

Abstract
The relationship between cancer and autoimmunity is complex. However, the incidence of solid tumors such as melanoma has increased significantly among patients with previous or newly diagnosed systemic autoimmune disease (AID). At the same time, immune checkpoint blockade (ICB) therapy of cancer induces de novo autoinflammation and exacerbates underlying AID, even without evident anti-tumor responses. Recently, systemic lupus erythematosus (SLE) activity was found to drive myeloid-derived suppressor cell (MDSC) formation in patients, a known barrier to healthy immune surveillance and successful cancer immunotherapy. Crosstalk between MDSCs and macrophages generally drives immune suppressive activity in the tumor microenvironment. However, it remains unclear how peripheral pre-generated MDSC under chronic inflammatory conditions modulates global macrophage immune functions and the impact it could have on existing tumors and underlying lupus nephritis. Here we show that pathogenic expansion of SLE-generated MDSCs by melanoma drives global macrophage polarization and simultaneously impacts the severity of lupus nephritis and tumor progression in SLE-prone mice. Molecular and functional data showed that MDSCs interact with autoimmune macrophages and inhibit cell surface expression of CD40 and the production of IL-27. Moreover, low CD40/IL-27 signaling in tumors correlated with high TAM infiltration and ICB therapy resistance both in murine and human melanoma exhibiting active IFNγ signatures. These results suggest that preventing global macrophage reprogramming induced by MDSC-mediated inhibition of CD40/IL-27 signaling provides a precision melanoma immunotherapy strategy, supporting an original and advantageous approach to treat solid tumors within established autoimmune landscapes.
INTRODUCTION
Myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages (TAMs) are known suppressors of T cell responses within the tumor microenvironment (TME) (1). Generally, MDSC amplify the immune suppressive activity of macrophages and dendritic cells via crosstalk at the TME and limit the efficacy of cancer immunotherapy (2). Although solid tumors drive myeloid cells dynamics to establish immune suppression and tolerance (1), existing or pre-existing (e.g. chronic inflammation) external factors can drive polarization of monocytes/macrophages and condition MDSC to adopt monocytic (M-MDSC) or polymorphonuclear (PMN-MDSC) phenotypes (3, 4). However, the functional status of MDSC is defined by the expression of co-inhibitory molecules (i.e., PD-1 and its-ligand (PD-L1)), co-stimulatory molecules (i.e., CD86 or CD40), and the production of soluble factors (i.e., arginase 1 (Arg1) and nitric oxide synthase 2) (5–7). Although TAMs and MDSC exert suppressive roles in cancer (2), their role in autoimmune diseases (AID), particularly in systemic lupus erythematous (SLE), is contentious. Generally, SLE is associated with generation of autoantibodies and deposition of immune complexes in peripheral tissues. However, recent studies also found that SLE disease activity drives formation of dysfunctional macrophages and MDSC in patients (4, 8), which can worsen lupus nephritis (4), the most common cause of morbidity (4, 8). The reasons for these outcomes are poorly understood; however, these data underscore the role of macrophages and MDSC over the severity of nephritis in SLE.
In parallel, the report of solid tumors among patients with previous or newly diagnosed systemic autoimmunity has increased significantly, particularly for melanoma patients (9, 10). In those patients, cancer survival and the efficacy of immune check point blockade (ICB), targeting either cytotoxic T lymphocyte-associated protein 4 (CTLA-4) or programmed death 1 (PD-1), is lower or about the same as that of the general population (10–12). Interestingly, clinical response or resistance to ICB depends on conserving IFNγ signaling in melanoma and immune cells, respectively (13, 14). Nevertheless, excessive IFNγ induction by ICB induces de novo autoinflammation and exacerbates underlying AID, even without anti-tumor responses (15), suggesting that off-target immune events are impacting the performance of ICB therapy. Although rare and not well understood, this is the case for development of nephrotoxicity among cancer patients receiving ICB therapy (16). Different from the antibody-mediated glomerular damage in SLE, acute tubular necrosis and interstitial nephritis are the most common causes of acute kidney injury resulting from ICB therapy (16). Interestingly, tubular renal damage is also observed in lupus nephritis (17). However, it remains unknown whether or how melanoma growth alters the severity of nephritis, and how such changes might influence AID severity or ICB therapy responses in autoimmunity. In this study, we identify the CD40/IL-27 axis as an autoimmune signature that reflects crosstalk between MDSC and macrophages during tumor progression and therapy response in SLE-prone mice. Mechanistically, we show that pathogenic expansion of SLE-generated MDSC by melanoma drives global macrophage polarization impacting simultaneously the severity of lupus nephritis and tumor progression in SLE-prone mice. We also demonstrate that CD40 agonists can reverse the deactivation of CD40/IL-27 and limit type 2-suppressive immune responses at the TME. These results suggest that preventing global macrophage reprograming via deactivation of CD40/IL-27 signaling delivers effective anti-tumor effects with limited renal toxicity, providing precise immunotherapy for melanoma within established autoimmune landscapes.
