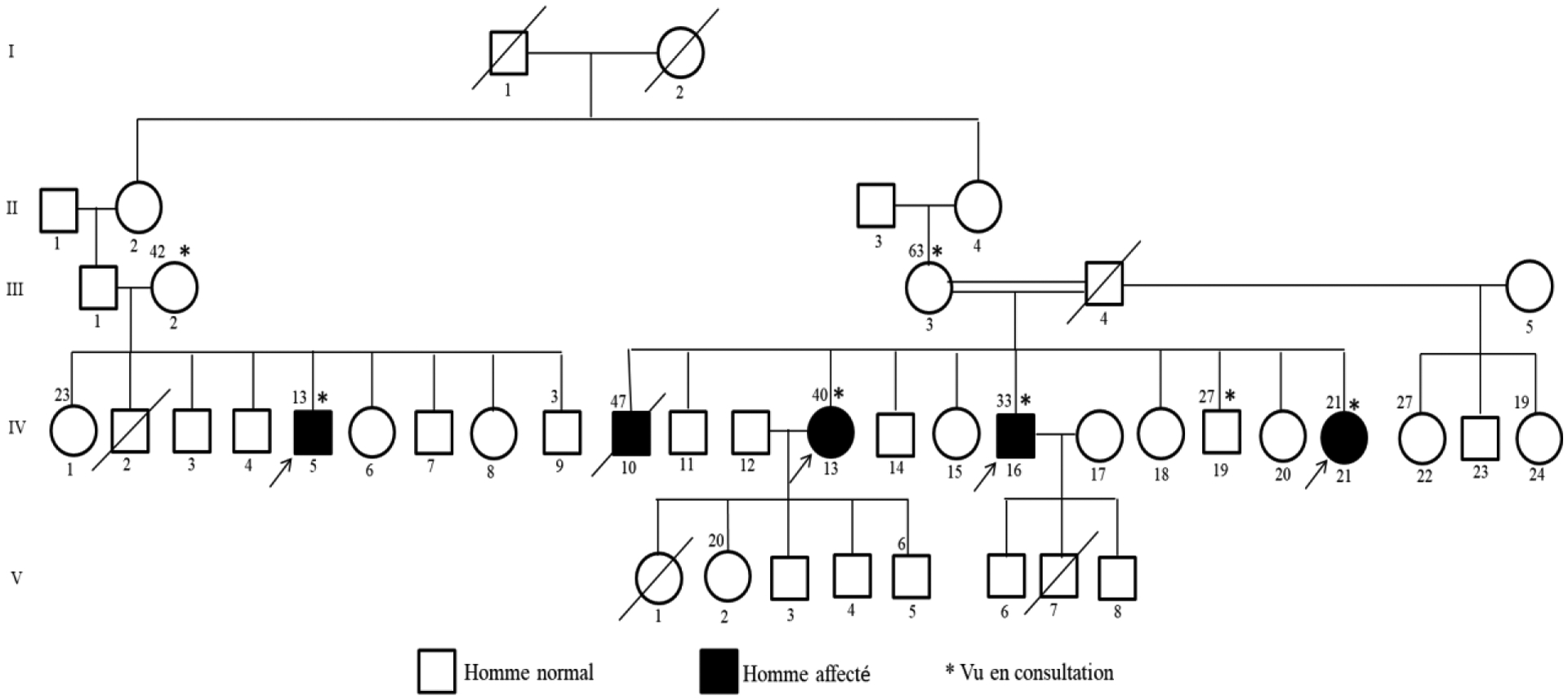
Pedigree d’une famille avec une suspicion de transmission autosomique récessive
L’âge moyen des patients lors de la première consultation était de 18,12 ans avec des extrêmes de 1 et 59 ans. La tranche d’âge de 11–20 ans était la plus représentée. Le sexe masculin était le plus représenté avec un sexe ratio de 1,58. L’ethnie Bambara était la plus représentée avec un total de 16 familles, suivie des malinkés et soninkés avec 8 familles chacun (Figure 2). Les familles étaient majoritairement originaires de la région de Sikasso, soit 45,6% (Figure 3).
