

Summary
White lupin is an old crop with renewed interest due to its seed high protein content and high nutritional value. Despite a long domestication history in the Mediterranean basin, modern breeding efforts have been fairly scarce. Recent sequencing of its genome has provided tools for further description of genetic resources but detailed characterization of genomic diversity is still missing. Here, we report the genome sequencing of 39 accessions that were used to establish a white lupin pangenome. We defined 32 068 core genes that are present in all individuals and 14 822 that are absent in some and may represent a gene pool for breeding for improved productivity, grain quality, and stress adaptation. We used this new pangenome resource to identify candidate genes for alkaloid synthesis, a key grain quality trait. The white lupin pangenome provides a novel genetic resource to better understand how domestication has shaped the genomic variability within this crop. Thus, this pangenome resource is an important step towards the effective and efficient genetic improvement of white lupin to help meet the rapidly growing demand for plant protein sources for human and animal consumption.
Keywords: white lupin, pangenome, domestication, plant diversity
Introduction
White lupin (Lupinus albus L.) is a pulse whose domestication started about 3000–4000 years ago in the Mediterranean region (Taylor et al., 2020). It is cultivated for its seeds that contain high levels of proteins and are used both for food and feed (Wolko et al., 2011). Wild forms of white lupin (var. graecus) can only be found in Greece and adjoining Balkan region, with the earliest evidence of its use as a green manure and grain crop come from that same region (Kurlovich, 2002). Early Greek farmers selected larger seeds and white flowers, and presumably, removal of seed dormancy (water permeable seeds) was the earliest domestication trait. Greek and Roman literature suggests that seed indehiscence (i.e. resistance to pod shattering) had not yet been incorporated by the first century A.D. (Gladstones, 1998).
Wild collections and landraces of white lupin have high content of quinolizidine alkaloids that accumulate in their seed, resulting in a bitter taste and possible toxicity. Lysine‐derived alkaloids are characteristic of the Tribe Genisteae (Kinghorn et al., 1988; Wink and Mohamed, 2003; van Wyk, 2003), a monophyletic basal clade of the Fabaceae family. Traditionally, these bitter compounds were removed from white lupin seeds by soaking in water, a practice that is still carried out today across the Mediterranean and Nile regions (Taylor et al., 2020). However, this is uneconomic on a broad scale, that is, for animal feed production, which motivated the identification of low‐alkaloid mutants in Germany in the 1930s, aided by advances in chemistry (Gladstones, 1998). Modern cultivars of white lupin incorporate low‐alkaloid genes, hence, the term ‘sweet’ lupins.
White lupin breeding efforts have rarely been intensive or sustained over long periods. As a result, white lupin yields remain low and highly variable in comparison to other pulses like soybean for which intensive and sustained breeding efforts have been made internationally. Although white lupin cultivation represents a promising crop for Europe, in a political context aiming towards plant protein independence from American soybean imports, the lack of well‐characterized genetic resources has hampered a fast deployment of white lupin as an alternative crop to soybean imports. The recent sequencing of the white lupin genome (Hufnagel et al., 2020; Xu et al., 2020) demonstrated a resurgence of interest for this ‘old’ crop. We believe that white lupin intra‐genomic diversity might reflect the early traces of its slow and sporadic domestication history.
Here, we report a pangenome for white lupin that reveals important aspects of the species diversity, single nucleotide polymorphisms (SNPs), and gene presence–absence variations (PAVs). We construct a species pangenome consisting of ‘core’ genes that are present in all individuals and ‘variable’ (soft‐core or shell) genes that are absent in some individuals (Golicz et al., 2016b; Vernikos et al., 2015). Building on this comprehensive dataset, we were able to identify footprints of selection for low seed alkaloid content and candidate genes in these regions. Our analyses provide new perspectives on white lupin intra‐species diversity and domestication history.
Results
De novo assembly and pangenome construction
We gathered a set of 39 white lupin accessions, including 25 modern cultivars, 10 landraces, and 4 wild accessions from 17 countries that broadly represented diversity across the species (Table S1). Genome sequences of 15 of these accessions were available from a previous report (Hufnagel et al., 2020), whereas 24 accessions have been sequenced within this study to obtain broader species representation. Short‐read sequences have been assembled de novo for each accession (28.5× mean depth 150 bp paired‐end reads; Table S2).
The de novo assembly for each accession produced a total of 14.9 Gb of contigs longer than 500 base pairs (bp) with an N50 value (the minimum contig length needed to cover 50% of the assembly) of 24 475 bp. These de novo assemblies showed a mean complete BUSCOs score of 96.3%, a value similar to the Amiga reference genome (97.7%). Assembly completeness assessed by BUSCO was higher than 91.7% for all accessions, and in case of three accessions (Kiev, P27174, and Magnus), the score was similar to the reference genome (Figure 1a).
Figure 1.
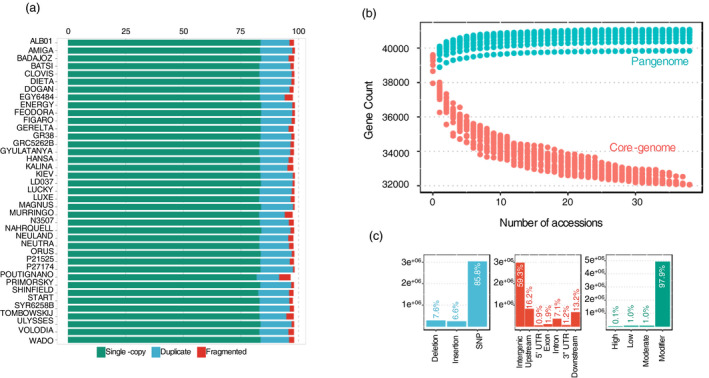
Pangenome of Lupinus albus. (a) BUSCO per cent completeness of all assemblies. All of the assemblies of this study have BUSCO completeness higher than 91.7%. A proportion of orthologs presented in single‐copy, duplicated, and genes that are fragmented in each assembly is shown. (b) Pangenome modelling. The modelling of the pangenome expansion predicts a closed pangenome with a total of 40 844 +/−289 genes. The core genome is predicted to contain 32 068 of these genes. (c) Distribution of variants along white lupin pangenome. Types of variations identified (blue); positioning of the variants in the genome in relation to the gene structures (red); impact of the variants (green).
The pangenome was built using a 'map‐to‐pan' approach (Hu et al., 2017) similar to the tomato pangenome (Gao et al., 2019). All de novo assembled contigs were compared with the reference genome to identify previously unknown sequences. A total of 270 Mb of nonreference sequence with identity <90% to the reference genome was obtained. After pangenome construction and removal of contaminants and overly repetitive sequences, we assembled an additional 3663 scaffolds, with a length greater than 2000 bp, for a total length of 11 733 253 bp. Using a threshold of a minimum 10× coverage, we identified 178 newly predicted protein‐coding genes, among which 61 could be annotated with gene ontology (GO) terms or Pfam domains (Dataset S1). The white lupin pangenome, including reference and nonreference genome sequences, had a total size of 462 705 661 bp and contained 38 446 protein‐coding genes. The total size of the constructed pangenome is compatible with nuclear DNA content estimates based on flow cytometry (Naganowska et al., 2003), which suggests that it represents the complete genome sequence of the species. We added to the White Lupin Genome portal (www.whitelupin.fr) dedicated user‐friendly tools for the exploitation of the pangenome, such as a BLAST tool for individual accessions, download of specific regions of accessions, and a genome browser mapping all the variants.