Materials and Methods
Mice
Mice were bred and housed at the NCI-Frederick animal facility. Type 1 IFNAR-deficient ARE (IFNγ/IFNAR1Bactin Cre) mice were generated by cross breeding ARE (IFN-gamma Cre 1821) and ifnar−/− (B6.Cg. IFNAR1tm1.2Eees) as reported (18, 19). The genotyping protocols for ARE and ifnar−/−ARE mice were performed as previously described (18, 19) and detailed in supplementary methods section. All animal care and use procedures were performed in accordance with protocols approved by the NCI Animal Care and Use Committee (Frederick, MD).
Tumor studies and tumor cell lines
Melanoma cell spheroids were grown alone or with 100 IU/ml of rIFNγ using the Cultrex 3D spheroid formation kit (Cultrex 3D, Trevigen, Gaithersburg, MD) following manufacturer’s instructions. Spheroid size was calculated from daily photographs taken with the Cytation 5 cell imaging reader (Biotek, Winooski, VT) and image J software (NIH, Bethesda, MD). Tumor xenografts were established in 18-22-week-old C57BL/6J WT, ARE+/−, ARE−/−, ifnar−/−ARE+/−, ifnar−/−ARE−/−, and Ifnar−/− mice either by subcutaneous (SQ) or intravenous (IV) challenge with 5 X 105 murine melanoma cells. The syngeneic melanoma cells lines were kind gifts from J. Weiss (B16F10, NCI, Frederick, MD), R. El Meskini (HcMel1274, NCI, Frederick, MD), and Dr. G. Merlino (MEL114433, and B2905, NCI, Bethesda, MD). Origin and genetic background of HcMel1274, MEL114433, and B2905 cells was reported separately (20). All cell lines used in this study were low passage, cultured 2 weeks prior to experiments, PCR tested for mycoplasma (Animal Molecular Diagnostics Laboratory, NCI-Frederick) and declared mycoplasma free. For SQ tumors, calipers were used twice weekly, and volume calculated using the formula: Volume= length x width^2/2 (mm2).
For anti-Ly6G studies, the anti-mouse monoclonal antibodies (mAb) against Ly6G (Clone 1A8, Biolegend, San Diego, CA) was administered intraperitoneally (IP). twice weekly at a dose of 200 ug per mouse starting 10 days prior to SQ injection. For cytokine neutralization studies, anti-IFNγ Ab (XMG-6) or an isotype control IgG (GL-113), kind gifts of G. Trinchieri (NCI, Bethesda, MD) were administered IP 2 days after IV challenge at a dose of 0.25 mg twice a week for 14 days. The mAb against IL-27p28 (20 ug per mouse, Clone MM27-7B1, Biolegend, San Diego, CA) or rmIL-27 (100 ng per animal; Biolegend) was administered IP twice weekly starting 7 days after SQ challenge. For single or combined immunotherapy, the following mAb (kind gifts from J. Weiss, NCI, Frederick MD): agonist anti-mouse CD40 (Clone FGK115B3, rat IgG2a) and mouse anti-PD1 (clone RPM1-14, rat IgG2a) were administered alone or combined IP twice weekly at a dose of 0.25 mg i.p. starting 7 days after SQ injection. Rat IgG2A antibodies from Bioxcell (Lebanon, NH) or Sigma Aldrich (St. Louis, MI) were used as isotype controls.
Cultured bone marrow derived myeloid cells (BMDM)
Mouse primary bone-marrow derived myeloid (BMDM) cells were obtained and cultured with either recombinant mouse (rm) macrophage-CSF (10 ng/ml M-CSF; Peprotech, Cranbury, NJ) or GM-CSF (Peprotech, Cranbury, NJ) for 7 days as reported previously (19). Immortalized BMDM cell lines from WT or ARE−/− mice were established by infecting primary bone marrow cells with the J2 recombinant retrovirus as described previously (21). In some instances, BMD cells were stimulated with LPS (Enzo Life Sciences, 500 ng/ml) for 18 h, rmIFNγ (Peprotech, Cranbury, NJ) at 2 IU/ml to 100 IU/ml for 10 minutes or 18 h; 25 – 50 ng/ml of rmIL-27 (R&D systems, Minneapolis, MN); or their combination for 18 h. All cells used were mycoplasma free as determined by PCR (Animal Molecular Diagnostics Laboratory, NCI-Frederick).
Immunoblotting, sample preparation and antibodies.
See supplementary methods for detailed preparation and antibodies.
Cytokine measurements
Serum samples or culture media were processed and subsequently frozen at −80 °C until use. Serum samples were analyzed either by luminescence (Luminex, Eve Technologies, Calgary, Canada) or electro chemiluminescence assays using the Meso-Scale Discovery (IFNγ or IL-27p28) or mouse custom Multiplex kit for 10 or 27 factors (Mesoscale Diagnostic, MSD). All samples were tested in duplicate and read on MSD QuickPlex SQ 120 imager/reader. All cytokine values were reported in units of pg/mL. See Supplementary Methods for further detail.