Figure 2:
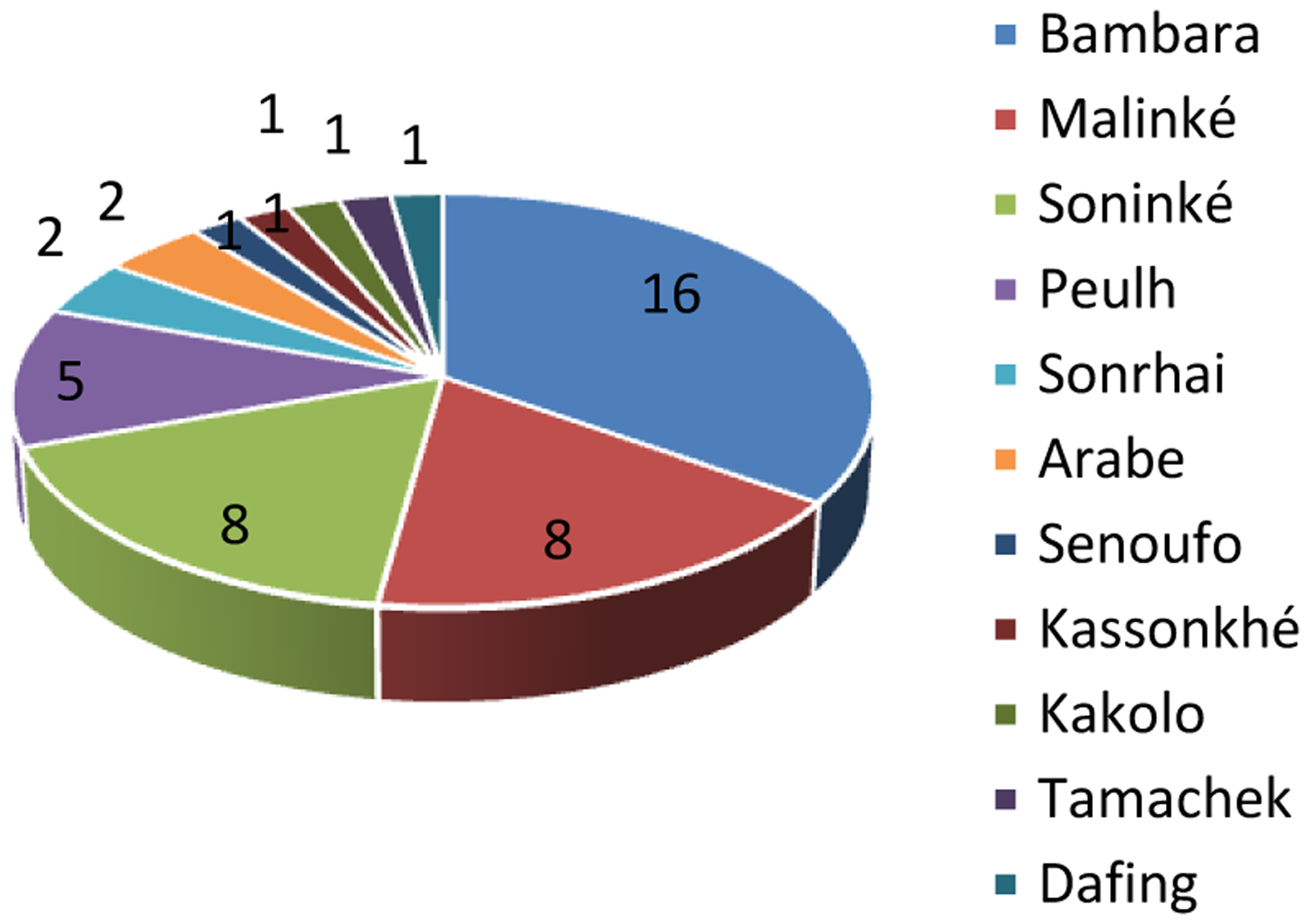
Répartition des familles en fonction de l’ethnie
Figure 3:
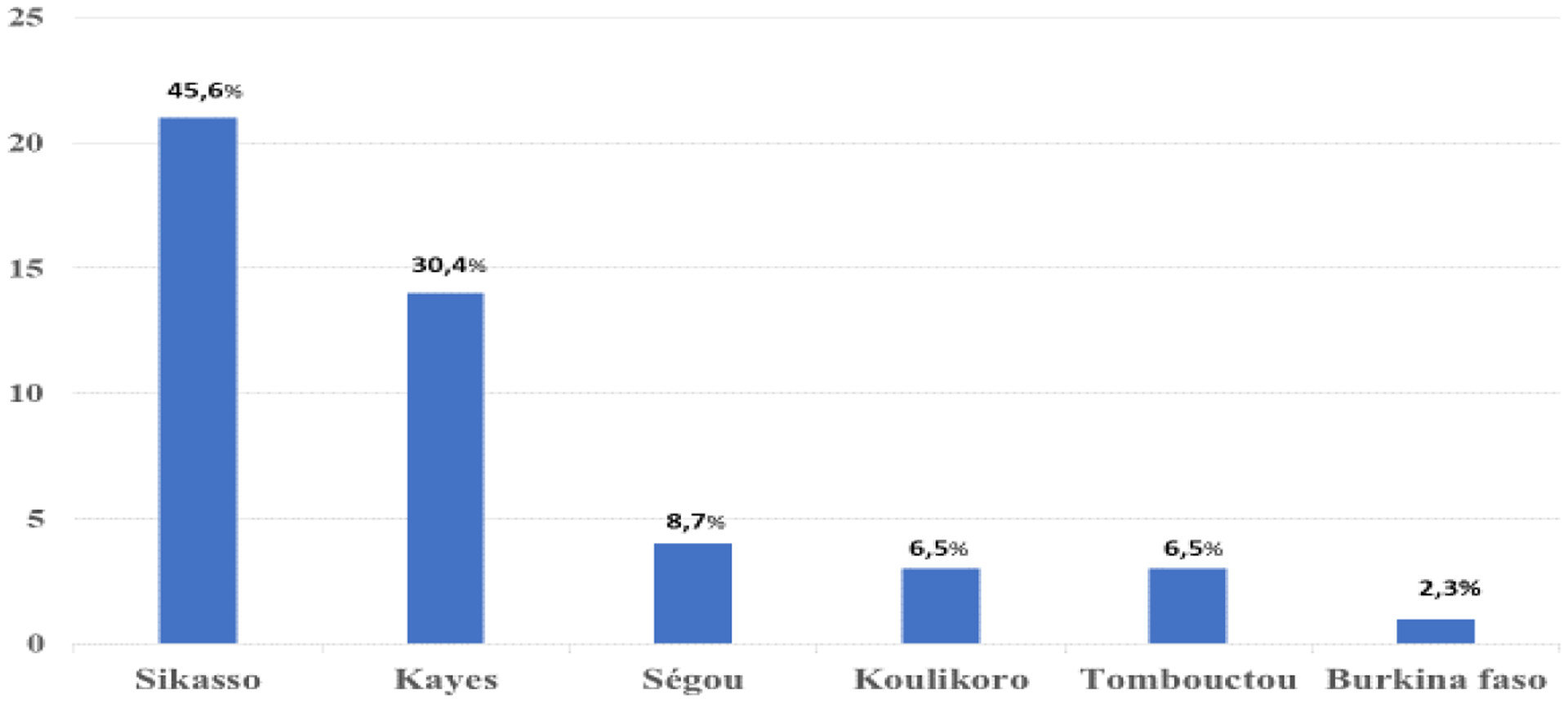
Répartition des familles en fonction de l’origine géographique
Caractéristiques cliniques
Un trouble de la marche était le motif de consultation dans98,5% des cas. L’âge moyen d’apparition des symptômes était de 11,10 ans avec des extrêmes de 1 et 54 ans. Le délai moyen de diagnostic était de 7,02 ans. Parmi nos patients, 97% avaient un déficit moteur proximal avec 36,3% qui étaient alités.