Core and variable genes
The presence or absence of each protein‐coding gene was predicted for each of the 39 accessions based on the mapping of reads from each accession to the pangenome assembly using SGSGeneLoss (Golicz et al., 2015). Likewise to other plants pangenomes (Gao et al., 2019; Golicz, Bayer, et al., 2016; Gordon et al., 2017; Montenegro et al., 2017; Yu et al., 2019; Zhao et al., 2020), we categorized genes in the white lupin pangenome according to their presence frequencies, using Markov clustering in the GET_HOMOLOGUES‐EST pipeline (Contreras‐Moreira et al., 2017). The majority of the genes, 32 068 (78.5%), are core genes shared by all the 39 accessions; 6,046 soft‐core (14.8%), being absent in 1 accession; and 8776 (21.4%) are shell, present in 2–37 accessions (Figure 1b). The size of the pangenome expanded with each additional accession to 38 446 genes, and extrapolation leads to a predicted pangenome size of 40 844 +/−289 genes (Figure 1b).
Single‐nucleotide polymorphism detection and annotation
To capture and broadly characterize white lupin diversity, we applied a strict variant discovery pipeline using GATK 4.1.0.0. A total of 9 442 876 raw SNPs were identified among the 39 accessions, 806 740 of which were recognized in the 24 newly assembled pangenome scaffolds. After filtering, 3 527 872 SNPs were retained in the 39 accessions, corresponding to a rate of 1 variant every 127 bp (Figure S1). The majority (85.8%) of the high‐quality variants are SNPs (3 027 761) and the other 501 111 variants detected are insertions and deletions (indels; Figure 1c—blue). Most variants (59.3%) are distributed on inter‐genic regions, 7.1% are within introns, and only 1.9% (96,576) of the variants are located in exons (Figure 1c—red). From the variants present in the CDS region, 4725 showed potentially large effects by causing start codon changes, premature stop codons, or elongated transcripts, and 50,478 are considered to produce a moderate effect by leading to codon changes in annotated genes. The frequency of these missense SNPs in the core gene set was 1 each 4.26 kb, which was lower than the variable gene set, with a rate of 1 for 1.84 kb. The rest of the variants lead to synonymous changes in proteins (low effect variants) or modifiers, causing changes outside the coding regions (Figure 1c—green). Collectively, this comprehensive dataset of the genome variation of white lupin provides a resource for biology and breeding of this species.
Population structure
To establish a phylogenetic benchmark for the analysis of the pangenome, we built a consensus maximum likelihood tree (Figure 1, 2) to infer the phylogenetic relationships for these L. albus accessions using a complete set of 3.5 m SNPs described above. This phylogenetic tree clustering supported six clades, which exhibited distinctive geographic origin and distinctive botanical features. In the Type 1 are grouped accessions with early‐flowering traits, including the Chilean agrogeotypes, and German and French accessions used in breeding programmes. This group also included the widely used cv. Kiev Mutant, which was generated by mutagenesis techniques, as well as other accessions that are derived from the same breeding program in Ukraine [Primorsky and Dieta, (Kurlovich, 2002)]. Type 2 is also composed by accessions with early flowering, a number of which have characteristics of Polish agroecotypes described by Kurlovich (Kurlovich, 2002) and are adapted to grow in Eastern Europe. One of the most representative accessions of this group is the cv. Kalina (Kurlovich, 2002), an old cultivar created in the Polish breeding program sharing similar genetic background with the broadly used Russian cultivar Start. Interestingly, Start is reported to carry different early‐flowering genes than Kiev Mutant (Adhikari et al., 2011). Type 2 also comprises two landraces from Syria and Israel/Palestine. Type 3 encompasses autumn‐sown genotypes with strong vernalization requirement and dwarf phenotype from the French breeding programme and the Algerian landrace ALB01. Algerian landraces are also reported to have a strong need of vernalization (Kurlovich, 2002). Type 4 comprises landraces from Iberian and Apennine Peninsula together with the described thermoneutral cultivars [i.e. Neutra (Wolko et al., 2011)]. Type 5 is composed only of Ethiopian landraces and Wild group is composed of the four graecus accessions of the panel, all wild accessions presenting small black‐speckled seeds and nondomesticated traits (hard seeds and shattering pods).
Figure 2.
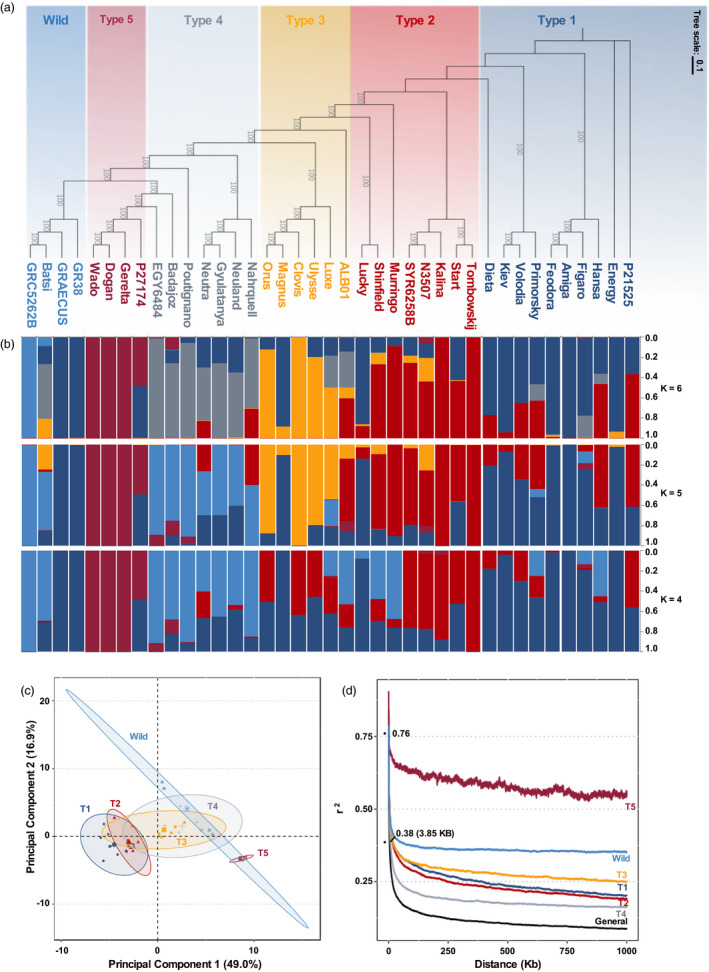
Phylogeny and population structure of 39 accessions of L. albus. (a) Maximum likelihood phylogenetic tree of white lupin constructed based on 3.5 m SNPs. The accessions are divided into six idiotypes. (b) Model‐based clustering analysis with different numbers of ancestral kinships (k = 4, 5, and 6). The y‐axis quantifies cluster membership and the x‐axis lists the different accessions. The positions of these accessions on the x‐axis are consistent with those in the phylogenetic tree. (c) Principal component analysis based on 3.5 m SNPs. The ellipses are discriminating the accessions of each idiotype groups. (d) Genome‐wide average LD decay estimated from different white lupin group. The decay of LD with physical distance between SNPs to half of the maximum values occurred at 3.85 kb (r 2 = 0.38) considering all accessions.