Antibodies and flow cytometry analysis
See supplementary methods for detailed list of antibodies and cell viability dyes. Spleen single-cell preparations were obtained and blocked prior to staining, as reported previously (19). For tumor single-cell preparations, the mouse tumor dissociation kit (Myltenyi Biotec, San Diego, CA) was used according to manufacturer’s instructions. For Foxp3 detection, a staining buffer set (eBioscience) was used following manufacturer’s instructions. Positive magnetic selection (AutoMACS Pro, Miltenyi) was used for isolation of Ly6Chi and Ly6Ghi splenocytes and tested for ability to suppress the proliferation of CFSE-labelled naïve T cells stimulated with CD3 and CD28 mAb. Percent inhibition of T cell proliferation was quantified for CD8+ and CD4+ T cells co-cultured with Ly6Ghi cells sorted from the spleen or whole BM using a weighted formula that quantified the percentage of T cells within each daughter population. The gaiting strategy for myeloid cell identification and expression analysis (MFI) is shown in Supplementary Fig. S2F. All flow cytometry experiments were performed using a BD LSRII Fortessa (BD Biosciences) or a BD FACSAria II (BD Biosciences) analyzer using BD FACS Diva Software (BD biosciences). All data analyses were performed using FlowJo 10 software (Tree Star, Ashland, CO).
Quantitative real-time PCR and Nano String data.
RNA from tissues and cultured cells was isolated using a RNeasy kit (Qiagen) and reversed transcribed to synthesize cDNA with Superscript III cDNA kit (Invitrogen) according to the manufacturer’s instructions. Gene expression studies were done as indicated in Supplementary methods using the following TaqMan (Applied Biosystems) gene probes: IFNγ (Mm01168134_m1), IL-27(Mm00461162_m1), IL-27ra (Mm00497259_m1), CD40 (Mm00441891_m1), NOS2 (Mm00440502_m1), and ARG1 (Mm00475988_m1). HPRT1 (Mm03024075_m1) or Beta-actin (Mm02619580_m1) was used to normalize and measure gene expression fold change differences.
Pathology and Histology studies
Mice underwent full necropsy and tissues, blood, and serum were collected for clinical and anatomic pathology assessment. Paraffin or optimal cutting temperature compound (OCT compound; Sakura Finetek) sections were stained with hematoxylin & eosin (H&E) or prepared for IHC and visualized with DAB, as described previously(19). For tumors, melanin pigment was bleached with 0.5% KOH/3% H2O2 and a 1% acetic acid rinse before antibody staining. Bright-field images were acquired either with SPOT 5.2 software using a Nikon Eclipse E600 microscope (Nikon) and SPOT RT3 camera (Digital Instruments) or with CellSens imaging software (Olympus of America, Center Valley, PA) using a DX41 microscope (Olympus) and DP71 camera (Olympus). Slides containing positive cells were digitized with either an Aperio ScanScope XT (Leica) or Axio Scan.Z1 scanner (Zeiss) at 200X. For additional details and the list of antibodies, please see supplementary methods.
Analysis of transcriptomic data of human melanoma
RPKM data were retrieved from published RNA sequencing results of pre-treatment melanoma from 2 Gene Expression Omnibus (GEO) public databases: GSE78220 (22) and GSE85898 (23). Access to de-identified human data for research purposes was granted to G. Merlino via CRADA agreement from published RNA sequencing result of pre-treatment melanoma from 42 patients treated with anti-CTLA4 (24). Previously, we identified which quantitates differentiation status of melanoma and predicts of patient outcome in response to immune checkpoint blockade (20). We used the expression of a reported 45-gene melanocytic plasticity signature (MPS) gene pattern to evaluate the intensity of cytokine signaling in human melanoma. The z-score of MPS genes across all genes were calculated in each patient’s melanoma samples as input to CytoSig, and the output is the z-score of each cytokine, representing the intensity of signaling on the melanoma. CytoSig is a web application that predicts the cytokine signaling activities based on a sample’s gene expression profile (https://cytosig.ccr.cancer.gov) under development in NCI. For each transcriptomic input, the expression levels of given genes were deconvoluted to the aggregated regulatory effects (induction or repression) of factors, based on the treatment response profiles included in the public data collection, and the output is the predicted activities of cytokine, chemokine, and growth factors, as reported (56). Technical information about CytoSig Platform is available (P. Jiang). The deconvolution for identifying cell types associated with each melanoma sample was performed following the instructions of CIBERTSORT at https://cibersort.stanford.edu/.
Statistical Analysis
Statistical analyses and graphical representations were performed using GraphPad Prism 8 software for Windows (Graph Pad Software). Differences between means were obtained either with an unpaired, 2-tailed Student’s T-test for individual comparisons and 1-way or 2-way ANOVA with Holm-Sidak correction for multiple group comparisons. Survival percentages were generated using the Kaplan-Meier method using the log-rank (Mantel-Cox) test for statistical analysis. All statistical tests were conducted at the 0.05 (alpha) level.