Un décollement scapulaire a été retrouvé dans 52,2%(Figure 4A), une hypertrophie des mollets (Figure 4B) dans 27% et des déformations articulaires et/ou rachidiennes dans 32%.
Figure 4:
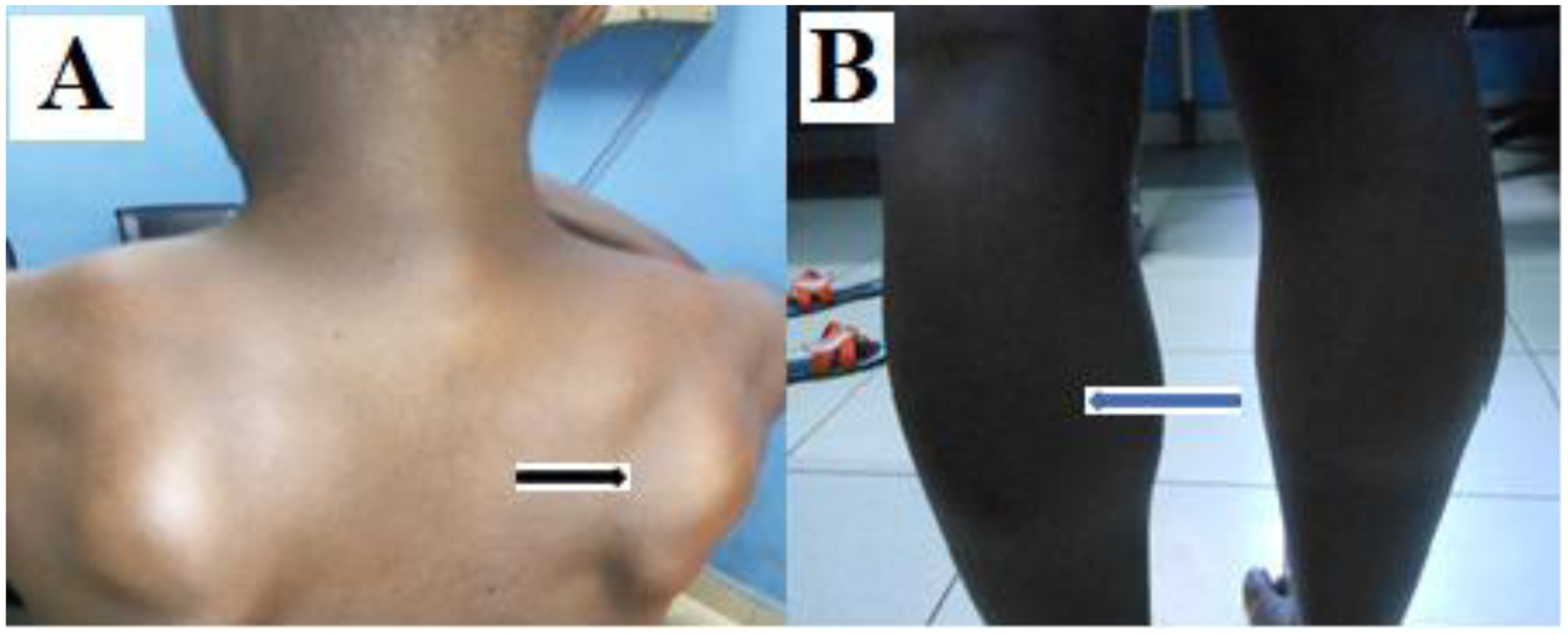
Caractéristiques phénotypiques de certains des patients montrant: A) décollement scapulaire (flèche noire) et B) hypertrophie des mollets (flèche bleue)
Les réflexes idiomusculaires étaient diminués ou abolis chez 89,5% des patients. Au total, 43 patients présentaient une marche dandinante, soit environ 64% des cas. À noter que 4,5% des patients manifestaient des signes cliniques d’atteinte cardiaque et la consultation cardiologique objectivait des bruits du cœur irréguliers et assourdis. La distribution des symptômes chez les patients évoquait une transmission autosomique récessive dans 67,4% des familles avec une notion de cas similaire dans la famille dans 55,2% des cas. (Tableau I)
Tableau I:
Résumé des signes cliniques et paracliniques retrouvés chez les patients
| Signes cliniques | Effectifs | Pourcentages |
|---|---|---|
| Déficit moteur proximal | 65 | 97 |
| Amyotrophie proximale et segmentaire | 35 | 52 |
| Hypertrophie des mollets | 18 | 27 |
| Réflexes idio-musculaires abolis | 48 | 72 |
| Déformations squelettiques | 21 | 32 |
| Troubles cardiaques | 3 | 4,5 |
| Marche dandinante | 43 | 64 |
| CPK élevés | 34 | 51 |
| EMG (tracé myogéne) | 26 | 40 |
Caractéristiques paracliniques
Les taux de CPK étaient élevés chez 51% des patients avec une moyenne de 992 UI et des extrêmes de 208 et de 6991 UI. Les autres enzymes musculaires réalisées étaient l’Aldolase et la LDH qui sont majoritairement revenues normales.
