

Abstract
Advanced therapy medicinal products (ATMPs) are innovative therapies that mainly target orphan diseases and high unmet medical needs. The uncertainty about the product's benefit-risk balance at the time of approval, the limitations of nonclinical development, and the complex quality aspects of those highly individualized advanced therapies are playing a key role in the clinical development, approval, and post-marketing setting for these therapies. This article reviews the current landscape of clinical development of advanced therapies, its challenges, and some of the efforts several stakeholders are conducting to move forward within this field. Progressive iteration of the science, methodologically sound clinical developments, establishing new standards for ATMPs development with the aim to ensure consistency in clinical development, and the reproducibility of knowledge is required, not only to increase the evidence generation for approval but to set principles to achieve translational success in this field.
Keywords: drug development, drug approval, research design, methods, clinical trials advanced therapies, cell- and tissue-based therapy, genetic therapy, jurisprudence
Graphical abstract
Most authorized ATMPs are based on small, open-label, uncontrolled, and single-arm pivotal trials using single and intermediate variables to evaluate outcomes. Health authorities are demanding methodologically sound clinical development to guide higher-quality evidence generation and decision-making. New standards for ATMP development are required to ensure consistency in clinical development.
Introduction
Advanced therapy medicinal products (ATMPs) are a medicinal class that includes gene therapy medicinal products, somatic cell therapy medicinal products, and tissue-engineered therapies1.2,3 The marketing approval of these therapies in the last years has been crucial to the growth of clinical research in this field.4 However, due to the current type of target diseases, i.e., orphan and unmet needs, and the inherent complexity of these products, less comprehensive clinical data have justified their approval. Here, we review and discuss the current landscape and challenges for clinical development and approval of advanced therapies, as well as the current efforts and potential future approaches to overcome these obstacles.
Level of clinical evidence at the time of marketing authorization
Until September 31, 2021, 19 advanced therapies were approved in the EU5.6 The key therapeutic areas mainly include hematological malignancies, monogenic diseases, and cartilage diseases (Table 1). The clinical development of these approved ATMPs for the authorized clinical indications was based on 25 pivotal trials. Most of these trials consisted of small, open-label, non-randomized, single-arm studies, comparing the efficacy with historical controls, and using intermediate variables to evaluate the primary efficacy outcome (Table 2)5. Other studies that analyzed ATMP clinical trials in an early development phase reported similar results.7 The type of current target diseases including orphan indications,8 unmet needs,9 and the presence of pediatric patient populations has justified more flexible clinical designs and methodologies using adaptive pathways and balancing the need for timely patient access through staggered approval (Table 2).10
Table 1.
Approved ATMPs in the EU and therapeutic indication
| Trade name | International non-proprietary name or common name | Pharmacotherapeutic group/ATC code | Therapeutic area (MeSH)/type of authorization |
|---|---|---|---|
| Gene therapy medicinal products | |||
| Kymriah (orphan medicine) | tisagenlecleucel | antineoplastic agents/L01XX71 | precursor B cell lymphoblastic leukemia-lymphoma lymphoma, large B cell, diffuse/standard approval |
| Yescarta (orphan medicine) | axicabtagene ciloleucel | antineoplastic agents/L01XX70 | lymphoma, large B cell, diffuse/standard approval |
| Tecartus (orphan medicine) | autologous peripheral blood T cells CD4 and CD8 selected and CD3 and CD28 activated transduced with retroviral vector expressing anti-CD19 CD28/CD3-zeta chimeric antigen receptor and cultured | antineoplastic agents/L01X | lymphoma, mantle cell/conditional approval |
| Imlygic | talimogene laherparepvec | antineoplastic agents/L01XX51 | melanoma/standard approval |
| Glybera (orphan medicine) | alipogene tiparvovec | lipid-modifying agents/C10AX10 | hyperlipoproteinemia type I/approval under exceptional circumstances |
| Strimvelis (orphan medicine) | autologous CD34+ enriched cell fraction that contains CD34+ cells transduced with retroviral vector that encodes for the human ADA cDNA sequence | immunostimulants/L03 | severe combined immunodeficiency/standard approval |
| Luxturna (orphan medicine) | voretigene neparvovec | ophthalmologicals, other ophthalmologicals/S01XA27 | Leber congenital amaurosis retinitis pigmentosa/standard approval |
| Zynteglo (orphan medicine) | betibeglogene autotemcel | other hematological agents/B06AX02 | β-thalassemia/conditional approval |
| Zolgensma (orphan medicine) | onasemnogene abeparvovec | other drugs for disorders of the musculoskeletal system/M09AX09 | muscular atrophy spinal/conditional approval |
| Libmeldy (orphan medicine) | atidarsagene autotemcel | other nervous system drugs/N07 | leukodystrophy, metachromatic/standard approval |
| Abecma (orphan medicine) | idecabtagene vicleucel | antineoplastic agents/L01 | multiple myeloma/conditional approval |
| Skysona (orphan medicine) | elivaldogene autotemcel | other nervous system drugs/N07 | adrenoleukodystrophy/standard approval |
| Somatic cell therapy medicinal products | |||
| Provenge | autologous peripheral blood mononuclear cells activated with prostatic acid phosphatase granulocyte-macrophage colony-stimulating factor (Sipuleucel-T) | other immunostimulants/L03AX17 | prostatic neoplasms/standard approval—withdrawn |
| Zalmoxis (orphan medicine) | allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low-affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2) | antineoplastic agents/L01 | hematopoietic stem cell transplantation graft versus host disease/conditional approval—withdrawn |
| Alofisel (orphan medicine) | darvadstrocel | immunosuppressants/L04 | rectal fistula/standard approval |
| Tissue-engineered medicinal products | |||
| Chondrocelect | characterized viable autologous cartilage cells expanded ex vivo expressing specific marker proteins | other drugs for disorders of the musculoskeletal system/M09AX02 | cartilage diseases/standard approval—withdrawn |
| MACI | matrix-applied characterized autologous cultured chondrocytes | other drugs for disorders of the musculoskeletal system/M09AX02 | fractures, cartilage/standard approval—withdrawn |
| Spherox | spheroids of human autologous matrix-associated chondrocytes | other drugs for disorders of the musculoskeletal system/M09AX02 | cartilage diseases/standard approval |
| Holoclar (orphan medicine) | ex vivo expanded autologous human corneal epithelial cells containing stem cells | ophthalmologicals/S01XA19 | stem cell corneal diseases/conditional approval |
Table 2.
Design features of pivotal clinical trials for the approved advanced therapy medicinal products in the EU
| Trade name | Pivotal study | Non-randomized | Non-controlled | Historical control | Intermediate endpoints | Population/no. of patients (enrolled) |
|---|---|---|---|---|---|---|
| Gene therapy medicinal products | ||||||
| Kymriah (ALL) | Phase II | ✓ | ✓ | ✓ | ✓ | Children/92 |
| Kymriah (DLBCL) | Phase II | ✓ | ✓ | ✓ | ✓ | Adults/147 |
| Yescarta | Phase I/II | ✓ | ✓ | ✓ | ✓ | Adults/111 |
| Tecartus | Phase II | ✓ | ✓ | Adults/105 | ||
| Imlygic | Phase III | ✓ | Adults/437 | |||
| Glybera | 3 Phase II/III | ✓ | ✓ | ✓ | Adults/45 | |
| Strimvelis | Phase I/II | ✓ | ✓ | ✓ | Children/12 | |
| Luxturna | Phase III | ✓ | Children and adults/31 | |||
| Zynteglo | Phase I/II and Phase III | ✓ | ✓ | ✓ | Children and adults/41 | |
| Zolgensma | Phase III | ✓ | ✓ | ✓ | Children/22 | |
| Libmeldy | Phase I/II | ✓ | ✓ | ✓ | Children/22 | |
| Skysona | Phase II/III | ✓ | ✓ | Children/32a | ||
| Abecma | Phase II | ✓ | ✓ | ✓ | ✓ | Adults/140 |
| Somatic cell therapy medicinal products | ||||||
| Provenge | Phase III | Adults/512 | ||||
| Zalmoxis | Phase I/II and Phase III | ✓ (Phase I/II) | ✓ (Phase I/II) | ✓ | Adults/71 | |
| Alofisel | Phase III | Adults/212 | ||||
| Tissue-engineered medicinal products | ||||||
| Chondrocelect | Phase III | ✓ | Adults/138 | |||
| MACI | Phase III | ✓ | Adults/144 | |||
| Spherox | Phase II and Phase III | ✓ (Phase II) | ✓ | Adults/177 | ||
| Holoclar | Observational retrospective | ✓ | ✓ | Adults/104a | ||
ALL, refractory B cell acute lymphoblastic leukemia; DLBCL, diffuse large B cell lymphoma.
Number of patients in the intervention arm.