RESULTS
Sensitivity of Melanoma to IFN influences tumor growth in ARE mice
Different from mouse models with whole gene deletions, the replacement of one (ARE+/−) or two (ARE−/−) alleles at the IFNγ 3’ UTR ARE region in the lupus-prone ARE model uniquely mimics the low and high disease activity observed in the relapse and remitting states of human systemic lupus erythematous (SLE) (19, 25). As ARE+/− and ARE−/− mice constitutively produce low and moderate IFNγ levels and co-activate Type 1 IFNs, we used these mice to investigate how melanoma models progressed under different pre-existent autoimmune conditions. Wild-type (WT), ARE+/− or ARE−/− were challenged intravenously (IV) or subcutaneously (SQ) with two pigmented IFNγ-sensitive models (B16F10 and B2905), or partially resistant HcMel1274 cells (20); (hereafter called HcMel) (Fig. 1A). The B2905 and Hc-Mel1274 models genotypically represents (BRAF/RAS/NF-1) wild-type and RAS-mutant human melanomas, respectively (9). Sensitivity of melanoma cells to IFNγ was confirmed in vitro by measuring proliferation and production of phosphorylated STAT1 (pSTAT1) after IFNγ exposure (Supplementary Figs. S1A - S1C). Compared to tumor growth and overall survival (OS) in WT mice, OS gain was modest, albeit significant (p=0.02), and associated with decreased tumor growth and metastases (pulmonary and/or extrapulmonary) only in B16F10- and B2905-bearing ARE+/− mice (Fig. 1B–1D; Supplementary Figs. S1D and S1E). These data suggest that melanoma cells intrinsic sensitivity to IFNs define the extent of anti-tumor effects in ARE mice, specifically at metastatic sites.
Figure 1. Disease status influenced survival and growth for IFN-sensitive melanoma cells in ARE mice.

(A) Experiment layout of melanoma models (B16, HcMel and M114433) and the cohorts of WT, ARE−/−, ARE+/−, ifnar−/−ARE+/−, ifnar−/−ARE−/−, and ifnar −/− mice (n=5-6 per group). (B-D) Survival studies for B16 and HcMel cells injected IV into separate cohorts of ARE mice (n=6 per group). (B). Survival curves (C-D) Number of metastasis per animal in lung (C) and (D) other organs from A. Each symbol represents the mean area in a slide containing all lung lobes. (E) Tumor growth curves of B16 after SQ injection separately in ARE mice and ifnar−/−ARE mice. (F) Percentage of T (CD4+, CD8+) and myeloid (F4/80+ macrophage and Ly6Ghi neutrophils) populations within tumors in E, as determined by FACS. Data are shown as mean ± SEM of two or three independent experiments. Statistical significance, two-way ANOVA unless specified. * p < 0.05, ** p < 0.01, *** p < 0.001.
Previously, it was reported that activation of type 1 IFNs and recognition of melanin-related antigens delayed tumor growth in melanoma models (26, 27). To rule out that these factors influenced the outcome in ARE mice, pigmented B16 and non-pigmented M114433 melanoma cells were injected SQ in type 1 IFNAR-sufficient (WT, ARE+/−, and ARE−/−) and type 1 IFNAR-deficient mice (ifnar−/−ARE+/−, ifnar−/−ARE−/−, and ifnar−/−) (Fig. 1A). The MEL114433 cells are derived from albino C57BL/6-Tyrcbrd mice and their expression of pigmentation antigens (e.g. tyrosinase or tyrosinase-related protein 2) is suppressed (20). However, like B16F10 cells (hereafter called B16), MEL114433 cells remained sensitive in vitro to IFNγ (Supplementary Fig. S1F). The ifnar−/− mice lack the common type 1 IFN receptor (ifnar) and make host cells insensitive to type 1 IFNs, whereas the type 1 IFNAR-deficient ARE mice produced constitutively high serum IFNγ levels but, unlike ARE mice, exhibit mild autoimmunity (18). B16 and MEL114433 cells grew faster in ifnar−/− mice than in WT mice (Fig. 1E; Supplementary Fig. S1F), yet these cells grew slower in ARE+/− and ifnar−/−ARE+/− mice ruling out a direct role of type 1 IFNs or melanin antigen recognition. Analysis of the immune landscape in B16 tumors using flow cytometry revealed that F4/80+ macrophages were predominant in tumors from ifnar−/−ARE+/− mice, whereas lymphoid CD8+ T cells were predominant in tumors from ifnar−/−ARE−/− mice (Fig. 1F; Supplementary Fig. S1G). As all tumors exhibited low infiltration of Ly6Ghi PMN (Fig. 1F) and CD8+ T cells constitutively produce IFNγ due to the ARE removal (19), these data suggest that the balance between melanoma cell sensitivity to IFNγ and the extent of myeloid cell infiltrates determine the anti-tumoral effect of the chronic expression of IFNγ in an autoimmune background.