Environ 40% des patients ont réalisé un EMG (Figure 5) qui a montré dans tous les cas la nature myogène sur lequel un grand nombre d’unités étaient recrutées (anormalement riche en unités motrices par rapport à l’effort fourni).
Figure 5:
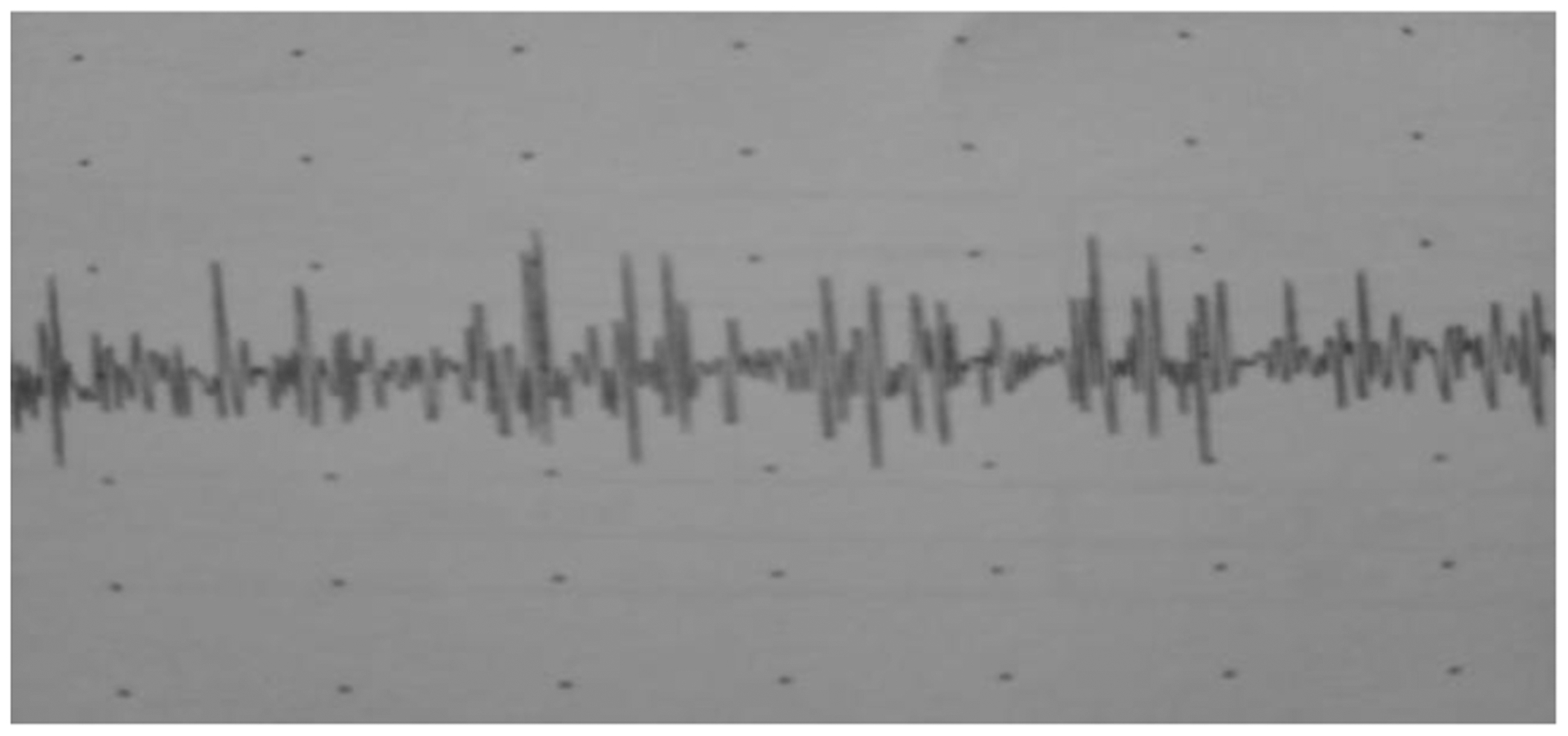
Tracé EMG montrant un tracé myogène
L’échographie cardiaque a été réalisée chez 64% de nos patients, seulement 4,5% sont revenus pathologiques montrant entre autres une fuite mitrale non significative sur valve épaissie ou une cardiomyopathie dilatée avec altération sévère de la fraction d’éjection. L’IRM et la biopsie musculaire n’ont pu être réalisées.
DISCUSSION
Dans notre étude l’ethnie Bambara était la plus représentée et la région de Sikasso était l’origine géographique la plus fréquente; ceci s’expliquerait par le fait qu’il s’agisse respectivement de l’ethnie la plus nombreuse et de la région la plus peuplée au Mali. Les troubles de la marche étaient le motif de consultation dans la quasi-totalité des cas soit 98,5%, ce qui se rapproche des 100% obtenu par Belgheti ZA au Maroc [4]. L’âge moyen d’apparition des symptômes était de 11,10 ans ce qui est similaire aux 10,6 ans obtenus par Monies D et al. en Arabie Saoudite [5]. Presque tous nos patientsprésentaient un déficit moteur proximal, ce qui est conforme aux résultats de Liang W et al. qui avaient trouvé 100% en Chine [6]. Les réflexes idio-musculaires étaient diminués ou abolis chez 89,5% des patients tandis que 79% d’entre eux présentaient des troubles trophiques à type d’amyotrophie ou d’hypertrophie. Ces résultats sont comparables avec les données actuelles de la littérature sur cette pathologie [7,8]. Le taux de CPK de 51% de nos patients était élevé. Ce qui