Although controlled randomized clinical trials are the standard for evidence generation in terms of efficacy and safety for regulatory decision-making, the treatment comparison with the standard of care (SOC) or placebo might have not been considered feasible and/or ethical in these cases. This is translated into less comprehensive clinical data at the time of marketing authorization (MA), and therefore, greater uncertainty about the product's benefit-risk balance.11 For instance, Zalmoxis authorization was mainly based on promising results of an open-label, non-randomized phase I–II study, supported by the preliminary efficacy and safety data from the first 17 patients of an ongoing phase III controlled study. The final results from this controlled study failed to confirm any benefit at post-marketing level and the drug had to be withdrawn.12 Another recent case is Kymriah, approved based on a phase II open-label and single-arm study and where the randomized post-marketing phase III trial that analyzed the drug against SOC failed to meet the primary efficacy endpoint, i.e., event-free survival.13,14 Nevertheless, the patient profile for this last study may differ from that included in the pivotal trial that led to its MA. It should be mentioned that although most of the products were approved based on single-arm designs, some of their competitors conducted controlled studies to support the MA for the same indication, e.g., Spinraza,15 or planned controlled post-marketing trials, e.g., Kymriah or Yescarta.11 By contrary for cell therapies, it should be noted that even though Zalmoxis and Alofisel were granted with an orphan designation, phase III studies were conducted including a comparator arm.16,17
With these types of flexible and expedited developments with the ATMPs, the current landscape of biological therapies has initiated a shift from traditional clinical developments to a highly product-specific one. Elsallab et al. conducted a matched comparison of the regulatory submissions between ATMPs (n = 17) and other biologicals (n = 17). The results showed that clinical studies for ATMPs did not meet the same strict standards for clinical evidence that were applied to other biological products. The evidence on the design, conduct, and outcome of ATMP clinical studies suffered from more objections when compared with other biologicals. Despite matching for the disease area and orphan status, ATMPs had more non-randomized, non-blinded trials and included significantly lower numbers of patients, raising doubts about the trial outcomes.18 How this non-robust data can affect the approval of advanced therapies has also been reviewed. Bravery and co-workers tried to answer the question whether ATMPs are more or less likely to be approved than other medicines. The results showed that for all medicine applications combined, there is a 76% success rate (n = 632) compared with 59% for ATMPs (n = 22), but for non-orphan ATMPs the chance of success seems to be lower, at only 50% (n = 10).19 Other studies also analyzed the evidence submitted to support the ATMPs MA by quantifying the objections raised by regulatory authorities during the assessment. The two more common issues included suitable quality and clinical data demonstrating the efficacy and safety.20,21 Barkholt et al. identified the “hotspots” in ATMP development analyzing the MA applications (MAAs) (n = 20) and all scientific advice given for ATMPs by the European Medicines Agency (EMA) (from 2009 to 2018). The clinical data package, the clinical results, the target indication, limited safety information and limited safety and efficacy follow-up, and risk management were the most common development issues and objections raised during the MAA procedure.21 Similar results were obtained by Bravery et al., where 74% of applications (n = 19 ATMP submissions) raised major objections to the clinical data package. This category covers issues such as lack of randomization, issues with the design, conduct of the clinical study, and/or choice of control group. It was found that failed products have more issues in this category (83% of applications; n = 6). The authors found that evidence submitted with the ATMP dossiers are in need of improvement.19 This point is also highlighted by the fact that those applications that have been granted with an accelerate assessment revert to standard timelines during the MA procedure due to the immaturity of the data and the major issues raised (n = 6 out of 7 EU approved ATMP granted with accelerate assessment; as of September 31st, 2021).2 Carvalho et al. analyzed and compared the major objections reported in the MAA assessment for approved ATMPs (n = 3) and non-approved ATMPs (n = 4).22 The most frequent objections for gene therapy medicinal products in terms of clinical efficacy were the lack or insufficient efficacy demonstration, the change or use of novel and non-validated primary endpoints, and efficacy claims based on non-prespecified post-hoc analysis. Regarding safety, the most common objections were the limited safety database and the risks associated with immunogenicity. Most deficiencies were addressed through the submission of additional data either during the MAA review or post-marketing setting.22
Efforts to overcome the clinical challenges faced by advanced therapies
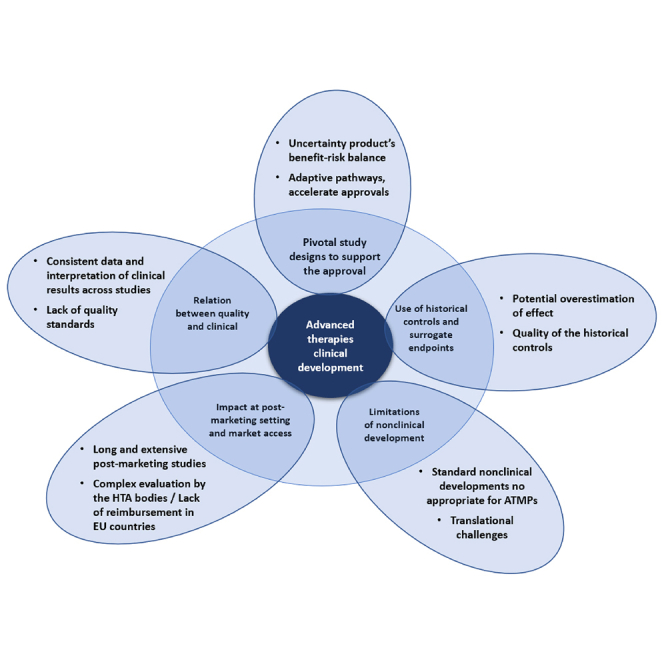
All these reported data support the fact that there is room for improvement in terms of clinical evidence generated to support the drug approval (Figure 1). A more efficient, consistent, and robust clinical development not only may give more chances to achieve MA and led to less objections by the agencies allowing for a quicker product launch, but it also may prevent post-marketing withdrawal anticipating the negative benefit/risk balance. It is recognized that clinical development for diseases that have a high unmet need and/or are orphan can be complex and can leverage the opportunities that regulatory bodies offer to speed up access and get an accelerated approval. However, given all the implications that expedited clinical developments might have—not only to the patients and payers but to the pharmaceutical companies—whenever feasible, the gold standard pivotal randomized clinical trials, clinically relevant endpoints, and longer follow-up should be performed.
Figure 1.

Current landscape of ATMPs clinical development
When randomized control designs are not feasible, alternative design options should be considered aimed to provide robust evidence. Many efforts have been carried out to launch methodological recommendations to address the shortcomings of conducting studies in small populations. The Small Population Clinical Trials Task Force within The International Rare Diseases Research Consortium investigated the use of non-conventional statistical methods on small population trials with the input of regulatory agencies.23 Three relevant European Commission-funded projects (i.e., ASTERIX, IDeAl, and InSPiRe) are promoting the development of new or improved statistical methodology for clinical trials for small population groups, as well as defining adequate randomization procedures, investigating adaptive designs, extrapolating dose-response information, among others.24, 25, 26
On the other hand, several innovative trial designs under the concept of master protocol are starting to change the landscape of clinical research.27, 28, 29 This approach uses a single infrastructure, trial design, and protocol to simultaneously evaluate multiple drugs and/or disease populations in multiple sub-studies, allowing for efficient and accelerated drug development. A master protocol provides an opportunity to increase data quality through shared standardized trial procedures and the use of centralized data capture systems.30 Within this concept there are different innovative typologies, i.e., basket, umbrella, and platform designs, which have been raised as a potential solution to improve clinical evidence robustness. Platform trials allow multiple interventions to be evaluated simultaneously against a common control group within a single master protocol. The treatments are tested for similar indications and with test products entering and leaving the study based on results. The control arm usually consisting of the SOC may change over time if newer drugs replace the SOC.31 Comparisons between each of the intervention arms and the control arm can be done to determine which is the best intervention option for a given disease. Yescarta and Kymriah are chimeric antigen receptor (CAR) T cell therapies approved for patients with refractory diffuse large B cell lymphoma (DLBCL) on the basis of ZUMA-1 and JULIET trials, respectively.32,33 In the absence of head-to-head trials, an indirect treatment comparison between both products was carried out. It was concluded that this comparative analysis is not feasible due to the substantial differences between the trials, e.g., timing of leukapheresis and enrollment, use of bridging chemotherapy (90% in JULIET versus 0% in ZUMA-1), different lymphodepleting regimens, different outcome definitions, etc.34 In addition, as previously mentioned, the comparison of Kymriah against SOC failed to meet the primary efficacy endpoint.35 To explore the option of a platform trial for these therapies would have allowed the comparisons between each of the intervention arms and the SOC, as well as efficiently sharing the same control group given that is an orphan disease. The same point can be raised in the case of spinal muscular atrophy (SMA), a rare disease. The SOC for SMA has improved over the last decade due to changes in care, as well as the fact that new promising drugs are becoming available, such as Zolgensma, Spinraza, or Evrysdi. The IQWiG, Germany's health technology appraisal institute, has carried out separate benefit assessments comparing these three new drugs, finding that Zolgensma offers no additional benefit compared with Spinraza for treating SMA. IQWiG pointed out that the differences between populations across different studies made indirect comparisons challenging and makes it difficult to understand which of the three products might be suitable in different situations.36 This type of innovative trials would allow a stratification into multiple subgroups depending on the SMA type and SMN2 gene copy number, with eligibility for each intervention arm defined by the intervention's mechanism of action. In addition, another advantage of conducting platform trials is the investigation of treatment combinations. For instance, during clinical development of Zolgensma, Spinraza treatment was started on parental request to determine if there was additional benefit from this combination therapy.37 Finally, it should be noted that master protocols for CAR T cell therapies have already been initiated in the field of ATMP, e.g., phase I proof-of-concept study in relapsed and refractory multiple myeloma and a phase II study in patients with metastatic or unresectable synovial sarcoma or myxoid/round cell liposarcoma.38,39 Although platform trials are usually focused on oncology, they also have been conducted in other disease settings such as Alzheimer's disease.40