We examined genetic structure by performing a Bayesian model‐based clustering analysis and found that the six population groups matched the maximum‐likelihood tree (Figure 2b). This presented evidence of significant admixture in some lines and a weak population structure, a pattern already seen in other studies of L. albus (Raman et al., 2014). This weak population structure is also seen through the population‐differentiation statistic (F ST). The F ST value between all six groups was 0.27; however, F ST between Type 1 and Type 2 are as low as 0.086, and Type 4 and Wild have an F ST of 0.092. Indeed, regarding the Bayesian model, in scenarios dividing the accessions in 4 or 5 sub‐populations (Figure 2b, K = 4 and K = 5), accessions from Type 4 are merged with the Wild group. On the other hand, Type 5 showed a strong differentiation from the other groups, with F ST values ranging from 0.34 to 0.46, with Type 4 and Type 3, respectively, which is corroborating with previous studies (Raman et al., 2014). Principal component analysis reinforced the similarity among some groups (Figure 2c). The first two principal components explain 65.9% of genotypic variance and highlights the overlap among certain groups, in particular, Type 1 and Type 2.
Differentiation of genetic diversity between the six groups was investigated further through analysis of decay of linkage disequilibrium (LD, Figure 2d). The decay of LD with physical distance between SNPs to half of the maximum values occurred at 3.85 Kb (r 2 = 0.38), consistent with a high level of diversity and partially outcrossing mode of reproduction in this species (Green et al., 1980). Type 4 group also showed a fast LD decay of 5.7 Kb (r 2 = 0.40) and Type 1–3 groups have an average LD decay of 10.5 Kb. Wild group showed a slower LD decay (38.1 Kb, r 2 = 0.39) when compared with the other white lupin groups, presumably an effect of the small number of wild accessions in the analysis. Nevertheless, these LD decay levels can still be considered fast compared with other plant species, for example, rice (∼75–150 Kb, [Mather et al., 2007)], soybean [˜340–580 Kb, (Hyten et al., 2007)], or wheat [˜7–12.4 Mb, (Molero et al., 2019)], and also self‐pollinated crops. The Type 5 group (Ethiopian landraces) only reached half of its LD decay after 1.5 Mb, reinforcing the high similarity of its accessions and a possible genetic isolation of this group (Raman et al., 2014). The average nucleotide diversity π per site (Tajima, 1983) showed that diversity was five times lower in Type 5 group (π = 0.068) compared to the general nucleotide diversity (π = 0.372), while for the Wild group, although is also composed of only four accessions, showed a nucleotide diversity π = 0.402.
Protein‐coding genes presence and absence characterization
Presence and absence variants (PAVs) are an important type of structural variation and play an important role in shaping genomes, therefore, contributing to phenotypic diversity (Marroni et al., 2014). The construction of a white lupin pangenome allowed identification of 1195 PAVs, representing protein‐coding genes that are absent in at least one of the accessions, being 1132 genes from the reference genome and 63 from the newly identified genes (Dataset S2–S3). We further examined if the phylogenetic groups have an influence in the number of PAVs and if the PAVs are homogeneous within the groups (Figure ). The wild accessions have a significantly higher number of newly identified genes, with the accessions GRAECUS and GR38 only missing four of them. The four wild accessions share 157 of the 178 newly identified genes in the pangenome (Figure 3a).
Figure 3.
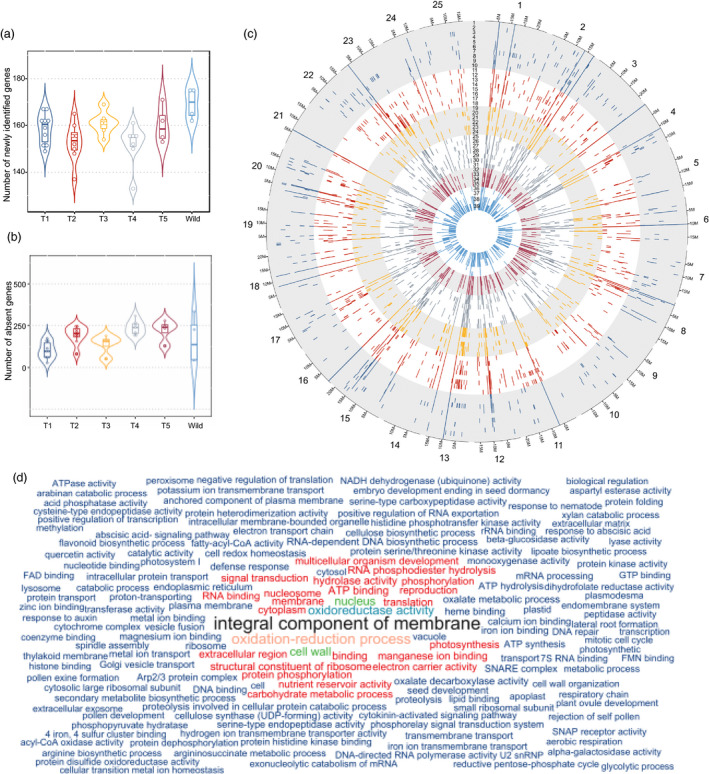
PAV of coding gene in L. albus. (a) Number of newly identified genes by phylogenetic group. (b) Number of absent genes by phylogenetic groups. (c) Positioning of absent genes in the 25 white lupin chromosomes in each 1 of the 39 accessions. Order of accessions from outer to inner track: 1‐AMIGA, 2‐FEODORA, 3‐FIGARO, 4‐ENERGY, 5‐KIEV MUTANT, 6‐HANSA, 7‐P21525, 8‐PRIMORSKY, 9‐DIETA, 10‐VOLODIA, 11‐START, 12‐N3507, 13‐TOMBOWSKIJ, 14‐KALINA, 15‐SYR6258B, 16‐LUCKY, 17‐MURRINGO, 18‐SHINFIELD, 19‐ALB01, 20‐LUXE, 21‐ULYSSE, 22‐MAGNUS, 23‐CLOVIS, 24‐ORUS, 25‐NAHRQUELL, 26‐GYUNLATANYA, 27‐NEULAND, 28‐NEUTRA, 29‐BADAJOZ, 30‐EGY6484B, 31‐POUTIGANO, 32‐P27174, 33‐GERELTA, 34‐DOGAN, 35‐WADO, 36‐GR38, 37‐GRAECUS, 38‐BATSI, and 39‐GRC5262B. The accessions’ colours reflect the six idiotypes. (d) Functional enrichment analysis of the variable genome. Graphical representation of enriched biological process (GOs). Size of the words and colours are proportional to their representativeness in the gene pool.
The number of missing genes within individual genomes ranges from 45 (Amiga—Type 1) to 348 genes (GRC5262B—Wild). Each group shares a median of 31 common lost genes among all its accessions and a total of 103 genes are absent in at least one accession of each group (Figure 3b). There are 137 genes that have been exclusively lost within accessions of the Wild group; however, only 30 genes are shared among all the graecus accessions. On the other hand, genomes of Ethiopian landraces (Type 5) share a total of 118 common missing genes, among which 39 are unique for this group. Remarkably, for this group, there is a concentration of lost genes on Chr17. This includes a set of nine tandem duplicated genes covering a region of 120 Kb (Figure S2). They are annotated as ‘Putative ferric‐chelate reductase (NADH)’ homologs of Arabidopsis gene FRO2, known for its role of iron uptake by the roots under stress condition (Connolly et al., 2003).
On checking the position of the PAVs on the chromosomes, we could identify some peculiarity regarding the PAVs within the groups. For example, on Chr13, there is a concentration of PAVs in the region of 5–10 Mb, which are missing from most accessions of Types 2–5 and Wild, but are present in the genomes of most Type 1 members. Similar pattern happens in the 3.6–6.4 Mb region of Chr04. Chr23 has the highest number of PAVs (78), a common feature of all the groups.