Melanoma-associated macrophage expansion limits OS gains in ARE−/− mice
To understand how melanoma progresses without survival gains in ARE−/− mice, we examined changes in the histological scores (HS) for lupus nephritis and the serum cytokine milieu between tumor-bearing and tumor-free WT and ARE−/− mice. HS for lupus nephritis assessed both glomerular and tubulointerstitial damage, as kidney disease predictors (8). Compared to HS in tumor-free ARE mice, molecular pathology analysis found no significant glomerular changes in B16-bearing ARE−/− mice despite sustained infiltration of CD3+ T cells and Ly6G+ polymorphonuclear (PMN) cells (Supplementary Fig. S2A - S2C). However, B16-bearing ARE−/− mice had a higher evidence of tubular renal damage, including dilated tubules containing eosinophilic material (Fig. 2A and 2B), and increased levels of factors known to advance nephritis, such as IL-10 (28), IL-12p70 (29), IL-17 (30), TNFα (31), and GM-SCF (Fig. 2C; Supplementary Fig. S2D). Interestingly, kidney infiltration of F4/80+ macrophages in B16-bearing mice was significantly higher than in tumor-free mice (Fig. 2D and 2E, Top), suggesting a pathogenic association between melanoma associated macrophages and tubular renal damage in ARE−/− mice.
Figure 2. Myeloid cells advance lupus nephritis and tumor growth in ARE−/− mice.

(A-C) Age-matched ARE mice were left tumor free or IV challenged with melanoma cells. Cohorts were euthanized at day 21. (A) Representative kidney H&E images for tubular renal damage (arrows, right) and overall grade (B). Upper bar indicates SLE-disease activity. (C) Serum levels of indicated factors per genotype (n = 4-5 mice/group). (D-E) Representative IHC images (D) and summary quantitation (E) of high F4/80 staining in kidneys (top), lung metastases (Center), and primary tumors (bottom) from B16-bearing ARE−/− mice. (F) Total number of positive cells per mm2 stained for the ratio iNOS+/CD206+ cells from D. (G) Intratumoral phosphorylated STAT-1 and STAT3 protein expression, by immunoblotting (n=3 per group), 2 independent experiments. GAPDH used as loading control. (H) Pro-tumoral conditions assessed by digital measure of necrotic areas (left) and CD31+ staining (right). (I) Statistical comparisons for intratumoral levels of IL-1b and IL-12p70, as measured by MSD, in samples from G. Every dot represents 2 technical replicates from one mouse per genotype. For A and D, scale bars: 200 um (Original magnification, 10 X). Data are shown as mean ± SEM. Two-way ANOVA, * p < 0.05; **, p < 0.01; and ***, p < 0.0001.
Apparently, circulating IFNγ levels related to AID status in ARE−/− mice could induce circulating monocytes towards inflammatory phenotypes that exacerbate kidney damage but limit tumor growth (1, 17). To test this, we characterized the myeloid population in tumors from ARE mice. Like in kidneys, IHC showed high infiltration of F4/80+ TAMs in lung metastases and primary tumors from ARE−/− mice compared to WT counterparts (Fig. 2D and 2E, middle and bottom, respectively). Nevertheless, F4/80+ TAMs from tumor-bearing ARE−/− mice exhibited a lower ratio of iNOS+/CD206+ cells, an index indicating more M2 than M1 macrophage polarization (Fig. 2F; Supplementary Fig. S2E). This high M2-polarization in ARE−/− tumors correlated with high IFN signaling (p-STAT1, and p-STAT3), large necrotic areas, and low CD31 vascular density staining (Fig. 2G and 2H). Moreover, unlike kidneys, FACS analysis, based on the expression of Ly6G and Ly6C antigens (Supplementary Fig. S2F), and multiplex cytokine (MSD) assays found low infiltration of CD11b+Ly6GhiLy6Clo PMN and low levels of PMN recruiting factors, e.g., CXCL2 (Supplementary Fig. S2G and S2I). Similar FACS data patterns from M114433-bearing ARE tumors supported these findings (Supplementary Fig. S2H). These data together with low levels of pro-inflammatory cytokines (such as IL-1b and IL-12p70; Fig. 2I), suggested that high F4/80+ TAM frequency drives immune suppression in ARE tumors. Collectively, aberrant global expansion of melanoma-associated macrophages likely limited OS in ARE−/− mice by compromising kidneys and tumors.
Melanoma impaired IL-27 production from ARE monocytes
The impact of macrophages on lupus nephritis and tumor growth suggested a pre-existing regulatory myeloid factor(s) with anti-inflammatory and anti-tumor properties was inactivated in ARE−/− mice. Apart from the low levels of VEGF in tumor-bearing ARE mice (Supplementary Fig. S2D), no other factor decreased in our multiplex panel analysis. However, compared to tumor-free mice, exacerbation of nephritis and sustained IL-17 levels in tumor-bearing ARE−/− mice resembled the Th-17 driven autoimmune susceptibility reported for IL-27 receptor alpha-deficient mice (IL-27ra−/−/ WSX-1−/−) (32). The IL-27ra subunit integrates the signal-transducing receptor for interleukin 27 (IL-27), a cytokine produced by myeloid cells (e.g. macrophages and dendritic cells) with IFN-like properties, as reported in SLE patients and lupus-prone mice (28). Although IL-27 limits development of Th17 cells and production of IL-17 (32), it was unknown whether a growing tumor could alter IL-27/IL-27ra signaling in ARE mice.
