
Figure 5.
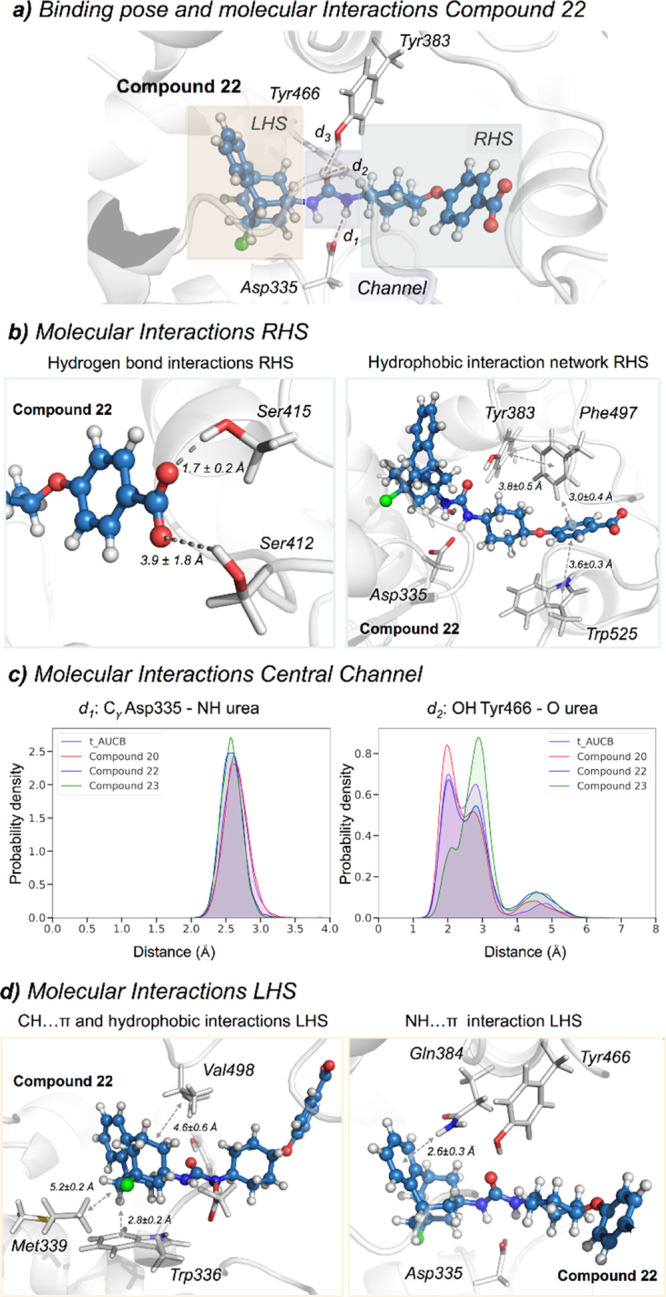
(a) Representative structure of 22 bound in the active site of sEH obtained from the most visited conformations along the MD simulations. The benzohomoadamantane moiety occupies the LHS pocket while the benzoic acid group lays in the RHS pocket. The central urea unit establishes hydrogen bonds with Asp335 (d1), Tyr466 (d2), and Tyr383 (d3). The PDB 5AM3 has been used as the starting point for all MD simulations. (b) Most relevant molecular interactions in the RHS. Average distances (in Å) obtained from the last 150 ns of MD simulations are represented. Hydrogen bonds between the oxygens of the carboxylate group of 22 and the hydrogen of the OH group of Ser412 and Ser415 are shown. The π–π stacking average distances are computed between the most proximal carbon atoms of each ring. The CH···π and hydrophobic interaction average distances are computed between the hydrogen atoms and the centroid of each aromatic ring (c) Histogram plots of the distance between the carboxylic group of the catalytic Asp335 and the amide groups of the inhibitor [d1(CγAsp335-NHINH)] and the distance between the carbonyl group of the urea inhibitor and the OH group of Tyr466 residue [d2(OHTyr466-OINH)] along the MD simulations of t-AUCB (purple), 20 (red), 22 (blue), and 23 (green). (d) Most relevant molecular interactions in the LHS. Average distances (in Å) obtained from the last 150 ns of MD simulations are represented. The CH···π interaction is calculated between the hydrogen of benzohomoadamantane unit and the centroid of the benzoid ring of Trp336. The NH···π interaction is monitored between the amide hydrogen of Gln384 and the center of the aromatic ring of the benzohomoadamantane scaffold.