Functional analysis of PAVs suggests enrichment of GO terms as ‘integral component of membrane’ (GO:0016021) and ‘oxidation‐reduction process’ (GO:0055114) (Figure 3d, Figure S3 and Data S2). These suggest an enrichment of genes and gene families coding for membrane receptors proteins or membrane transporters. Other GO terms suggest that some of the genes may be involved in cell wall remodelling (‘cell wall’—GO:0005618 and ‘cell wall organization’—GO:0071555). Genes with these functions are frequently linked to biotic and abiotic stress responses (Novaković et al., 2018; Osakabe et al., 2013). PAV genes related to abiotic and biotic stress responses have been observed in several plant species (Bayer et al., 2019; Gao et al., 2019; Gordon et al., 2017; Montenegro et al., 2017; Shen et al., 2015; Zhao et al., 2020; Zhou et al., 2017) and these may reflect the evolution for adaptive traits related to each agroecotype. Moreover, the presence/absence of these stress response‐related genes may also be partially due to whole‐genome triplication event on white lupin genome (Hufnagel et al., 2020), which caused an overlapping roles in various loci.
Footprints of selection and alleles identification in candidate genes
To demonstrate the power of white lupin pangenome to address basic research questions, we used it to detect possible footprints of selection and to identify alleles in candidate genes underlying major QTLs. Firstly, to examine potential selective signals during white lupin domestication and breeding, we scanned white lupin genome searching for regions with marked reductions in nucleotide diversity (Figure 4a).
Figure 4.
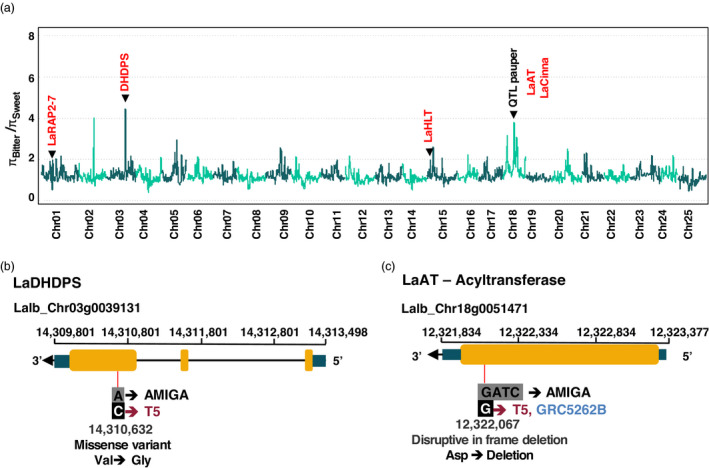
Footprints of selection in the white lupin genome. (a) Nucleotide diversity (π) comparison between bitter and sweet accessions. A major QTL previously reported for alkaloid accumulation and candidate genes (red) that overlapped with selective sweeps are marked. (b) Candidate gene located on chromosome 3. The gene LaDHDPS is homologue of L. angustifolius gene in the iucundus QTL. Type 5 accessions, originated from Ethiopia, have a SNP in the third exon that causes a mis‐sense variant. (c) Candidate gene located on chromosome 18. The gene LaAT is located inside QTL Pauper and Type 5 accessions and wild accessions GRC5262B have a deletion that causes a disruption in frame of its exon.
The domestication and breeding efforts in white lupin have focused on searching for accessions with reduced seed alkaloid content, reduced time to flower, as well as reduced branching. Therefore, we combined all the accessions representing cultivars and breeding lines, which have low‐alkaloids seeds (sweet), and compared them with landrace and wild accessions that have high‐alkaloid seeds (bitter) (Figure 4a, Table S3). A selective sweep affecting only the sweet white lupin accessions would be expected to leave a typical low‐polymorphism and high‐divergence signal around the region of the selected genes. We measured the sweep on the nucleotide diversity [π value (Tajima, 1983)] by comparing the two groups (πBitter/πSweet) over 250‐kb windows. We identified 333 putative selection sweeps associated with the breeding of the low‐alkaloid accessions (πBitter/πSweet > 1.512, 0.90 percentile). We observed a prominent peak on chromosome 18, in the region of a previously reported major white lupin QTL for low‐alkaloid content, which corresponds to the pauper locus (Książkiewicz et al., 2017; Lin et al., 2009).
Interestingly, other peaks with high sweeps of diversity are present, indicating that other genomic regions may be implicated with this trait and may carry other important genes of these pathways. There are 14 peaks in the 0.99 percentile (πBitter/πSweet > 2.324) that are distributed along 7 distinct chromosomes. Furthermore, they highlight specific genomic regions of sweet accessions that have been selected during domestication and breeding.
We searched for homologs of alkaloid‐related genes discovered in the close relative narrow‐leafed lupin and we identified that some of them co‐localized in peak regions (Figure 4a). A homolog of the L. angustifolius gene RAP2‐7, a transcription factor from the APETALA2/ethylene response (AP2/ERF) family (Kroc et al., 2019), was co‐located with a peak in the chromosome 1 (LaRAP2‐7, Lalb_Chr01g0010481). Likewise, a homolog LaDHDPS (Lalb_Chr03g0039131), which encodes a chloroplast 4‐hydroxy‐tetrahydrodipicolinate synthase, is located in a peak on chromosome 3. Both genes are in syntenic regions of narrow‐leafed lupin genome and are inside or in proximity of iucundus alkaloid QTL in narrow‐leafed lupin. Interestingly, the gene LaDHDPS presented a mis‐sense SNP in the third exon that is causing an impacting amino acid change in the enzyme (Figure 4b). The alternative allele C is present only in the Type 5 accessions, which are bitter.
Similarly, the gene LaHLT (Lalb_Chr15g0077591, Hufnagel et al., 2020), a acyltransferase, is located on a peak on chromosome 15. Another acyltransferase (LaAT, Lalb_Chr18g0051471) is located in the region of the pauper QTL, and is a candidate gene to be underlying this major loci (Książkiewicz et al., 2017). Interestingly, a small deletion is present inside the single exon of this gene, which causes a disruption in its reading frame in accessions Type 5 and in wild accession GRC5262B (Figure 4c). However, there are 66 genes described within this pauper QTL region (Hufnagel et al., 2020), including a cinnamoyl‐CoA reductase (LaCinna, Lalb_Chr18g0051351), which seems to be involved in the synthesis of derived compounds or conjugates of quinolizidine alkaloids in Lupinus angustifolius (Czepiel et al., 2021).
Discussion
A pangenome is a complete set of genes for a species, including core genes that are present in all individuals, and variable genes that are absent in one or more individuals (Golicz et al., 2016a). We generated a de novo assembly for 38 white lupin accessions and, taking advantage of a good reference assembly for the species (Hufnagel et al., 2020), we constructed a L. albus pangenome by iteratively and randomly sampling these sequenced accessions. This dataset is a representative of the diversity of the species, containing wild accessions, landraces, and cultivars of white lupin from across their respective distributions. As a result, we estimate that this white lupin pangenome assembly effectively encompasses the complete sequence for the genome of the species, with 462.7 Mb sequence and containing 38 446 protein‐coding genes. The finding that 21.5% of genes in the pangenome exhibit varying degrees of genic presence/absence variants (PAVs) highlights the diverse genetic feature of white lupin and the significant improvement of the reference genome by including genomic information of other accessions and discovery of new genes. Remarkably, the white lupin pangenome showed a high content of core genes (78.5%), as compared with other plant species such as tomato [74.2%, (Gao et al., 2019)], Arabidopsis thaliana [70%, (Contreras‐Moreira et al., 2017)], bread wheat [64%, (Montenegro et al., 2017)], sesame [58%, (Yu et al., 2019)], and wild soybean [49%, (Li et al., 2014)], which might be a reflection of its domestication history and modest breeding efforts to date.