To determine a role for IL-27 in the host response, we first assessed mRNA expression in tissues and serum levels of IL-27p28 (hereafter referred as IL-27) in tumor-free ARE−/− and WT mice. IL-27p28 is a biologically active subunit of IL-27 (33). IL-27 mRNA and circulating levels were significantly increased in ARE−/− mice compared to counterparts in WT mice (Fig. 3A; Supplementary Table S1; Supplementary Fig. S3A and S3B), suggesting that IL-27 has an important physiological role in ARE mice. Because type 1 IFNs could also regulate IL-27 in ARE mice (28, 33), circulating levels of IFNγ and IL-27 were compared in type 1 IFNAR-deficient and sufficient ARE mice. Circulating levels of IL-27 significantly correlated with IFNγ levels in tumor-free mice (Fig. 3B and 3C), supporting the premise that IFNγ regulates IL-27 production in ARE mice. Surprisingly, when compared to tumor-free counterparts, the serum levels of IL-27, not IFNγ, significantly decreased in tumor-bearing ARE−/−, ifnar−/−ARE+/−, and ifnar−/−ARE−/− mice (Fig. 3B), indicating an alteration in IFNγ, not type 1 IFN, regulation of IL-27 in blood myeloid cells. To this end, neutralization of IFNγ with an anti-IFNγ mAb in vivo further reduced circulating IL-27 levels and blood monocyte counts in tumor-bearing ARE+/− mice compared to isotype controls (Supplementary Fig. S3C- S3E), consistent with IFNγ activated monocytes being the source of IL-27. Indeed, we confirmed that BM-derived macrophages from ARE+/− mice exposed ex-vivo with LPS produced IL-27 (Supplementary Fig. S3F), supporting that high levels of IL-27 in ARE mice results from high frequency and production from activated monocytes/macrophages.
Figure 3. Melanoma impairs IFNγ-regulated IL-27/IL-27ra signaling in Ly6C+ monocytes.

(A) Nanostring analytics of IL-27p28 mRNA alone (left) or compared to IFNγ in bone marrow (BM), kidney (Ki), liver (li), lymph node (LN), spleen (Sp), and Thymus (Thy) from tumor-free ARE−/− and WT mice (n=4-5 mice per genotype). (B-C) Individual serum levels of IL-27 (top) compared to IFNγ (bottom) levels in age matched tumor free (B), or in correlation (C) determined by MDS (pro-inflammatory panel) (n = 6 mice per genotype). (D-F) Representative dot plots (D) and statistical comparison of the percentage population (E) and the expression of IL-27ra (F) in Ly6Chi monocytes examined by flow cytometry from D in B16-bearing and tumor-free ARE mice (n=5-6 per genotype). Pooled data in B and E from 2 independent experiments. Data are shown as mean ± SEM. Two-way ANOVA: *, p < 0.05; **, p < 0.01; ***, p < 0.0001, and ****, p < 0.00001
To address how the monocyte/macrophage population relates with IL-27/IL-27ra signaling, FACS analysis was used to compare splenocyte populations and IL-27ra expression in tumor-bearing and tumor-free ARE mice. Surprisingly, the frequency of CD11b+Ly6GloLy6Chi monocytes and expression of IL-27ra decreased, albeit significantly in ARE+/−, ifnar−/−ARE+/−, and ifnar−/−ARE−/− tumor-bearing mice (Fig. 3D to 3F), suggesting that IL-27 levels and IL-27ra signaling are directly associated to the size of the monocyte population during tumor progression. These data together with the conserved expression of IL-27ra in CD4+, CD8+ or CD4+CD25+FoxP3+ T cells due to IFNγ autoregulation from the ARE replacement (Supplementary Fig. S3G), supported the model that low IL-27 levels indicated fewer activated Ly6Chi monocytes and aberrant expansion of M2-like macrophages in tumor-bearing ARE mice.
Melanoma expands pre-existent PMN-MDSC and induces M2 polarization in ARE mice
It is known that IL-27 controls neutrophil polarization via IL-27ra (34). However, unlike monocytes, FACs analysis found that low IL-27 levels in tumor-bearing ARE mice was followed by significant expansion of CD11b+Ly6GhiLy6Clo PMNs expressing lower levels of IL-27ra than in tumor-free ARE+/− counterparts (Fig. 4A and 4B). Subsequent PCR analysis confirmed that expanded CD11b+GR1+ PMN from ARE+/− mice featured higher arginase 1 (ARG1) mRNA and lower nitric oxide 2 (NOS2) mRNA transcripts than WT counterparts (Fig. 4C), consistent with a suppressive phenotype. Moreover, Ly6Ghi PMN were found in high numbers and localized near iNOS+PD-L1+F4/80+ macrophages in lymph nodes and spleen of ARE mice, as shown by IHC (Fig. 4D, Supplementary Fig. S4A), suggesting that PMN engage macrophages in peripheral lymphoid organs and drive M2-macrophage polarization, a role attributed to MDSC (7).
