
Figure 1.
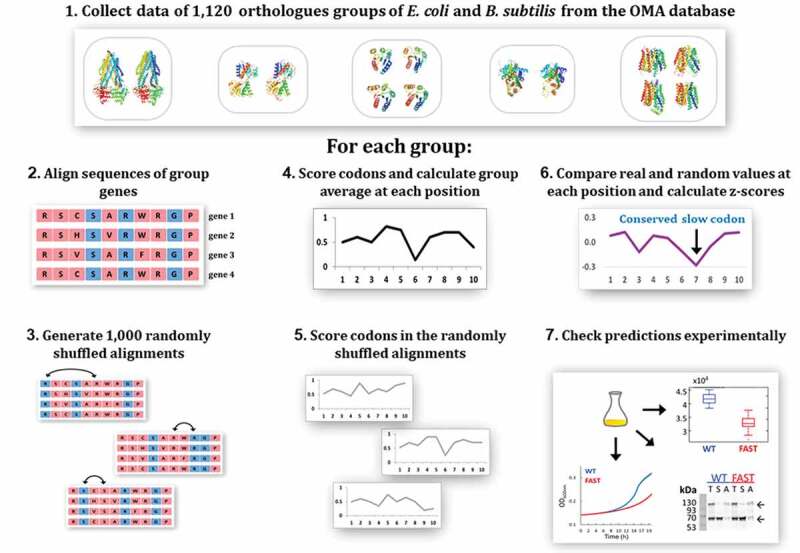
Overview of the methodology. 1. A list of 1,120 orthologue groups containing genes from both E. coli and B. subtilis was retrieved from the OMA database. For each one of these groups, steps 2–6 were performed. 2. The genes in the group were aligned using Clustal Omega. 3. A set of 1,000 randomized variants of the alignment was generated by shuffling synonymous codons along the sequence. 4. Codons in each gene were scored according to their predicted translation rate. The group average at each codon position was calculated. 5. Codon scoring and calculation of group average at each position was also performed for each of the 1,000 randomly-shuffled variants. 6. The ‘real’ codon score at each position was compared to the set of values received in that position for the 1,000 random variants and a z-score was calculated. A threshold of z-score = −2.8 was chosen as the bar under which a real codon score was considered to be significantly lower than control. Codons meeting this criterion are predicted by our model to have a functional role. 7. Out of the 57 homologous groups predicted by our model to contain functionally important slow codons, 8 were chosen for experimental validation. One gene from each of these groups was modified to replace the important slow codons with synonymous fast ones, and the modified gene was compared to the wild type gene in a set of experiments. See main text for details