The domestication of white lupin started during the Bronze Age (Gladstones, 1998), and the ancestral history of this species is different from other major crops such as rice, maize, sorghum, tomato, and soybeans, which are more ancient (Diamond, 2002). The early cultivated forms have the same Balkan region distribution as that of its wild ancestor types (graecus). L. albus domestication was slow with potentially centuries between acquisition of each domestication trait, which may explain why there is not a more pronounced genetic differentiation among wild, landrace, and cultivated types (Wolko et al., 2011). Our pangenome highlights that there was no genomic bottleneck breeding associated with this specie, with no big gene loss between cultivated and wild accessions. This is echoed in the lack of population structure presented within these accessions and in the low LD extent, which generally reduce the diversity and change allele frequencies either to fixation or intermediate frequencies (Hamblin and Jannink, 2011). Despite being a largely self‐pollinating crop [with an out‐crossing rate reported as 8–10% (Green et al., 1980)], white lupin showed a remarkably low degree of LD (<4 kb), even lower than the wild population of its relative, narrow‐leafed lupin that showed a decay of LD after 19.01 Kb (Mousavi‐Derazmahalleh, Nevado, et al., 2018). One distinction between these closely related species is that narrow‐leafed lupin is almost exclusively self‐pollinating and so the modest levels of outcrossing in white lupin may be a key factor governing the differences in LD between these two species. Having a low LD and weak population structure together means that association mapping is likely to be particularly powerful in white lupin, in contrast to the more highly structured and high LD species narrow‐leafed lupin, where association studies have so far proved rather weak (Mousavi‐Derazmahalleh, Bayer, et al., 2018; Mousavi‐Derazmahalleh, Nevado, et al., 2018).
Type 5 accessions, from Ethiopia, are the only group that showed a strong genetic differentiation from the others, with F ST values higher than 0.3. Such a distinct separation is evidence that the Ethiopian accessions have evolved in isolation and the genetic differences are probably due to ancient founder effects. The differences of Type 5 group are also highlighted by the PAVs. Together with the Wild group, Ethiopian landraces carry most of the new identified genes and also miss a large number of genes of the reference genome (Figure 3). Moreover, it is a highly homogeneous group, with all accessions sharing a large number of these lost genes. The loss of these genes might be an adaptive response for the local environment. For instance, the loss of the nine tandem duplicated homologue AtFRO2 on Chr17 might be an adaptive response to highland Ethiopian soils that are iron rich (Eyasu, 2016). A more detailed look into the PAVs among the different groups may be useful to better understand their specificities.
Our analysis brings a high resolution to the within‐species diversity. Using the pangenome dataset, we performed genome‐wide comparisons of the assemblies, enabling the characterization of more than 3 million complex variants, including many large‐effect coding variants that should be helpful in pinpointing causal variations in QTLs for important traits and in future genome‐wide association studies. In particular, our study demonstrated that 4725 genes were found to contain important coding variation in at least one accession and might have important biological functions underlying the variation in complex traits.
We used the power of pangenome to identify allelic differences that are responsible for phenotypic variation. By performing a genome‐wide analysis, we detected that nucleotide diversity was quite variable across the genome. The efforts of breeding in white lupin have been focused on combining domestication traits such as soft and white seeds and reduced pod shattering, which were already available from ancient times, with that of reduced alkaloids, increased yield, and the reduction in flowering time and excessive branching (Wolko et al., 2011). Looking for differences in nucleotide diversity across the genome among breeding accessions and comparing with landraces/wild accessions, we could detect some selective sweeps that reduced diversity in cultivated versus wild types. In these peaks, there is an important decrease in nucleotide diversity within the breeding lines and they represent marks of selection (Figure 4). However, although the sweeps of diversity co‐localize with an identified major QTL for low‐alkaloid content, there are other peaks along the chromosomes. Indeed, other sources of natural low‐alkaloid mutations were already described besides pauper (i.e. exiguus, mitis, nutricius, and reductus), but they have not been identified so far. These regions should be explored in order to find genes underlying phenotypic traits that have been selected directly or indirectly during domestication and breeding of white lupin. For instance, white lupin is known for thriving in soils with low nutrient availability by producing specialized root structures called cluster roots (Lambers et al., 2012). In a previous work, we demonstrated that the breeding accessions have an earlier establishment of the root system through lateral and cluster root formation that was indirectly selected (Hufnagel et al., 2020). By looking closer into these chromosome regions with low nucleotide diversity and high genetic differentiation, we might be able to find genes with important roles in the root architecture of white lupin. Hence, integration of the information from studies of gene function and the high density of variants described in this pangenome can provide a complementary approach to forward genetic studies and can contribute to develop the research and breeding of white lupin.
Conclusion
In summary, the white lupin pangenome comprises a wealth of information on genetic variation that has yet to be fully exploited by researchers and breeders. Although there is a large collection of white lupin accessions available in gene banks worldwide, they barely have been explored and genetically characterized. This pangenome represents a comprehensive and important resource to facilitate the exploration of white lupin as a legume model for future functional studies and molecular breeding.
Methods
Genome sequences of white lupin accessions
We retrieved the genome sequencing data of 15 white lupin accessions that were published previously (Hufnagel et al., 2020), including 11 modern cultivars, 1 landrace, and 2 wild accessions. They were sequenced using Illumina technology using paired‐end 2 × 150 bp short reads with average sequencing depth of 45.99×. It included Illumina genome data of 64.47× depth for the reference cultivar ‘Amiga’. Genome sequences of additional 24 accessions were generated here, including 12 modern cultivars, 9 landraces, and 2 wild accessions. Young leaves of individual plants were used to extract genomic DNA of each accession using the QIAGEN DNeasy Plant Mini kit following the supplier’s recommendations. The accessions were sequenced using Illumina technology using paired‐end 2 × 150 bp short reads (Macrogen, South Korea). It was generated a total of 196.85 Gb of data with average sequencing depth of 19.1x. (Table S2).
De novo genome assembly and pangenome construction
Reads were processed to trim adapters and low‐quality sequences using Cutadapt 1.15 (Bolger et al., 2014) with parameters ‘‐‐pair‐filter=any ‐q20,20 ‐m 35’ and the forward and reverse Illumina TruSeq Adapters. The final high‐quality, cleaned Illumina reads from each sample were de novo assembled using Spades 3.13.0 (Bankevich et al., 2012) with k‐mer size of 21,33,55,77,99,121. The assembled contigs were then aligned to the white lupin reference genome (Hufnagel et al., 2020) (GenBank accession no.: WOCE00000000, http://www.whitelupin.fr.), using the steps 7 and 8 of the EUPAN Pipeline (Hu et al., 2017), in order to extract contigs that were not aligning to the reference. Then, redundancy in the extracted contigs has been reduced using CD‐hit 4.8.1 with default parameters. The resulting contigs were then searched against the NCBI nt nucleotide database using blastn 2.10 (Camacho et al., 2009). Sequences with best hits from outside the Eudicots, or covered by known plant mitochondrial or chloroplast genomes, were possible contaminations and were, therefore, removed. The BUSCO v 3.1.0 (Waterhouse et al., 2018) was run on the set of predicted transcripts to access the completeness of each genome assembly.