Even though still limited, with the current experience of the approved ATMPs, the regulatory agencies are launching recommendations on the types of study designs and methodologies that can support the MA more robustly. This fact might lead to a shift on the current trend clinical designs based on uncontrolled pivotal studies or with historical control comparisons with randomized controlled trials. After the approval of the CAR T cell products, the EMA has published guidance on the clinical development for CAR T cell products.41 The recommendations include the performance of confirmatory trials with a randomized controlled design allowing the comparison with a reference product, e.g., high-dose chemotherapy followed by autologous stem cell transplantation. In the guideline, it is recognized that refractory settings are clinically very different from early settings, which in some cases may justify different requirements in terms of level of evidence for MAA. However, it is emphasized that even for those cases where late-stage refractory disease is targeted or where reference therapies are not available, a randomized controlled trial design should be followed, and an uncontrolled single-arm one would be exceptionally accepted.42 In parallel, the Food and Drug Administration (FDA) has also launched several guidelines for the development of ATMPs aimed at certain types of conditions. For instance, to support the standard approval of a gene therapy for hemophilia, the FDA recommends a non-inferiority clinical trial design, to compare the primary efficacy endpoint with that of current prophylaxis therapies, using within-subject comparison trial.43 In the case of gene therapies aimed at retinal disorders, inclusion of a randomized, concurrent parallel control group (placebo or active) is recommended whenever possible. Given that for these study designs, the intravitreal injection of the vehicle alone could be feasible but not ethical, other possibilities suggested including alternative dosing regimens or dose levels.44 The new guidance on gene therapy for neurodegenerative diseases comprises different study design alternatives depending on the indication, study population, or where the disease course is well characterized. For studies involving placebo, FDA recommends add-on designs or randomized, concurrent-controlled, double-blind crossover trials when possible.45 On the other hand, it is recognized that the typical paradigm of clinical development, i.e., phase I, II, and III, is shifting for advanced therapies and adaptive designs are becoming common. Regulatory agencies are also in consequence releasing new recommendations on innovative designs as well as advice programs to ensure that these adaptive approaches are as solid as possible.46, 47, 48 Finally, analytical tools, such as matching-adjusted indirect comparisons and network meta-analyses, have also been introduced for regulatory submissions and health technology assessments (HTAs) allowing comparisons.49
Use of historical controls
When a randomized clinical trial is not possible, the historical controls can be used to supplement a control arm. Different sources of external control can be used: retrospective data, prospective natural history, external data from completed trials, data from pragmatic trials, observational studies, or registries.50 The use of historical controls to compare the treatment effect have been highly used so far for the EU approved ATMPs (8 out of 19 approved therapies, as of September 31, 2021), above all in the case of gene therapies (7 out of 12 approved products) (Table 1).
Strimvelis, Kymriah, Luxturna, Zolgensma, Libmeldy, and Skysona target orphan diseases for pediatric population and all of them contextualized the results of the pivotal study with different types of historical controls. For Strimvelis and Libmeldy, the hypothesis of the study was based on demonstrating superiority over a historical control group, which was considered acceptable given the rarity of the disease. For Strimvelis, while the primary endpoint based on survival was compared with this historical reference, other efficacy endpoints were considered as within-subject, between pre- and post-treatment assessments. The historical control used was based on the outcomes obtained in a multicenter retrospective study (between August 1981 and March 2009) including 106 patients with adenosine deaminase-deficient severe combined immunodeficiency from 16 international centers.51 The main study for Libmeldy was conducted in a single center, as well as the concurrent natural history cohort. Both natural history cohort (n = 31) or untreated sibling data (n = 11), were used as controls to compare the treatment effect for the co-primary endpoint. It was considered by the assessors that a comparison with a matched sibling appears to have the least variability and the comparison between pre-symptomatic subjects versus their affected siblings is considered the most informative.52 In the case of Kymriah for relapsed or refractory B cell acute lymphoblastic leukemia (ALL), the single-arm design was planned to test for an improvement in overall remission rate relative to historical control rates from two previous studies performed for the same indication with other products (clofarabine and blinatumomab approved in 2007 and 2015 by the EMA, respectively).53 Luxturna’s trial randomized patients to a control or to intervention arm, given that for most inherited retinal dystrophies, natural history data were limited. The control group became eligible to receive the product 1 year after their baseline evaluations. Nevertheless, a natural history study that consisted of a retrospective medical chart review (from July 2014 to February 2016) was also submitted as a supportive by the applicant to further support the MAA (n = 70).54 For Zolgensma, two natural history studies were used for comparison; one retrospective and prospective study using data from the Paediatric Neuromuscular Clinical Research database (inclusion of the patients ranged from May 2005 to April 2009), and another prospective, multi-center natural history study (from November 2012 to September 2014) that enrolled 26 SMA infants.37,55 Similarly, in the case of Skysona, both data from a retrospective natural history study (data collected between 2011 and 2012; n = 137) and a retrospective and prospective data collection study (from 2015 to December 2019; n = 59) were used.56
Yescarta, Zalmoxis, Kymriah, and Abecma target adult orphan indications and also used historical controls to compare product's efficacy. For Yescarta, a retrospective, patient-level, pooled analysis from two randomized phase III clinical and retrospective databases was conducted to support the results from the pivotal study (SCHOLAR-1),57 and for Kymriah, the pivotal efficacy results were compared with three historical datasets (SCHOLAR-1, the pooled CORAL extensions, and the open-label, randomized PIX301 trial). For Zalmoxis, at the time of approval, there was neither approved therapy nor widely accepted SOC. Therefore, the treatment effect could only be compared versus historical control data from either a large retrospective survey (between January 1995 and December 2004) or single-center experiences.16,58 For additional comparisons with historical control data from patients, the European Blood and Marrow Transplant society patient database was used to better define the product's clinical benefit.16 For Abecma, the results were compared with a matched real-world historical control that consisted of a non-interventional, retrospective study (n = 190) as well as reported literature.59
The relevance of historical data is sometimes questioned and could lead to an overestimation of effect. The limitations of historical controls are well known; comparability of the population, potential changes in SOC, lack of standardized diagnostic criteria or equivalent outcome measures, and variability in follow-up procedures.60, 61, 62 The standardization and quality of the data collection, the selection of an applicable approach to account for biases, to plan for an extensive sensitivity analyses to demonstrate the robustness of the results, or the use quantifiable and objective outcomes are some of the measures that would improve the quality of the historical controls.63 The case of Abecma is an example of the historical control limitations. The real-world evidence study was found to be inconclusive by the FDA to provide context or comparison for the outcome of the pivotal study. The missing data, differences in follow-up and response assessment, population heterogeneity, and bias in endpoint assessment, hampered the comparison.64,65 When similarity can be proven between arms, the use of a historical control replacing the concurrent control arm can be the alternative source of data in a context of life-threatening disease with no treatment available. In other scenarios, a clear justification for a non-randomized trial is needed and an early dialogue with regulatory agencies at the design stage is highly encouraged to avoid potential problems during the clinical development plan and final authorization. According to recent FDA recommendations, the use of historical controls is discouraged but it might be considered appropriate only under very exceptional circumstances where: the product targets a rare and serious neurodegenerative disease for which there is an unmet medical need, the disease course is well documented, highly predictable and can be objectively measured and verified, the study population and the historical controls are suitably comparable, and the expected treatment effect is large and self-evident.45
It is known that registries provide an important source of information on diseases, patients, SOC, or outcomes of treatments, in particular for rare diseases or patients treated with ATMPs. In this sense, there have been some proposals to overcome the current challenges in using registries data such as interoperability and patient privacy improvement, standardization of data and terminology, better reporting of clinical trial outcomes, and other measures to maximize registry use in drug and therapeutic development to support evidence-based clinical decision-making.66 EMA's initiative for patient registries, launched in September 2015, is focused on supporting a systematic and standardized approach to their use for regulatory purposes.67 The need for individual patient data is a key factor to conduct better historical comparison. For instance, for Kymriah in refractory ALL indication, external control was used for comparison with data pooled from the three main Kymriah trials, despite confounding patient populations and matching on few variables.33,68 For Kymriah and Yescarta in DLBCL indication, the treatment effect was compared with SCHOLAR-1 sponsored by Kite Pharma (MA holder of Yescarta).57 The acceptance of comparison between Yescarta pivotal results and the SCHOLAR-1 study was attributed to the availability of individual patient data, enabling the company to match patients in both trials.68,69 However, for Kymriah given that only published data of SCHOLAR-1 was available for comparisons, the data from the pooled CORAL extensions study was accepted by the agency as a more suitable comparator than SCHOLAR-1 due to similarities in the populations enrolled and the objective response rate results obtained.33,57,68
Use of surrogate endpoints
Another important factor observed in the pivotal studies of the approved ATMPs is the use of surrogate variables instead of a clinically relevant final endpoint. Results from surrogate endpoints are common in accelerated approvals and allow for clinical trials with shorter follow-up periods and smaller sample sizes.70,71 It has been reported that the pivotal trial evidence supporting MA for products granted conditional MA or accelerated assessment was based dominantly on non-validated surrogate endpoints.72 This point can be translated into lower likelihood identifying safety issues (especially if they are rare) and long-term observations of safety adverse events. It has been reviewed that surrogate endpoints might lead to erroneous, or even harmful conclusions, since they might fail to fully capture the complete risk-benefit profile.73 On the other hand, this type of endpoints is ethically preferable, especially when clinical events are rare/delayed in slowly progressive diseases or when there is a high unmet need, as well as practically preferable since the short-term assessment helps to avoid non-compliance and missing data, increasing effectiveness and reliability of the study.73, 74, 75 The acceptability of the surrogate endpoints needs to be based on their biological plausibility and empirical evidence, and should be validated with evidence that goes beyond showing a statistical association between the surrogate and clinical endpoints.73,74,76
In the case of all approved gene therapies that target cancer diseases, the proportion of patients with objective overall response rate (ORR) was used as the intermediate primary variable. In these cases, ORR was an acceptable endpoint given that the studies that support the MA consist of phase II exploratory trials and given that an accelerated approval was granted. According to the most recent version of the EMA guideline on anticancer drugs, for confirmatory trials the overall survival, progression-/event-/disease-free survival would be considered as adequate primary endpoints. However, selected patient-reported outcomes, such as symptom control, could also constitute clinically relevant and valid primary endpoints, provided high data quality are ensured.77 In addition, and if available, the use of validated biomarkers should be considered to allow a clinical trial to identify and differentiate between drug responders and non-responders.