diffère des 76,67% trouvés par Wang L et al. en Chine [6]. En effet, chez certains patients nous n’avons pas pu doser les CPK pour diverses raisons telles que les décès prématurés ou l’inaccessibilité du lieu de résidence. Aussi, certains de ces patients étaient à un stade très avancé de la maladie, ce qui pourrait biaiser les taux de CPK qui, à ce stade, pourraient revenir bas du fait de la destruction massive des muscles. Toutes les EMG réalisées montraient un tracé myogène: tracé trop riche en unités motrices, avec des amplitudes faibles. Les vitesses de conductions des potentiels moteurs et sensitifs étaient normales. L’échographie cardiaque de 4,5% des patients était pathologique, montrant chez certains des signes de cardiomyopathie dilatée. Ceci est conforme aux connaissances sur les manifestations cardiaques de certaines myopathies des ceintures. Néanmoins, ce faible pourcentage peut s’expliquer par le fait que non seulement l’atteinte cardiaque n’est pas décrite dans toutes les formes de myopathie des ceintures, mais aussi, dans celles concernées, ces manifestations n’apparaissent qu’à un stade très avancé de la maladie [9,10].
CONCLUSION
Notre étude confirme la présence des dystrophies musculaires des ceintures au Mali et apporte pour la première fois une description clinique et paraclinique détaillée dans cette région. Elle confirme une prédominance des formes récessives qui s’expliquerait par le taux élevé de consanguinité dans nos pays comme précédemment rapporté [11]. Une étude moléculaire pourrait retrouver des variantes génétiques non décrites précédemment et confirmer ainsi l’hétérogénéité génétique de cette population comme rapportée dans l’étude d’autres pathologies neurologiques [12–14].