Annotation of the white lupin pangenome
A custom repeat library was constructed by screening the pangenome and the white lupin reference genome using RepeatModeler (http://www.repeatmasker.org/RepeatModeler/), and used to screen the nonreference genome to identify repeat sequences using RepeatMasker (http://www.repeatmasker. org/). Contigs with more than 98% of repetitive sequences were removed from the annotation pipeline. Protein‐coding genes were predicted from nonreference genome using MAKER2 (Yandell and Holt, 2011). Ab initio gene prediction was performed using Augustus (Stanke and Morgenstern, 2005) and SNAP (Korf, 2004). Augustus (Stanke et al., 2008) has been previously trained for white lupin as described in the documentation, and SNAP was trained for two rounds based on already assembled transcriptome of white lupin, as described in maker2 documentation. In addition, protein sequences of white lupin, Medicago truncatula, and the Viridiplantae subset of Swissprot were used as evidence. Finally, gene predictions based on ab initio approaches, and transcript and protein evidence were integrated using the MAKER2 pipeline. A set of high‐confidence gene models supported by transcript and/or protein evidence were generated by MAKER2. In order to remove possible remaining contamination, all high‐confidence, maker‐generated, protein sequences were aligned against the nr databases, and sequences with best hits from outside Eudicots or with best hit inside chloroplastic and mitochondrial sequences were removed. Genes that matched white lupin reference sequences were also removed in the same way.
In parallel, contigs with a length superior to 2 Kb from the whole assembly of the 39 lupin accession were annotated using the Egnep 1.5.1 pipeline (Sallet et al., 2014). RepeatMasker was used to detect and remove contigs constitute by more than 98% of known repeat sequences based on the previously built white lupin repetitive‐element sequences database. The white lupin transcriptome (Hufnagel et al., 2020) was used as ESTs evidence, using a minimum identity percentage of 95%, along with the proteome of white lupin, Medicago truncatula, and the Viridiplantae subset of the Swissprot database, with weight of 0.4, 0.3, and 0.3, respectively. Resulting predicted proteins were searched against REXdb in order to remove possible transposable elements. The resulting genes prediction was again scanned with repeat masker, and genes composed of more than 90% of detected repetitive sequences were removed from further analyses in order to control false positive.
Gene presence/absence variation and pangenome modelling
Reads were processed to trim adapters and low‐quality sequences using Cutadapt 1.15 (Bolger et al., 2014) with parameters ‘‐‐pair‐filter=any ‐q20,20 ‐m 35’ and the forward and reverse Illumina TruSeq adapters. Resulting high‐quality reads were then aligned to the pangenome using BWA‐MEM (Li and Durbin, 2010) with default parameters. Picard tools were used to remove possible PCR and optical duplicates, and reads considered as not properly paired were removed using samtools view. The presence or absence of each gene in each accession was determined using SGSGeneLoss (Golicz et al., 2015). In brief, for a given gene in a given accession, if <10% of its exon regions were covered by at least five reads (minCov = 5, lostCutoff = 0.1), this gene was treated as absent in that accession, otherwise it was considered present. The parameters used for the new set of gene discovered in the pangenome were different: minCov = 10 and lostCutoff = 0.8. For more precise pangenome studies, taking into account all the genes discovered in all the different varieties, GET_HOMOLOGUES_EST was used on the whole CDS and proteome of the whole 39 varieties with parameters ‘‐R 123545 ‐P ‐M ‐c ‐z ‐A ‐t 2’ to detect clusters of genes shared by at least two varieties.
SNP discovery and annotation
Cutadapt (Martin, 2011) was used to remove Illumina Truseq adapters from the sequencing data and to remove bases with a quality score lower than 30, in both 5’ and 3’ end of the reads. Reads with a length lower than 35 were discarded. We then used BWA‐MEM version 0.7.17 (Li and Durbin, 2010) to map the re‐sequencing reads from all 39 genotypes to the white lupin reference genome. PCR and optical duplicates were detected and removed using Picard Tools. After that, GATK 4 HaplotypeCaller tool was used in emit‐ref‐confidence GVCF mode to produce one gvcf file per sample. These files were merged using GATK Combine GVCFs. Finally, GATK GenotypeGVCFs were used to produce a vcf file containing variants from all the 39 samples. This identified a total of 9 442 876 SNPs/indel. After filtering for minimum allele frequency of 0.15 and heterozygosity frequency of 0–0.2, 3 527 872 SNPs were retained for further analysis.
Evolutionary analysis
A maximum‐likelihood phylogenetic tree was constructed based on 3 121 673 parsimony‐informative SNPs with 1000 bootstraps using IQ‐TREE (Nguyen et al., 2015), using ModelFinder (Kalyaanamoorthy et al., 2017) option. Then, a phylogenetic tree was prepared using the iTOL v 4.3 (Letunic and Bork, 2016).
Population structure based on the same set of SNPs was investigated using STRUCTURE (Hubisz et al., 2009). Thirty independent runs for each K from 1 to 15 were performed with an admixture model at 50 000 Markov chain Monte Carlo (MCMC) iterations and a 10 000 burn‐in period. Principal component analysis using this SNP dataset was performed using the function ‘princomp’ in R (http://www.R‐project.org/). The linkage disequilibrium (LD) pattern was computed using PopLDdecay v3.40 (Zhang et al., 2019). LD decay was measured on the basis of the r 2 value and the corresponding distance between two given SNPs.
Selective sweep analyses
To detect genomic regions affected by domestication, we used the same set of 3 121 673 SNPs using Tassel (Bradbury et al., 2007). The level of genetic diversity (π) was measured with a window size of 2000 SNPs and a step size of the same length, generating windows of approximately 250 kb. Genome regions affected by selection or domestication should have substantially lower diversity in sweet white lupin accessions (cultivars and breeding lines) than the diversity in bitter accessions (landraces and wild types). The accession EGY6484B was removed from the analysis due to its inconclusive status of bitterness. Windows with the top 10% highest ratios of πBitter/πSweet (≥1.512) were selected as candidate selection and domestication sweeps.
Conflict of interests
The authors declare that they have no competing interests.
Authors' contributions
A.S. developed bioinformatic resources and performed pangenome assembly. J.T. and F.D. performed DNA extraction and experiments. M.N., H.S., L.Y., and M.K. provided genetic material. B.H. performed data analysis. B.H., M.K., M.N., and B.P. designed experiments and wrote the article.
Funding
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Starting Grant LUPINROOTS—grant agreement No. 637420 to B.P.) and from the Innovate UK project 133048 (Ethiopian Lupins for Food and Feed) to H.S.
Data Availability Statement
The detailed methods and datasets supporting the conclusions of this report are included within the article and its additional files. All deep sequencing data reported in this study have been submitted to the NCBI. The datasets generated and analysed during the current study are available from the corresponding author upon request. Full genomic and raw sequence data are publicly available for download on the White Lupin genome portal [www.whitelupin.fr/pangenome] that contains a Genome Browser, Expression tools, and a Sequence retriever dedicated to the pangenome. The pangenome project and raw data have been deposited at DDBJ/ENA/GenBank under the accession PRJNA608889.
Supporting information
Figure S1 Variants distribution across the Lupinus albus genome.
Figure S2 Deletion of nine tandem duplicated genes on Chr17 of Ethiopian accessions of Lupinus albus.
Figure S3 Functional enrichment analysis of variable genome of Lupinus albus.
Table S1 Accessions of white lupin used in the pangenome construction.
Table S2 Summary of re‐sequencing of white lupin accessions using short reads produced in this report.
Table S3 Class and alkaloid status of pangenome white lupin accessions.
Acknowledgements
We thank J. B. Magnin‐Robert (INRA Agroécologie, Dijon, France), Nathalie Harzic (Jouffray‐Drillaud, France), Paolo Annicchiarico (Council for Agricultural Research and Economics, Milan, Italy), and David McNaughton (Soya UK, UK) for providing seeds from the different accessions. This work was supported by CIRAD–UMR AGAP HPC Data centre of the South Green bioinformatics platform (http://www.southgreen.fr/).