Figure 4. Melanoma expands pre-existing PMN-MDSC in ARE mice.

(A-B) Summary quantitation of Ly6Ghi PMN (A) expressing IL-27ra (B) in tumor-free and B16-bearing WT and ARE mice. (C) RT-qPCR analysis show higher Arginase 1 (Arg1) mRNA than NOS2 mRNA in isolated CD11b+GR1+ cells from B16-bearing ARE+/− mice compared to counterparts in WT mice. (n=3). (D) Summary quantitation of Ly6G+ cells within indicated tissues from tumor-bearing WT and ARE+/− mice (n= 6 organs per genotype). (E-J) Statistical comparison of the percentage populations and expression (mean florescence intensity; MFI) of indicated markers in CD45.2+Ly6CloLy6Ghi PMN from tumor-free ARE (E, F) and type 1 Ifnar-deficient ARE mice (H, I). (G, J) Representative histograms show that Ly6G+ cells inhibited proliferation of CD3-activated CD4+ T cells, as evidenced by overlay CFSE signaling (green) from control (grey), in ARE (G) and type 1 Ifnar-deficient ARE mice (J). Data are shown as mean ± SEM. Two-way ANOVA: *, p < 0.05; **, p < 0.01; ***, p < 0.0001, and ****, p < 0.00001
Recently, it was reported that deletion of the type 1 IFN receptor (ifnar) is not sufficient to convert myeloid cells to MDSCs (35). However, in the absence of type 1 IFN priming, immune cells exhibit weakened responses to other cytokines, including IFNγ (36). As SLE generate PMN-MDSCs (4), we hypothesized that IFNγ can induce formation of PMN-MDSCs independent of type 1 IFN signaling to regulate M2 polarization and tumor growth in ARE mice. To confirm this, FACS was used to characterize the expression of MDSC-associated surface markers and IL-27ra in splenocytes from tumor-free ARE, WT, ifnar−/−ARE+/−, and ifnar−/−ARE−/− mice. CD11b+Ly6GhiLy6Clo PMN expressed higher levels of co-inhibitory factors, including TGFβ (p < 0.01), PD-L1 (p < 0.01), and CXCR2 (p < 0.001) than WT counterparts (Fig. 4E to 4I; Supplementary Fig. S4B). Although ARE+/−, ifnar−/−ARE+/−, and ifnar−/−ARE−/− spleens had a lower population of CD11b+Ly6GhiLy6Clo PMN than WT counterparts (Fig. 4E and 4H), we examined ex-vivo whether PMN suppress activated T cell proliferation. As expected, isolated PMN from ARE spleens strongly suppressed T cells proliferation compared to WT counterparts, while PMN cells from ifnar−/−ARE+/−, and ifnar−/−ARE−/− mice achieved only 50% suppression (Fig. 4G and 4J; Supplementary Fig. S4C and S4D). These data confirmed that CD11b+Ly6GhiLy6Clo are PMN-MDSCs induced by chronic exposure to IFNγ in tumor-free ARE mice.
In contrast, compared to WT counterparts, CD11b+Ly6GloLy6Chi ARE spleen monocytes overexpressed IFNγ-regulated co-stimulatory molecules reported in mature activated monocytes (37), such as: CD80, CD40, and MHCII (Supplementary Fig. S4E and S4F). Interestingly, spleen Ly6Chi monocytes from ARE mice also expressed higher IL-27ra than bone marrow (BM) counterparts (Supplementary Fig. S4G), supporting the relationship between IL-27 signaling and the spleen monocyte population. Collectively, these data suggested that melanoma expanded a pre-existent population of IFNγ-generated PMN-MDSC in ARE mice, which facilitated peripheral polarization of Ly6Chi monocytes into M2-macrophages at the TME.
PMN-MDSC impairs IL-27 production via inhibition of CD40 expression in ARE monocytes
Previously, we reported that macrophage depletion worsened lupus nephritis in ARE mice (19). Then, to confirm in vivo whether melanoma-associated expansion of PMN-MDSC leads to peripheral decline of IL-27, mAb against Ly6G (anti-Ly6G) or against the IL-27p28 subunit (anti-IL-27) were injected IP into 7 day-tumor bearing ARE+/− mice twice a week for 14 days. Elimination of PMN-MDSC with anti-Ly6G in WT and ARE+/− mice reduced tumor growth by decreasing CD206+ expression in polarized TAM, instead of increasing infiltration of CD3+ T cells (Fig. 5A; Supplementary Fig. S5A and S5B), as demonstrated by low intratumoral populations of CD11b+F4/80+ (p < 0.05) and high Ly6Chi monocytes compared to isotype-treated mice (Fig. 5B and 5C). This outcome involved systemic increases in circulating Ly6ChiCD40+ TAM precursors and levels of IL-27 compared to isotype controls (Fig. 5D and 5E), suggesting that CD40+ monocytes produce IL-27 in ARE mice. Indeed, in vitro stimulation of ARE−/− macrophages (KOM) to IFNγ and IL-27, not IL-27 alone, induced the highest expression of CD40 mRNA compared to WT macrophages (WTM) (Supplementary Fig. S5C). However, IL-27 alone was insufficient to upregulate IFNγ-induced genes such as CD40, CIITA, and CXCL9 in KOM (Supplementary Fig. S5D). These data indicated that IL-27 redundantly supports IFNγ-CD40 regulation in monocyte/macrophages.