Although surrogate variables are not always ideal, it is not trivial to select either a final and/or surrogate primary efficacy endpoint for an ATMP, which can accurately predict or correlate with clinical benefit in the studied indication. For instance, in the case of Luxturna, the applicant had to develop a novel clinical meaningful endpoint to assess the drug effect through a mobility test.54 For Zolgensma, although the survival endpoint was used as a final co-primary outcome, survival with no motor milestone achievement would have not probably been considered as clinically meaningful outcome in the treatment of SMA. Moreover, performance and socialization at school age around 5–6 years was suggested by the experts as long-term data to be captured relating to efficacy.37,78 For those patients with lipoprotein lipase (LPL) deficiency, the most severe associated complication is pancreatitis. The hypothesis that Glybera could improve chylomicron particle metabolism and then reduce the pancreatitis in these patients could not be substantiated by clinical data at the time of MA.79
With increasing pressure for an early access to therapies, the use of surrogates is likely to increase. The key guidelines of the European network for HTA, which have been adopted by many European HTA agencies, state a preference for using final patient-relevant outcomes, but the need for surrogate endpoints is also recognized.48,80 Therefore, it is recommended to use surrogate variables only to those that have been validated appropriately, to avoid uncertainty on coverage decisions on health technologies, as well as to ensure less objections during the MAA assessment.81 It has been reported that only few HTAs have provided specific methodological guidance on the statistical methods that should be used for the validation and assessment of acceptability of surrogate endpoints, and there is still lack of methodological consensus around the level of evidence necessary for the validation of these endpoints. In consequence, efforts on better harmonization are currently being conducted to minimize different access for patients across different jurisdictions.80,82 On the other hand, to guide the developers, the FDA has recently published a list of accepted surrogate endpoints that were the basis of approval of a medicinal or a biological products under both the accelerated and standard pathways.83 Finally, the validation of a surrogate endpoint is not a straightforward process, given that the association between surrogate and final outcome usually is demonstrated by randomized controlled trials, or epidemiological/observational studies. Therefore, as discussed by Ciani et al., extension of follow-up studies, as well as the natural history studies combined with analyses on baseline data, emerging large data networks, or previous conducted trials on the disease might help to identify adequate surrogate variables.81
Limitations of nonclinical development
Properly designed nonclinical studies can reduce the clinical uncertainty and support a positive risk-benefit ratio. However, the traditional and standardized approaches for nonclinical toxicity testing are often not appropriate for evaluating the safety of gene and cell therapy products, and several challenges are also associated with the nonclinical development of ATMPs.84, 85, 86 General nonclinical studies and toxicity studies may be unable to detect the effects relevant for human efficacy and safety. Some examples include Glybera, the proof-of-concept demonstrated reduction in plasma triglycerides related to LPL activity of treated animals, and this was used as the primary pharmacodynamic measure to show activity. However, the applicant failed to adequately demonstrate pharmacokinetic and pharmacodynamic properties of the product in the clinical setting, since LPL plasma activity could not be consistently demonstrated, and no sustained TG decrease could be observed.79 Associated CAR T cell toxicities, such as cytokine release syndrome, neurological toxicity, on target/off tumor events were not fully anticipated by nonclinical studies either.86,87 For Zolgensma, the different cardiovascular safety profile observed in the preclinical and clinical stage was attributed to a difference in transduceability at individual cardiomyocyte levels between mice and humans.37 Finally, adeno-associated virus (AAV)-related toxicities are currently being discussed by the agencies.88 Dorsal root ganglion pathology has been observed in nonhuman primates but it is still unclear if it is translated to the clinical setting in human beings.37,89 Similarly, although AAV integration associated with hepatocellular carcinoma (HCC) was observed in neonatal mice, there has been minimal evidence of HCC occurring in patients receiving gene therapies.89
An iterative approach was suggested to be informative, for example, when early clinical experience identifies unexpected adverse reactions then additional preclinical studies may provide a mechanistic basis for mitigation measures.86,90 On the other hand, the need for standards to enable cross-comparisons of, and confidence in, testing results, or ensure techniques that are consistently implemented for the nonclinical studies so that data can be compared, would allow to increase and share knowledge in the field, e.g., biodistribution studies.91 Finally, a risk-based approach during product development to design a tailor-made ATMP development program is usually recommended to determine the extent of quality, nonclinical and clinical data necessary for an MA and to justify any deviation from the requirements, i.e., as defined in annex I, part IV of Directive (2001)/83/EC.92,93
Interplay between clinical evidence and product quality
Not only does the limited clinical evidence at the MAA stage impact the approval decision, but the quality development and lack of quality standards for these products is a key challenge.94 Several factors limit the achievement of consistent data and adequate interpretation of clinical results across studies: the uniqueness of each product, the heterogeneity of this novel group of products, the variability in the pipeline of clinical development and approaches chosen, the divergent manufacturing strategies, and the different tests/assays applied during clinical development and its validation.75,95
The quality of manufacturing can affect the clinical outcome, and issues within the quality module of MAA dossier might be directly related with the acceptability of the clinical package. Issues are mostly related to validation of the analytical methods, design and control of the manufacturing process, and comparability.18 The comparability of manufacturing processes remains one of the major issues and was raised during assessment of the majority of the approved products.96 When a process change is required, for instance, to increase production volume for a phase III trial or commercialization, questions of comparability between processes during the MAA review and how this can affect the clinical safety and efficacy outcomes are common. This point can imply the requirement of generating additional clinical data or impair the validity of previously generated data, as was the case for Kymriah or Zolgensma.37,96 Not only the comparability between manufacturing processes, but batch to batch inconsistency, which might contribute to the heterogeneity of clinical response, has been observed for some approved therapies.37 The inadequate comparability assessments, coupled with the difficulty of potency assays, can also impact key clinical aspects, such as the consistency of doses administered during the clinical development.37,97 For cell therapies, the mechanisms to study cell activity are complex and poorly understood and the cell counts may vary over time, which makes it difficult to establish standard, effective doses, and routes of administration in clinical trials. This might lead to inconsistent trial results that are hard to interpret and replicate across studies.91 For some approved gene therapies, uncertainties with regard to control of the effective dose, without a stable reference standard to control the potency of the product have been also observed.37
Standardization of manufacturing may be difficult given proprietary platforms, but some common processes, such as common operational steps, product characterization, design and validation of processes, and testing could be achieved to improve some of these issues.98 Previous experience available in humans with similar products and with similar standards that allow performing a comparison with valid pooling data would help to improve the current translation challenges in the ATMP field, e.g., AAV-based gene therapies, CAR T cell therapies, autologous cultured chondrocytes, or mesenchymal adult stem cells. For instance, it has been stated that longitudinal investigations of anti-CAR immune responses through the same validated assays would be particularly important in understanding how immunogenicity can lead to treatment failure. For the three approved CAR T cell therapies, there were huge differences in the reported percentages of patients with pre-existing antibodies and it was suggested that this fact could reflect the different assays used for detection.99 Similarly, pre-existing immunity and immunogenicity toward the vector or transgene are the largest challenge for AAV-based gene therapies given that it can interfere with therapeutic efficacy if not identified and managed optimally.100 Common ways to test tissue engineering product integrity, including tensile strength and suture retention, to ensure that these products meet safety thresholds for use in clinical environments, has also been raised.91