Hufnagel, B. , Soriano, A. , Taylor, J. , Divol, F. , Kroc, M. , Sanders, H. , Yeheyis, L. , Nelson, M. and Péret, B. (2021) Pangenome of white lupin provides insights into the diversity of the species. Plant Biotechnol. J., 10.1111/pbi.13678
Contributor Information
Bárbara Hufnagel, Email: barbara.hufnagel@supagro.fr.
Benjamin Péret, Email: benjamin.peret@cnrs.fr.
References
- Adhikari, K. , Buirchell, B. , Yan, G. and Sweetingham, M. (2011) Two complementary dominant genes control flowering time in albus lupin (Lupinus albus L.). Plant Breed. 130, 496–499. [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A.A. , Dvorkin, M. , Kulikov, A.S. , Lesin, V.M. et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. J. Comput. Biol. 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer, P.E. , Golicz, A.A. , Tirnaz, S. , Chan, C.K.K. , Edwards, D. and Batley, J. (2019) Variation in abundance of predicted resistance genes in the Brassica oleracea pangenome. Plant Biotechnol. J. 17, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. and Madden, T.L. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, E.L. , Campbell, N.H. , Grotz, N. , Prichard, C.L. and Guerinot, M.L. (2003) Overexpression of the FRO2 ferric chelate reductase confers tolerance to growth on low iron and uncovers posttranscriptional control. Plant Physiol. 133, 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras‐Moreira, B. , Cantalapiedra, C.P. , García‐Pereira, M.J. , Gordon, S.P. , Vogel, J.P. , Igartua, E. et al. (2017) Analysis of plant pan‐genomes and transcriptomes with Get_HOMOLOGUES‐EST, a clustering solution for sequences of the same species. Front. Plant Sci. 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czepiel, K. , Krajewski, P. , Wilczura, P. , Bielecka, P. , Święcicki, W. and Kroc, M. (2021) Expression profiles of alkaloid‐related genes across the organs of narrow‐leafed lupin (Lupinus angustifolius l.) and in response to anthracnose infection. Int. J. Mol. Sci. 22, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, J. (2002) Evolution, consequences and future of plant and animal domestication. Nature, 418, 700–707. [DOI] [PubMed] [Google Scholar]
- Eyasu, E. (2016) Soils of the Ethiopian Highlands: Geomorphology and Properties. The Nertherlands: CASCAPE Project, ALTERA, Wageningen University and Research Centre. 385 pp. [Google Scholar]
- Gao, L. , Gonda, I. , Sun, H. , Ma, Q. , Bao, K. , Tieman, D.M. et al. (2019) The tomato pan‐genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 51, 1044–1051. [DOI] [PubMed] [Google Scholar]
- Gladstones, J.S. (1998) Distribution, origin. taxonomy, history and importance. In: Lupins as Crop Plants: Biology. Production and Uti‐lization ( Gladstones, J.S. , Atkins, C. and Hamblin, J. , eds), pp. 1–39. Oxon, New York: CAB International. [Google Scholar]
- Golicz, A.A. , Batley, J. and Edwards, D. (2016) Towards plant pangenomics. Plant Biotechnol. J. 14, 1099–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz, A.A. , Bayer, P.E. , Barker, G.C. , Edger, P.P. , Kim, H. , Martinez, P.A. et al. (2016) The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 7, 13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz, A.A. , Martinez, P.A. , Zander, M. , Patel, D.A. , Van De Wouw, A.P. , Visendi, P. et al. (2015) Gene loss in the fungal canola pathogen Leptosphaeria maculans . Funct. Integr. Genomics, 15, 189–196. [DOI] [PubMed] [Google Scholar]
- Gordon, S.P. , Contreras‐Moreira, B. , Woods, D.P. , Des Marais, D.L. , Burgess, D. , Shu, S. et al. (2017) Extensive gene content variation in the Brachypodium distachyon pan‐genome correlates with population structure. Nat. Commun. 8, 2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, A. , Brown, A. and Oram, R. (1980) Determination of outcrossing rate in a breeding population of Lupinus albus L. (White Lupin). Plant Breed. 84, 181–191. [Google Scholar]
- Hamblin, M.T. and Jannink, J.‐L. (2011) Factors affecting the power of haplotype markers in association studies. Plant Genome J. 4, 145. [Google Scholar]
- Hu, Z. , Sun, C. , Lu, K.‐C. , Chu, X. , Zhao, Y. , Lu, J. , Shi, J. et al. (2017) EUPAN enables pan‐genome studies of a large number of eukaryotic genomes. Bioinformatics, 33, 2408–2409. [DOI] [PubMed] [Google Scholar]
- Hubisz, M.J. , Falush, D. , Stephens, M. and Pritchard, J.K. (2009) Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel, B. , Marques, A. , Soriano, A. , Marquès, L. , Divol, F. , Doumas, P. et al. (2020) High‐quality genome sequence of white lupin provides insight into soil exploration and seed quality. Nat. Commun. 11, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyten, D.L. , Choi, I.‐Y. , Song, Q. , Shoemaker, R.C. , Nelson, R.L. , Costa, J.M. , Specht, J.E. et al. (2007) Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics, 175, 1937–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy, S. , Minh, B.Q. , Wong, T.K.F. , von Haeseler, A. and Jermiin, L.S. (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods, 14, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinghorn, A.D. , Hussain, R.A. , Robbins, E.F. , Balandrin, M.F. , Stirton, C.H. and Evans, S.V. (1988) Alkaloid distribution in seeds of Ormosia, Pericopsis and Haplormosia. Phytochemistry, 27, 439–444. [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5. 10.1186/1471-2105-5-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroc, M. , Koczyk, G. , Kamel, K.A. , Czepiel, K. , Fedorowicz‐Strońska, O. , Krajewski, P. , Kosińska, J. et al. (2019) Transcriptome‐derived investigation of biosynthesis of quinolizidine alkaloids in narrow‐leafed lupin (Lupinus angustifolius L.) highlights candidate genes linked to iucundus locus. Sci. Rep. 9, 2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Książkiewicz, M. , Nazzicari, N. , Yang, H. , Nelson, M.N. , Renshaw, D. , Rychel, S. , Ferrari, B. et al. (2017) A high‐density consensus linkage map of white lupin highlights synteny with narrow‐leafed lupin and provides markers tagging key agronomic traits. Sci. Rep. 7, 15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurlovich, B.S. ed. (2002) Lupins: Geography, Classification, Genetic Resources and Breeding. St. Petersburg: Publishing House “Intan.” [Google Scholar]
- Lambers, H. , Bishop, J.G. , Hopper, S.D. , Laliberté, E. and Zúñiga‐Feest, A. (2012) Phosphorus‐mobilization ecosystem engineering: the roles of cluster roots and carboxylate exudation in young P‐limited ecosystems. Ann. Bot. 110, 329–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. and Bork, P. (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2010) Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics, 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y.H. , Zhou, G. , Ma, J. , Jiang, W. , Jin, L.G. , Zhang, Z. et al. (2014) De novo assembly of soybean wild relatives for pan‐genome analysis of diversity and agronomic traits. Nat. Biotechnol. 32, 1045–1052. [DOI] [PubMed] [Google Scholar]
- Lin, R. , Renshaw, D. , Luckett, D. , Clements, J. , Yan, G. , Adhikari, K. , Buirchell, B. et al. (2009) Development of a sequence‐specific PCR marker linked to the gene “pauper” conferring low‐alkaloids in white lupin (Lupinus albus L.) for marker assisted selection. Mol. Breed. 23, 153–161. [Google Scholar]
- Marroni, F. , Pinosio, S. and Morgante, M. (2014) Structural variation and genome complexity: is dispensable really dispensable? Curr. Opin. Plant Biol. 18, 31–36. [DOI] [PubMed] [Google Scholar]