Remerciements
Ce travail a été financé par les fonds U01HG007044 du National Institute of NeurologicalDisorders and Stroke des USA, administrés par National HumanGenomeResearch Institute faisant partie du NIH Common Fund H3Africa Initiative.
RÉFÉRENCES
- 1.Miladi N, Bourguignon JP, Hentati F. Cognitive and psychological profile of aTunisian population of limbgirdlemusculardystrophy. NeuromusculDisord, 1999; 9: 352–354. [DOI] [PubMed] [Google Scholar]
- 2.Nabli F, Farhat E, Kefi M, Amouri R, Hentati F. Les dystrophies musculaires en Tunisie: une expérience de 10 ans. La myopathie en Tunisie. Journées de Neurologie de Langue Française, Lyon, 2010. http://archives.jnlf.fr/data/ModuleProgramme/PageSite/2010-1/Resume/7131.asp [Google Scholar]
- 3.Meilleur KG, Coulibaly S, Traoré M, Landouré G, La Pean, Sangaré M et al. Genetic testing and counselling for hereditary neurological diseases in Mali. J Community Genet, 2011; 2: 33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belgheti ZA. Les dystrophies musculaires progressives de l’enfant (à propos de 63 cas). Mémoire, Octobre 2014. Faculté de Médecine et de Pharmacie de Fès, Maroc. http://scolarite.fmp-usmba.ac.ma/cdim/mediatheque/memoires/e_memoires/91-14.pdf [Google Scholar]
- 5.Monies D, Alhindi HN, Almuhaizea MA, Abouelhoda M, Alazami AM, Goljan E et al. A first-line diagnostic assay for limb-girdle muscular dystrophy and other myopathies. Hum Genomics, 2016; 10(1): 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L, Zhang VW, Li S, Li H, Sun Y, Li J et al. The clinical spectrum and genetic variability of limb-girdle muscular dystrophy in a cohort of Chinese patients. Orphanet J Rare Dis, 2018; 13(1): 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Govindarajan R, Shepard KM, Jones Lk Jr. Diagnosis and treatment of limb girdle and distal dystrophies. Neurol Clin Pract, 2016; 5(5): 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Myoinfo (AFM-Téléthon), Urtizberea JA. Avancées dans les myopathies des ceintures. Avancées de la recherche, Savoir & Comprendre. Evry: AFM-Téléthon, 2019; 25 p [Google Scholar]
- 9.Ghaoui R, Benavides T, Lek M, Waddell LB, Kaur S,North KN et al. TOR1AIP1 as a cause of cardic failure and recessive limb girdle muscular dystrophy. NeuromusculDisord, 2016; 26(8): 500–503. [DOI] [PubMed] [Google Scholar]
- 10.Pozsgai ER, Griffin DA, Heller KN, Mendell JR, Rodino-Klapac LR. Systemic AAV-mediated beta sarcoglycan delivery targeting cardiac and skeletal muscle ameliorates histological and functional deficit in LGMD 2E mice. MolTher, 2017; 25(4): 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sangaré M, Hendrickson B, Sango HA, Chen K, Nofziger J, Amara A et al. Genetics of low spinal muscular atrophy carrier frequency in sub-Saharan Africa. Ann Neurol, 2014; 75(4): 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landouré G, Mochel F, Meilleur KG, Ly M, Sangaré M, Bocoum N et al. Novel mutation in the ATM gene in a Malian family with ataxia telangiectasia. J Neurol, 2012; 260(1): 324–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guinto CO, Diarra S, Diallo S, Cissé L, Coulibaly T, Diallo SH et al. A novel mutation in KIF5A in a Malian family with spastic paraplegia and sensory loss. Ann Clin TranslNeurol, 2017; 4(4): 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yalcouyé A, Diallo SH, Coulibaly T, Cissé L, Diallo S, Samassékou O et al. A novel mutation in the GARS gene in a Malian family with Charcot-Marie-Tooth disease. Mol Genet Genomic Med, 2019; 7: e782. [DOI] [PMC free article] [PubMed] [Google Scholar]