The quality requirements are not reduced due to accelerated access routes, and it is under debate that greater standardization and harmonization across regulatory authorities could reduce the burden on companies to ensure compliance at every phase of the development and commercialization process.101,102 Several organizations are working to assemble and define standards and the convergence of common requirements.101,103, 104, 105 Although it should be recognized how challenging standardization is given the diversity in the cell and gene therapy space and its rapid progress, the standard needs have already been identified.91,98 Examples from a quality standpoint include: (1) create management systems for processing and handling cells, establish cell collection requirements that ensure consistency, safety, and comparability in the final products, (2) identify potential commonalities across manufacturing processes and create broadly applicable guidelines, or (3) establish guidelines to harmonize manufacturers' characterization, design, and validation processes to lower barriers. From nonclinical and clinical standpoint, it has been proposed: (1) to establish consensus on which biodistribution approaches are most applicable, (2) to implement a standard approach to pre-existing immunity assay development, selection, and evaluation to enhance patient safety and quality of clinical trial data, or (3) initiate cell counting methods/technologies, optimal timing for dose assessment, and qualify routes of administration and dose preparation methods to select safe and effective doses, among others.91
Impact at the post-marketing setting and market access
Pre-registration randomized clinical trials are not always representative of patient populations in the routine practice due to the strict patient inclusion and exclusion criteria, and the strict intervention protocols.35,106, 107, 108, 109 Therefore, the generation of evidence throughout the medicine's life cycle is essential to gain more information about its effectiveness and safety in a more diverse clinical setting, to improve healthcare quality, and to provide information to either complement initial evidence or to verify whether the MA should be maintained as granted, varied, suspended, or revoked. In the case of ATMPs, real-world evidence plays a major role and is essential to confirm the benefit-risk profile given the imprecise clinical data available at the time of MA. This point might be translated into the need to perform long and extensive post-marketing studies.110,111
It has been reported that post-authorization studies for the approved ATMPs consist both in interventional studies (some of them ongoing at the time of MA) and observational studies. The profile of the planned interventional trials to further assess effectiveness resemble pre-market trials in terms of design, i.e., using single-arm designs, reduced sample sizes, and are focused on a narrow study population.11 In some cases, generation of evidence post-launch can be particularly challenging, especially when it requires long-term follow-up, since participants may be lost during the trial due to different causes (i.e., cure of the disease, depression, among others) or may be reluctant to participate when the pharmaceutical is already launched. The latter is more evident when the study is randomized.112 On the other hand, the burden that the clinical post-marketing requirements imply, along with the extensive manufacturing commitments, could hamper market access. This was the case for Glybera, where the extremely limited use of the product and the costs of post-marketing requirements, including maintaining the commercial manufacturing capabilities, led to its withdrawal after two years on the European market.113,114
The insufficient evidence available on comparative clinical effectiveness or clinical benefits hinder the determination of appropriate pricing and payment schemes. The decision on price and reimbursement requires an exhaustive study of the evidence generated during product development, the relative effectiveness and safety, the patient-reported outcomes (including quality of life), and the cost-effectiveness and budget impact to finally assess its place in therapy. At this stage, HTA bodies (HTAb) have an important role. The scientific evidence of the product and its potential contribution in the therapeutic management of the disease is deeply studied in EU countries, but the recommendations from the HTAb may differ among them, above all for orphan drugs.115 HTAb-specific requirements can be related to the acceptability of the endpoints used, the control arm, the inclusion and exclusion criteria, and, at the end, the generalization of the results obtained in clinical practice.76 When the product clinical data are limited, to determine all the aforementioned is complex and usually translates to long negotiations between the MA holder and the health authorities. This negotiation may be one of the reasons for the time elapse between MA and final drug prescription and this represents a major concern for healthcare systems, patients, and industry. The difficulty of accessing the market once the product is authorized highlights the differences in the answers that a regulatory agency and a healthcare system are seeking in clinical trials.116
Finally, there is industry pressure for corporate pharma and its investors to ensure the return of drug development investment. With a high expected value, but with immature evidence and high prices requested, the complexity of negotiations between industry and payers is becoming common, and sometimes the non-reimbursement has been justified. Managed entry agreements have been a solution to this challenge. Commercial arrangements have been frequently used in European countries either financial (discounts and rebates) or outcome-based to finally release a product into the market. Provenge, MACI, and ChondroCelect were withdrawn because of poor commercial performance and lack of reimbursement in EU countries.2,95,114 The limited use of the product, the costs of post-marketing requirements, including clinical trials and maintaining commercial manufacturing capabilities, are other factors that contributed to ATMP withdrawal.113,114
To avoid costly corrections in late clinical development and a weak market access value dossier (a document that provides evidence-based messages in communicating product value), a comprehensive risk assessment must be carried out before committing to a particular pivotal trial design. The development strategy for an ATMP should also include parallel EMA-HTAb advice regarding optimization of evidence generation of in the EU, to discuss different design options during clinical development, their applicability with respect to efficiency and risk of bias, and the potential post-launch generation of evidence. The same approach is recommended through FDA interactions in the case of the United States, such as special protocols assessments.2 These discussions, along with the potential implementation of the advice, could reduce the risk of benefit-risk uncertainty and production of data that would be inadequate to support the company's future reimbursement request.117,118 In addition, the company's retrospective analysis from the drug pipeline development and failures during different phases of clinical trials will have led them to improve its research and development workflow in terms of learning, strategy, costs, and performance.119,120 For instance, Alofisel (sponsored by TiGenix) set a model of iterative strategy that enabled MA through improving late clinical development with the lessons learnt from the previous autologous cell therapy, sponsored by Cellerix.121
Conclusions
ATMPs are innovative therapies that mainly target orphan diseases and high unmet medical needs. The level of generated clinical evidence and the quality aspects of advanced therapies playing a key role in the development, approval, and post-marketing setting for these therapies. This article describes the current landscape of clinical development of advanced therapies, its challenges, and some of the potential solutions that are currently under discussion. Most authorized ATMPs are based on adaptive, small, open-label, uncontrolled, and single-arm pivotal trials. Flexibility on conventional regulatory requirements has been widely implemented by regulators, especially for low prevalence, life-threatening, or seriously debilitating diseases. Progressive iteration of the science, establishing new standards for ATMP development with the aim to ensure consistency in clinical development, and the reproducibility of knowledge is required not only to increase the generation of evidence for approval but to set principles to achieve translational success in this field. Although there is a trend toward an adaptive approach to licensing or a life-cycle approach, after the experience with the approvals of ATMPs so far, regulators and global working groups are developing and releasing new recommendations to promote an approach to clinical development that is methodologically sound and thus significantly more relevant. It remains to be seen how clinical development of ATMPs will evolve, but it is recommended that the industry stakeholders should strive to understand and try to apply the recommendations of relevant parties to better succeed in market access.
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author contributions
Acquisition of data, analysis and interpretation of data, C.I.-L.; drafting and revising the manuscript, C.I.-L.; reviewed and edited the manuscript, C.I.-L., A.A., M.O., and A.V. All authors have approved the final article.
Declaration of interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The findings and conclusions in this article should not be construed to represent any agency determination or policy.