- Martin, M. (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J. 17, 10. [Google Scholar]
- Mather, K.A. , Caicedo, A.L. , Polato, N.R. , Olsen, K.M. , McCouch, S. and Purugganan, M.D. (2007) The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics, 177, 2223–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero, G. , Joynson, R. , Pinera‐Chavez, F.J. , Gardiner, L. , Rivera‐Amado, C. , Hall, A. and Reynolds, M.P. (2019) Elucidating the genetic basis of biomass accumulation and radiation use efficiency in spring wheat and its role in yield potential. Plant Biotechnol. J. 17, 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenegro, J.D. , Golicz, A.A. , Bayer, P.E. , Hurgobin, B. , Lee, H. , Chan, C.‐K.‐K. et al. (2017) The pangenome of hexaploid bread wheat. Plant J. 90, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Mousavi‐Derazmahalleh, M. , Bayer, P.E. , Nevado, B. , Hurgobin, B. , Filatov, D. , Kilian, A. et al. 2018) Exploring the genetic and adaptive diversity of a pan‐Mediterranean crop wild relative: narrow‐leafed lupin. Theor. Appl. Genet. 131, 887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi‐Derazmahalleh, M. , Nevado, B. , Bayer, P.E. , Filatov, D.A. , Hane, J.K. , Edwards, D. et al. (2018) The western Mediterranean region provided the founder population of domesticated narrow‐leafed lupin. Theor. Appl. Genet. 131, 2543–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganowska, B. , Wolko, B. , Śliwińska, E. and Kaczmarek, Z. (2003) Nuclear DNA content variation and species relationships in the genus Lupinus (Fabaceae). Ann. Bot. 92, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt, H.A. , von Haeseler, A. and Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novaković, L. , Guo, T. , Bacic, A. , Sampathkumar, A. and Johnson, K.L. (2018) Hitting the wall‐sensing and signaling pathways involved in plant cell wall remodeling in response to abiotic stress. Plants (Basel, Switzerland), 7, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakabe, Y. , Yamaguchi‐Shinozaki, K. , Shinozaki, K. and Tran, L.S.P. (2013) Sensing the environment: Key roles of membrane‐localized kinases in plant perception and response to abiotic stress. J. Exp. Bot. 64, 445–458. [DOI] [PubMed] [Google Scholar]
- Raman, R. , Cowley, R.B. , Raman, H. and Luckett, D.J. (2014) Analyses using SSR and DArT molecular markers reveal that ethiopian accessions of white lupin (Lupinus albus L.) represent a unique genepool. Open J. Genet. 04, 87–98. [Google Scholar]
- Sallet, E. , Gouzy, J. and Schiex, T. (2014) EuGene‐PP: a next‐generation automated annotation pipeline for prokaryotic genomes. Bioinformatics, 30, 2659–2661. [DOI] [PubMed] [Google Scholar]
- Shen, X. , Liu, Z.Q. , Mocoeur, A. , Xia, Y. and Jing, H.C. (2015) PAV markers in Sorghum bicolour: genome pattern, affected genes and pathways, and genetic linkage map construction. Theor. Appl. Genet. 128, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Diekhans, M. , Baertsch, R. and Haussler, D. (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, 24, 637–644. [DOI] [PubMed] [Google Scholar]
- Stanke, M. and Morgenstern, B. (2005) AUGUSTUS: a web server for gene prediction in eukaryotes that allows user‐defined constraints. Nucleic Acids Res. 33, W465–W467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F. (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics, 105, 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J.L. , De Angelis, G. and Nelson, M.N. (2020) How have narrow‐leafed lupin genomic resources enhanced our understanding of lupin domestication?. In The Lupin Genome, ( Singh, K. B. , Kamphuis, L. G. & Nelson, M. N. eds.), pp. 95–108. Cham: Springer. [Google Scholar]
- Vernikos, G. , Medini, D. , Riley, D.R. and Tettelin, H. (2015) Ten years of pan‐genome analyses. Curr. Opin. Microbiol. 23, 148–154. [DOI] [PubMed] [Google Scholar]
- Waterhouse, R.M. , Seppey, M. , Simao, F.A. , Manni, M. , Ioannidis, P. , Klioutchnikov, G. et al. (2018) BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wink, M. and Mohamed, G.I.A. (2003) Evolution of chemical defense traits in the Leguminosae: mapping of distribution patterns of secondary metabolites on a molecular phylogeny inferred from nucleotide sequences of the rbcL gene. Biochem. Syst. Ecol. 31, 897–917. [Google Scholar]
- Wolko, B. , Clements, J.C. , Naganowska, B. , Nelson, M. and Huaan, Y. (2011) Lupinus ( Kole, C. , ed.). Wild Crop Relatives: Genomic and Breeding Resources. Berlin, Heidelberg: Springer Berlin Heidelberg. pp. 153–206. [Google Scholar]
- van Wyk, B.‐E. (2003) The value of chemosystematics in clarifying relationships in the genistoid tribes of papilionoid legumes. Biochem. Syst. Ecol. 31, 875–884. [Google Scholar]
- Xu, W. , Zhang, Q. , Yuan, W. , Xu, F. , Muhammad Aslam, M. , Miao, R. et al. (2020) The genome evolution and low‐phosphorus adaptation in white lupin. Nat. Commun. 11, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell, M. and Holt, C. (2011) MAKER2: an annotation pipeline and genome‐database management tool for second‐generation genome projects. BMC Bioinformatics, 12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J. , Golicz, A.A. , Lu, K. , Dossa, K. , Zhang, Y. , Chen, J. et al. (2019) Insight into the evolution and functional characteristics of the pan‐genome assembly from sesame landraces and modern cultivars. Plant Biotechnol. J. 17, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Dong, S.S. , Xu, J.Y. , He, W.M. and Yang, T.L. (2019) PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics, 35, 1786–1788. [DOI] [PubMed] [Google Scholar]
- Zhao J., Bayer P. E., Ruperao P., Saxena R. K., Khan A. W., Golicz A. A., Nguyen H. T., Batley J., Edwards D., Varshney R. K.. (2020) Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnology Journal 18(9), 1946–1954. 10.1111/pbi.13354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Silverstein, K.A.T. , Ramaraj, T. , Guhlin, J. , Denny, R. , Liu, J. , Farmer, A.D. et al. (2017) Exploring structural variation and gene family architecture with De Novo assemblies of 15 Medicago genomes. BMC Genom. 18, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Variants distribution across the Lupinus albus genome.
Figure S2 Deletion of nine tandem duplicated genes on Chr17 of Ethiopian accessions of Lupinus albus.
Figure S3 Functional enrichment analysis of variable genome of Lupinus albus.
Table S1 Accessions of white lupin used in the pangenome construction.
Table S2 Summary of re‐sequencing of white lupin accessions using short reads produced in this report.
Table S3 Class and alkaloid status of pangenome white lupin accessions.
Data Availability Statement
The detailed methods and datasets supporting the conclusions of this report are included within the article and its additional files. All deep sequencing data reported in this study have been submitted to the NCBI. The datasets generated and analysed during the current study are available from the corresponding author upon request. Full genomic and raw sequence data are publicly available for download on the White Lupin genome portal [www.whitelupin.fr/pangenome] that contains a Genome Browser, Expression tools, and a Sequence retriever dedicated to the pangenome. The pangenome project and raw data have been deposited at DDBJ/ENA/GenBank under the accession PRJNA608889.