References
- 1.Iglesias-Lopez C., et al. Regulatory Framework for Advanced Therapy Medicinal Products in Europe and United States. Front Pharmacol. 2019;10:921 doi: 10.3389/fphar.2019.00921. Erratum in: Front Pharmacol. 2020;11:766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iglesias-Lopez C., Obach M., Vallano A., Agustí A. Comparison of regulatory pathways for the approval of advanced therapies in the European Union and the United States. Cytotherapy. 2021;23:261–274. doi: 10.1016/j.jcyt.2020.11.008. [DOI] [PubMed] [Google Scholar]
- 3.European Commission Regulation (EC) No 1394/2007 of the European parliament and the council of 13 November 2007 on advanced therapy medicinal products and amending directive 2001/83/EC and regulation (EC) No 726/2004. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32007R1394&from=EN
- 4.Alamo J.E., Timón M., Gómez-Platero C.G., Abad C.D., Gonzalez M.V., Martín-de la Sierra-San Agustín M.Á. Clinical trials of advanced therapy investigational medicinal products in Spain: preparing for the European clinical trials regulation. Cell Gene Ther. Insights. 2019;5:1431–1449. [Google Scholar]
- 5.Iglesias-Lopez C., Agustí A., Vallano A., Obach M. Methodological Characteristics of Clinical Trials Supporting the Marketing Authorisation of Advanced Therapies in the European Union. Front. Pharmacol. 2021;12:773712 doi: 10.3389/fphar.2021.773712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.European Medicines Agency Medicines. 2021. https://www.ema.europa.eu/en/medicines
- 7.Hanna E., Rémuzat C., Auquier P., Toumi M. Advanced therapy medicinal products: current and future perspectives. J. Mark. Access Health Policy. 2016;4:31036. doi: 10.3402/jmahp.v4.31036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.European Medicines Agency Orphan designation. 2021. https://www.ema.europa.eu/en/glossary/orphan-designation
- 9.European Medicines Agency Unmet medical need. 2019. https://www.ema.europa.eu/en/documents/presentation/presentation-unmet-medical-need-introduction-definitions-stakeholder-perceptions-jllinares-garcia_en.pdf
- 10.European Medicines Agency . European Medicines Agency; 2021. Adaptive Pathways.https://www.ema.europa.eu/en/human-regulatory/research-development/adaptive-pathways [Google Scholar]
- 11.Fritsche E., Elsallab M., Schaden M., Hey S.P., Abou-El-Enein M. Post-marketing safety and efficacy surveillance of cell and gene therapies in the EU: a critical review. Cell Gene Ther. Insights. 2019;5:1505–1521. [Google Scholar]
- 12.Pharma Intelligence Disappointing end for MolMed’s Zalmoxis cell therapy in EU. 2019. https://pink.pharmaintelligence.informa.com/PS140998/Disappointing-End-For-MolMeds-Zalmoxis-Cell-Therapy-In-EU
- 13.Novartis' Press Release Novartis’ Kymriah fails to meet primary goal in non-Hodgkin lymphoma trial. 2021. https://www.clinicaltrialsarena.com/news/novartis-kymriah-lymphoma-trial/
- 14.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2018;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 15.Mercuri E., Darras B.T., Chiriboga C.A., Day J.W., Campbell C., Connolly A.M., Iannaccone S.T., Kirschner J., Kuntz N.L., Saito K., et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018;378:625–635. doi: 10.1056/NEJMoa1710504. [DOI] [PubMed] [Google Scholar]
- 16.European Medicines Agency Committee for medicinal products for human use (CHMP) assessment report: Zalmoxis. 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002801/WC500212588.pdf
- 17.European Medicines Agency Committee for medicinal products for human use (CHMP) assessment report: Alofisel. 2017. https://www.ema.europa.eu/en/documents/assessment-report/alofisel-epar-public-assessment-report_en.pdf
- 18.Elsallab M., Bravery C.A., Kurtz A., Abou-El-Enein M. Mitigating deficiencies in evidence during regulatory assessments of advanced therapies: a comparative study with other biologicals. Mol. Ther. Methods Clin. Dev. 2020;18:269–279. doi: 10.1016/j.omtm.2020.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ball O., Robinson S., Bravery C. EU market authorisation strategy: lessons from the first 22 ATMP submitted to the EMA. Cell Gene Ther. Insights. 2019;5:759–791. [Google Scholar]
- 20.de Wilde S., Coppens D.G.M., Hoekman J., de Bruin M.L., Leufkens H.G.M., Guchelaar H.J., Meij P. EU decision-making for marketing authorization of advanced therapy medicinal products: a case study. Drug Discov. Today. 2018;23:1328–1333. doi: 10.1016/j.drudis.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 21.Barkholt L., Voltz-Girolt C., Raine J., Salmonson T., Schüssler-Lenz M. European regulatory experience with advanced therapy medicinal products. Nat. Rev. Drug Discov. 2018;18:8–9. doi: 10.1038/nrd.2018.200. [DOI] [PubMed] [Google Scholar]
- 22.Carvalho M., Martins A.P., Sepodes B. Hurdles in gene therapy regulatory approval: a retrospective analysis of European Marketing Authorization Applications. Drug Discov. Today. 2019;24:823–828. doi: 10.1016/j.drudis.2018.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Day S., Jonker A.H., Lau L.P.L., Hilgers R.D., Irony I., Larsson K., Roes K.C., Stallard N. Recommendations for the design of small population clinical trials. Orphanet J. Rare Dis. 2018;13:195. doi: 10.1186/s13023-018-0931-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ASTERIX Project Advances in small trials design for regulatory innovation and eXcellence. 2021. http://www.asterix-fp7.eu/
- 25.IDEAL Project Integrated design and analysis of clinical trials in small population groups. 2021. https://www.ideal.rwth-aachen.de/
- 26.Friede T., Posch M., Zohar S., Alberti C., Benda N., Comets E., Day S., Dmitrienko A., Graf A., Kürsad Günhan B., et al. 2018. Recent Advances in Methodology for Clinical Trials in Small Populations: The InSPiRe Project. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park, J.J.H., Siden, E., Zoratti, M.J., Dron, L., Harari, O., Singer, J., Lester, R.T., Thorlund, K., and Mills, E.J. Systematic review of basket trials, umbrella trials, and platform trials: a landscape analysis of master protocols.Trials 20:572. [DOI] [PMC free article] [PubMed]
- 28.Bogin V. Master protocols: new directions in drug discovery. Contemp. Clin. Trials Commun. 2020;18:100568. doi: 10.1016/j.conctc.2020.100568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woodcock J., LaVange L.M. Master protocols to study multiple therapies, multiple diseases, or both. N. Engl. J. Med. 2017;377:62–70. doi: 10.1056/NEJMra1510062. [DOI] [PubMed] [Google Scholar]
- 30.Food and Drug Administration . 2018. Master Protocols: Efficient Clinical Trial Design Strategies to Expedite Development of Oncology Drugs and Biologics Guidance for Industry.https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm [Google Scholar]
- 31.Park J.J.H., Harari O., Dron L., Lester R.T., Thorlund K., Mills M. An overview of platform trials with a checklist for clinical readers. J. Clin. Epidemiol. 2020;125:1–8. doi: 10.1016/j.jclinepi.2020.04.025. [DOI] [PubMed] [Google Scholar]
- 32.European Medicines Agency Committee for medicinal products for human use (CHMP) assessment report: Yescarta. 2018. https://www.ema.europa.eu/en/documents/assessment-report/yescarta-epar-public-assessment-report_en.pdf
- 33.European Medicines Agency Committee for medicinal products for human use (CHMP) assessment report: Kymirah. 2018. https://www.ema.europa.eu/en/documents/assessment-report/kymriah-epar-public-assessment-report_en.pdf
- 34.Zhang J., Li J., Ma Q., Yang H., Signorovitch J., Wu E. A review of two regulatory approved anti-CD19 CAR T-cell therapies in diffuse large B-cell lymphoma: why are indirect treatment comparisons not feasible? Adv. Ther. 2020;37:3040. doi: 10.1007/s12325-020-01397-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Novartis Press Release Kymriah® demonstrates consistent efficacy and safety outcomes in US patients when used in real-world setting. 2019. https://www.novartis.com/news/media-releases/novartis-kymriah-demonstrates-consistent-efficacy-and-safety-outcomes-us-patients-when-used-real-world-setting
- 36.Pink Sheet Zolgensma provides No proven benefit over Spinraza in SMA, says German HTA. 2021. https://pink.pharmaintelligence.informa.com/PS144878/Zolgensma-Provides-No-Proven-Benefit-Over-Spinraza-In-SMA-Says-German-HTA?vid=Pharma&processId=d4166a13-c73f-4efd-80c8-aa5b34bfb9e8
- 37.European Medicines Agency . Zolgensma; 2020. Committee for Medicinal Products for human Use (CHMP) Assessment Report.https://www.ema.europa.eu/en/documents/assessment-report/zolgensma-epar-public-assessment-report_en.pdf [Google Scholar]
- 38.ClinitalTrial.gov Master protocol for the phase 1 study of cell therapies in multiple myeloma. 2021. https://clinicaltrials.gov/ct2/show/NCT04155749
- 39.Clinicaltrials.gov Master protocol to assess the safety and antitumor activity of genetically engineered T cells in NY-ESO-1 and/or LAGE-1a positive solid tumors - full text view - ClinicalTrials.gov. 2021. https://clinicaltrials.gov/ct2/show/NCT03967223?term=master+protocol+AND+cell+therapies&draw=2&rank=2
- 40.Bateman R.J., Benzinger T.L., Berry S., Clifford D.B., Duggan C., Fagan A.M., Fanning K., Farlow M.R., Hassenstab J., McDade E.M., et al. The DIAN-TU next generation Alzheimer’s prevention trial: adaptive design and disease progression model. Alzheimers Dement. 2017;13:8–19. doi: 10.1016/j.jalz.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.European Medicines Agency Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells. 2020. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-medicinal-products-containing-genetically-modified_en-0.pdf
- 42.Rodríguez-Otero P., Prósper F., Alfonso A., Paiva B., San Miguel J.F.S. CAR T-cells in multiple myeloma are ready for prime time. J. Clin. Med. 2020;9:3577. doi: 10.3390/jcm9113577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Food and Drug Administration Human gene therapy for hemophilia; guidance for industry. 2020. https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-
- 44.Food and Drug Administration Human gene therapy for retinal disorders; guidance for industry. 2020. https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-
- 45.Food and Drug Administration Human gene therapy for neurodegenerative diseases; draft guidance for industry. 2021. https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-
- 46.Food and Drug Administration Adaptive designs for clinical trials of drugs and biologics guidance for industry. 2019. https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htmand/or
- 47.Food and drug administration complex innovative trial design pilot meeting program. https://www.fda.gov/drugs/development-resources/complex-innovative-trial-design-pilot-meeting-program [DOI] [PubMed]
- 48.Brenda Gaydos A.K., Frank Miller M.P., Vandemeulebroecke M., Wang S.J. Perspective on adaptive designs: 4 years European Medicines Agency reflection paper, 1 year draft US FDA guidance – where are we now? Clin. Invest. 2012;2:235–240. [Google Scholar]
- 49.EUnetHTA Comparators and comparisons: direct and indirect comparisons. 2015. https://www.eunethta.eu/wp-content/uploads/2018/01/Comparators-Comparisons-Direct-and-indirect-comparisons_Amended-JA1-Guideline_Final-Nov-2015.pdf?x50316
- 50.EUnetHTA Criteria for the choice of the most appropriate comparator(s) summary of current policies and best practice recommendations. 2015. https://eunethta.eu/wp-content/uploads/2018/03/Criteria_WP7-SG3-GL-choice_of_comparator_amend2015.pdf
- 51.Hassan A., Booth C., Brightwell A., Allwood Z., Veys P., Rao K., Hönig M., Friedrich W., Gennery A., Slatter M., et al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood. 2012;120:3615–3624. doi: 10.1182/blood-2011-12-396879. [DOI] [PubMed] [Google Scholar]
- 52.European Medicines Agency . Libmeldy; 2021. Committee for Medicinal Products for Human Use (CHMP) Assessment Report.https://www.ema.europa.eu/en/documents/assessment-report/libmeldy-epar-public-assessment-report_en.pdf EMA/584450/2020. [Google Scholar]
- 53.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kolb S.J., Coffey C.S., Yankey J.W., Krosschell K., Arnold W.D., Rutkove S.B., Swoboda K.J., Reyna S.P., Sakonju A., Darras B.T., et al. Baseline results of the NeuroNEXT spinal muscular atrophy infant biomarker study. Ann. Clin. Transl. Neurol. 2016;3:132. doi: 10.1002/acn3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.European Medicines Agency Committee for advanced therapies (CAT) committee for medicinal products for human use (CHMP) assessment report: Skysona. 2021. https://www.ema.europa.eu/en/documents/assessment-report/skysona-epar-public-assessment-report_en.pdf
- 57.Crump M., Neelapu S.S., Farooq U., Van Den Neste E., Kuruvilla J., Westin J., Link B.K., Hay A., Cerhan J.R., Zhu L., et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130:1800–1808. doi: 10.1182/blood-2017-03-769620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ciceri F., Labopin M., Aversa F., Rowe J.M., Bunjes D., Lewalle P., Nagler A., Di Bartolomeo P., Lacerda J.F., Stangheilini M.T.L., et al. A survey of fully haploidentical hematopoietic stem cell transplantation in adults with high-risk acute leukemia: a risk factor analysis of outcomes for patients in remission at transplantation. Blood. 2008;112:3574–3581. doi: 10.1182/blood-2008-02-140095. [DOI] [PubMed] [Google Scholar]
- 59.European Medicines Agency Committee for medicinal products for human use (CHMP) assessment report: Abecma. 2021. https://www.ema.europa.eu/en/documents/assessment-report/abecma-epar-public-assessment-report_en.pdf
- 60.Walton M.K. Historical controls for clinical trials contemplation on use in drug development. 2012. https://events-support.com/Documents/Walton_Marc.pdf
- 61.Food and Drug Administration Rare diseases: natural history studies for drug development guidance for industry - draft guidance. 2019. https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
- 62.Food and Drug Administration . 2018. Framework for FDA’s Real-World Evidence Program. [Google Scholar]
- 63.Ghadessi M., Tang R., Zhou J., Liu R., Wang C., Toyoizumi K., Mei C., Zhang L., Deng C.Q., Beckman R.A. A roadmap to using historical controls in clinical trials - by drug information association adaptive design scientific working group (DIA-ADSWG) Orphanet J. Rare Dis. 2020;15:69. doi: 10.1186/s13023-020-1332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Food and Drug Administration ABECMA (idecabtagene vicleucel). BLA clinical review memorandum. 2021. https://www.fda.gov/vaccines-blood-biologics/abecma-idecabtagene-vicleucel
- 65.Celgene Protocol NDS-MM-003. Celgene Corporation. 2018:22821866–22822175. https://www.pei.de/SharedDocs/Downloads/DE/awb/nis-0401-0500/0461-beoplan.pdf?__blob=publicationFile&v=4 [Google Scholar]
- 66.Abou-El-Enein M., Grainger D.W., Kili S. Registry contributions to strengthen cell and gene therapeutic evidence. Mol. Ther. 2018;26:1172–1176. doi: 10.1016/j.ymthe.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.European Medicines Agency Patient registry initiative- strategy and mandate of the cross-committee task force. 2017. www.ema.europa.eu/contact
- 68.Elsallab M., Levine B.L., Wayne A.S., Abou-El-Enein M. CAR T-cell product performance in haematological malignancies before and after marketing authorisation. Lancet Oncol. 2020;21:e104–e116. doi: 10.1016/S1470-2045(19)30729-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.NICE Single technology appraisal. Axicabtagene ciloleucel for treating diffuse large B-cell lymphoma and primary mediastinal B-cell lymphoma after 2 or more systemic therapies [ID1115] 2018. https://www.nice.org.uk/guidance/ta559/evidence/final-appraisal-determination-committee-papers-pdf-6661404974
- 70.ICH ICH topic E 8 general considerations for clinical trials step 5 note for guidance on general considerations for clinical trials. 1998. http://www.emea.eu.int
- 71.Gutman S.I., Piper M., Grant M.D. Agency Healthc. Res. Qual; 2013. Background - Progression-free Survival: What Does it Mean for Psychological Well-Being or Quality of Life?https://www.ncbi.nlm.nih.gov/books/NBK137763/ [PubMed] [Google Scholar]
- 72.Bruce C.S., Brhlikova P., Heath J., McGettigan P. The use of validated and nonvalidated surrogate endpoints in two European Medicines Agency expedited approval pathways: a cross-sectional study of products authorised 2011–2018. PLoS Med. 2019;16:e1002873. doi: 10.1371/journal.pmed.1002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pazdur R. Endpoints for assessing drug activity in clinical trials. Oncologist. 2008;13:19–21. doi: 10.1634/theoncologist.13-S2-19. [DOI] [PubMed] [Google Scholar]
- 74.Food and Drug Administration Clinical trial endpoints for the approval of cancer drugs and biologics guidance for industry. 2018. https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htmand/https://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
- 75.ten Ham R.M.T., Hoekman J., Hövels A.M., Broekmans A.W., Leufkens H.G.M., Klungel O.H. Challenges in advanced therapy medicinal product development: a survey among companies in Europe. Mol. Ther. Methods Clin. Dev. 2018;11:121–130. doi: 10.1016/j.omtm.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.EUnetHTA Guideline on endpoints used for relative effectiveness assessment of pharmaceuticals: surrogate endpoints. 2015. https://eunethta.eu/wp-content/uploads/2018/03/surrogate_endpoints.pdf
- 77.European Medicines Agency Guideline on the clinical evaluation of anticancer medicinal products. 2019. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-evaluation-anticancer-medicinal-products-man-revision-6_en.pdf
- 78.Del Rosario C., Slevin M., Molloy E.J., Quigley J., Nixon E. How to use the Bayley scales of infant and toddler development. Arch. Dis. Child. Educ. Pract. Ed. 2020;106:108–112. doi: 10.1136/archdischild-2020-319063. [DOI] [PubMed] [Google Scholar]
- 79.European Medicines Agency Committee for medicinal products for human use (CHMP) assessment report: Glybera. 2012. www.ema.europa.eu
- 80.Grigore B., Ciani O., Dams F., Federici C., de Groot S., Möllenkamp M., Rabbe S., Shatrov K., Zemplenyi A., Taylor R.S. Surrogate endpoints in health technology assessment: an international review of methodological guidelines. Pharmacoeconomics. 2020;38:1055–1070. doi: 10.1007/s40273-020-00935-1. [DOI] [PubMed] [Google Scholar]
- 81.Ciani O., Buyse M., Drummond M., Rasi G., Saad E.D., Taylor R.S. Time to review the role of surrogate end points in health policy: state of the art and the way forward. Value Health. 2017;20:487–495. doi: 10.1016/j.jval.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 82.European parliament and European council proposal of regulation of the European parliament and of the council on health technology assessment and amending directive 2011/24/EU. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A52018PC0051
- 83.Food and Drug Administration Table of surrogate endpoints that were the basis of drug approval or licensure. https://www.fda.gov/drugs/development-resources/table-surrogate-endpoints-were-basis-drug-approval-or-licensure
- 84.FDA CBER Guidance for industry preclinical assessment of investigational cellular and gene therapy products. 2009. http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
- 85.Méndez-Hermida F. Basic non-clinical requirements for registration of new drugs. Approaches to the non-clinical development of advanced therapy medicinal products SME workshop: focus on non-clinical aspects. https://www.ema.europa.eu/en/documents/presentation/presentation-approaches-non-clinical-development-advanced-therapy-medicinal-products-fernando_en.pdf
- 86.Exley A.R., Rantell K., James M. Clinical development of cell therapies for cancer: the regulators’ perspective. Eur. J. Cancer. 2020;138:41–53. doi: 10.1016/j.ejca.2020.07.006. [DOI] [PubMed] [Google Scholar]
- 87.Sun S., Hao H., Yang G., Zhang Y., Fu Y. Immunotherapy with CAR-modified T cells: toxicities and overcoming strategies. J. Immunol. Res. 2018;2018:23861. doi: 10.1155/2018/2386187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pink Sheet Gene therapy AAV vector toxicities get US FDA’s attention. 2021. https://pink.pharmaintelligence.informa.com/PS144702/Gene-Therapy-AAV-Vector-Toxicities-Get-US-FDAs-Attention
- 89.Bolt M.W., Brady J.T., Whiteley L.O., Khan K.N. Development challenges associated with rAAV-based gene therapies. J. Toxicol. Sci. 2021;46:57–68. doi: 10.2131/jts.46.57. [DOI] [PubMed] [Google Scholar]
- 90.Taraseviciute A., Tkachev V., Ponce R., Turtle C.J., Snyder J.M., Liggitt H.D., Myerson D., Gonzalez-Cuyar L., Baldessari A., English C., et al. Chimeric antigen receptor T cell-mediated neurotoxicity in nonhuman primates. Cancer Discov. 2018;8:750–763. doi: 10.1158/2159-8290.CD-17-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nexight Group Community perspectived: need standards in regenerative medicine. 2020. https://www.standardscoordinatingbody.org/needed
- 92.European Medicines Agency Guideline on the risk-based approach according to Annex I, part IV of directive 2001/83/EC applied to advanced therapy medicinal products. 2013. www.ema.europa.eu
- 93.Kooijman M., van Meer P.J.K., Gispen-de Wied C.C., Moors E.H.M., Hekkert M.P., Schellekens H. The risk-based approach to ATMP development - generally accepted by regulators but infrequently used by companies. Regul. Toxicol. Pharmacol. 2013;67:221–225. doi: 10.1016/j.yrtph.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 94.IMI Online consultation on advanced therapies | summary of feedback received. 2017. https://www.imi.europa.eu/sites/default/files/archive/uploads/documents/ATMPconsultation2016/ATMP_consultation_feedbacksummary.pdf
- 95.Abou-El-Enein M., Hey S.P. Cell and gene therapy trials: are we facing an ‘evidence crisis’? EClinicalMedicine. 2019;7:13–14. doi: 10.1016/j.eclinm.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cockroft A., Wilson A. Comparability: what we can learn from the review of advanced therapy medicinal products. Regen. Med. 2021;16(7):655–667. doi: 10.2217/rme-2021-0026. [DOI] [PubMed] [Google Scholar]
- 97.Tavridou A., Rogers D., Bonelli M., Schiel A., Hidalgo-Simon H.A. Towards a better use of scientific advice for developers of advanced therapies. Br. J. Clin. Pharmacol. 2021;87:2459–2464. doi: 10.1111/bcp.14672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McNiece I.K., Wacker K.K., Kurtzberg J., Warkentin P.I. Standardization, workforce development and advocacy in cell and gene therapies: a summary of the 2020 Regenerative Medicine Interchange. Cytotherapy. 2021;23:886–893. doi: 10.1016/j.jcyt.2021.02.004. [DOI] [PubMed] [Google Scholar]
- 99.Wagner D.L., Fritsche E., Pulsipher M.A., Ahmed N., Hamieh M., Hegde M., Ruella M., Savoldo B., Shah N.N., Turtle C.J., et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021;18:379–393. doi: 10.1038/s41571-021-00476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mingozzi F., High K.A. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.The Medicine Maker How to standardize advanced therapy manufacture. 2021. https://themedicinemaker.com/manufacture/how-to-standardize-advanced-therapy-manufacture
- 102.Drago D., Foss-Campbell B., Wonnacott K., Barrett D., Ndu A. Global regulatory progress in delivering on the promise of gene therapies for unmet medical needs. Mol. Ther. Methods Clin. Dev. 2021;21:524–529. doi: 10.1016/j.omtm.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee M.H., Au P., Hyde J., Gacchina Johnson C., Heidaran M., Karandish S., Boxer L., Mendicino M., Yoon D., Tull L., et al. Translation of regenerative medicine products into the clinic in the United States: FDA perspective. Transl. Regen. Med. 2015:49–74. [Google Scholar]
- 104.Morrow D., Ussi A., Migliaccio G. Addressing pressing needs in the development of advanced therapies. Front. Bioeng. Biotechnol. 2017;5:55. doi: 10.3389/fbioe.2017.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Creating a roadmap for the development and manufacture of gene therapies. https://www.cellandgene.com/doc/creating-a-roadmap-for-the-development-and-manufacture-of-gene-therapies-0001
- 106.Blonde L., Khunti K., Harris S.B., Meizinger C., Skolnik N.S. Interpretation and impact of real-world clinical data for the practicing clinician. Adv. Ther. 2018;35(11):1763–1774. doi: 10.1007/s12325-018-0805-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abou-El-Enein M., Duda G.N., Gruskin E.A., Grainger D.W. Strategies for derisking translational processes for biomedical technologies. Trends Biotechnol. 2017;35:100–108. doi: 10.1016/j.tibtech.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 108.Jacobson C.A., Hunter B.D., Redd R., Rodig S.J., Chen P.H., Wright K., Lipschitz M., Ritz J., Kamihara Y., Armand P., et al. Axicabtagene ciloleucel in the non-trial setting: outcomes and correlates of response, resistance, and toxicity. J. Clin. Oncol. 2020;38:3095–3106. doi: 10.1200/JCO.19.02103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jacobson C.A., Hunter B., Armand P., Kamihara Y., Ritz J., Rodig S.J., Wright K., Lipschitz M., Redd R.A., Maus M.V., et al. Axicabtagene ciloleucel in the real world: outcomes and predictors of response, resistance and toxicity. Blood. 2018;132:92. [Google Scholar]
- 110.Ermisch M., Bucsics A., Bonanno P.V., Arickx F., Bybau A., Bochenek T., van de Casteele M., Diogene E., Fürst J., Garuoliene K., et al. Payers’ views of the changes arising through the possible adoption of adaptive pathways. Front. Pharmacol. 2016;7:305. doi: 10.3389/fphar.2016.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Joppi R., Gerardi C., Bertele V., Garattini S. Letting post-marketing bridge the evidence gap: the case of orphan drugs. BMJ. 2016;353:i2978. doi: 10.1136/bmj.i2978. [DOI] [PubMed] [Google Scholar]
- 112.Joint Briefing Paper "Adaptive licensing” or “adaptive pathways”: deregulation under the guise of earlier access executive. 2015. http://english.prescrire.org/Docu/DOCSEUROPE/
- 113.Genetic Engineering & Biotechnology News uniQure announces it will not seek marketing authorization renewal for Glybera in Europe. 2017. https://www.genengnews.com/topics/genome-editing/uniqure-says-it-will-not-pursue-ec-marketing-renewal-for-glybera-gene-therapy/
- 114.Jarosławski S., Toumi M. Sipuleucel-T (Provenge®) - autopsy of an innovative paradigm change in cancer treatment: why a single-product biotech company failed to capitalize on its breakthrough invention. BioDrugs. 2015;29:301–307. doi: 10.1007/s40259-015-0140-7. [DOI] [PubMed] [Google Scholar]
- 115.Abou-El-Enein M., Elsanhoury A., Reinke P. Overcoming challenges facing advanced therapies in the EU market. Cell Stem Cell. 2016;19:293–297. doi: 10.1016/j.stem.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 116.Dabbous M., Chachoua L., Caban A., Toumi M. Managed entry agreements: policy analysis from the European perspective. Value Health. 2020;23:425–433. doi: 10.1016/j.jval.2019.12.008. [DOI] [PubMed] [Google Scholar]
- 117.Moseley J., Vamvakas S., Berntgen M., Cave A., Kurz X., Arlett P., Acha V., Bennett S., Cohet C., Corriol-Rohou S., et al. Regulatory and health technology assessment advice on postlicensing and postlaunch evidence generation is a foundation for lifecycle data collection for medicines. Br. J. Clin. Pharmacol. 2020;86:1034–1051. doi: 10.1111/bcp.14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.European Medicines Agency Parallel consultation with regulators and health technology assessment bodies. 2021. https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance/parallel-consultation-regulators-health-technology-assessment-bodies
- 119.Bryant L.M., Christopher D.M., Giles A.R., Hinderer C., Rodriguez J.L., Smith J.B., Traxler E.A., Tycko J., Wojno A.P., Wilson J.M. Lessons learned from the clinical development and market authorization of Glybera. Hum. Gene Ther. Clin. Dev. 2013;24:55–64. doi: 10.1089/humc.2013.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dolgos H., Trusheim M., Gross D., Halle J.P., Ogden J., Osterwalder B., Sedman E., Rossetti L. Translational medicine guide transforms drug development processes: the recent Merck experience. Drug Discov. Today. 2016;21:517–526. doi: 10.1016/j.drudis.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 121.Yu T.T.L., Gupta P., Vincent Ronfard A.A.V., Bayon Y. Recent progress in European advanced therapy medicinal products and beyond. Front. Bioeng. Biotechnol. 2018;6:130. doi: 10.3389/fbioe.2018.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]