Figure 5. Decreased CD40/IL-27 regulation on Ly6Chi monocytes by MDSC promotes tumor progression in ARE mice.

Seven days post SQ challenge with B16 cells WT and ARE+/− mice received either anti-Ly6G mAb, anti-IL-27p28 (E-H), or rmIL-27 (I-K) i.p. twice a week (arrow). (A-E) For anti-Ly6G mAb, (A) Tumor growth curves (n=4, per group). (B-C) Percentage population of TAMs (B) and CD45.2+Ly6Chi Ly6Glo monocytes (C), assessed by FACS. (D) Frequency of Ly6GloLy6Chi and CD11b+CD40+ population in blood, as determined by FACS. (E) Serum levels of IL-27 before (pre) and after (post) B16 challenge (n=4), as determined by MSD. (F-H) For anti-IL-27p28, (G) Tumor growth Curves (n = 5 per intervention). (G-H) Percentage of spleen LY6G+ and Ly6Chi populations total (G) or expressing CD40+ (H), as determined by FACS. (I-K) For rmIL-27, (I) Tumor growth Curves (n = 3-4 per intervention). (J-K) Percentage of tumor infiltrating CD8+ and CD4+ T populations total (J) and spleen LY6G+ and Ly6Chi (K), as determined by FACS. Data are shown as mean ± SEM. Two-way ANOVA was used, except for final tumor measurements (Two-tailed Student’s T test), * p < 0.05, ** p < 0.01, *** p < 0.001.
Next, we tested how IL-27 manipulation impacts PMN-MDSC and tumor growth in ARE mice. As expected, neutralization of the IL-27p28 subunit (anti-IL-27) or administration of rmIL-27 (100 ng/ml) significantly accelerated or delayed tumor growth in ARE mice, respectively (Fig. 5F and 5I). However, OS gains could be limited by rapid tumor growth or unexpected death in the anti-IL-27- or rmIL-27-treated groups, respectively. Analysis of tumor immune composition by FACS found that persistently high F4/80+ TAM infiltration limited significant TME changes in anti-IL-27 treated mice compared to isotype-treated controls (Supplementary Fig. S5E and S5F). Interestingly, neutralization of IL-27p28 expanded the spleen populations of CD11b+Ly6Ghi PMN and Ly6Chi monocytes with low CD40 expression from ARE+/− mice compared to isotype-treated controls (Fig. 5G and 5H) resulting in lower circulating levels of monocyte pro-inflammatory cytokines IL-1 and IL-12p40 in ARE+/− mice (Supplementary Fig. S5G). By contrast, rmIL-27-treated tumors featured significantly high infiltration of CD4+ T cells along with low F4/80+ and Ly6G+ infiltrates compared to isotype-treated controls (Fig. 5J; supplementary Fig. S5H). More importantly, rmIL-27 prevented expansion of PMN-MDSC and Ly6Chi monocytes in spleens of ARE+/− mice (Fig. 5K; Supplementary Fig. S5I). These data, together with the conserved expression of IL-27ra in CD4+, CD8+ or CD4+CD25+FoxP3+ T cells due to IFNγ autoregulation from the ARE replacement (Supplementary Fig. S3G), support the model that tumor manipulation of IL-27 levels impact PMN-MDSC populations to reprogram SLE macrophage polarization via CD40/IL-27 expression in ARE mice.
Agonist CD40 antibodies prevent suppression of CD40/IL-27 regulation in ARE mice
As anti-PD1 responses were limited but showed less immune related toxicity than CTLA-4 antibodies in melanoma patients with pre-existing AD (10, 12), we explored whether therapeutic enhancement of CD40/IL-27 in monocytes could achieve tumor responses or enhance anti-PD1 response in ARE mice. To this end, we tested an agonist anti-CD40 mAb (herein anti-CD40) for the ability to ligate CD40 and reprogram tumor infiltrating myeloid cells (38). After 7 days post SQ-challenge with B16 cells, WT and ARE+/− mice received either an isotype control IgG, anti-CD40, anti-PD-1, or their combination (herein anti-PD1/CD40). The efficacy of anti-CD40 and anti-PD1 was monitored with serum levels of cytokines associated with efficient CD40 ligation and T cell activation, including IL-12, IL-1, IL-2, and IFNγ. Overall, monotherapies were well tolerated in survival studies, as evidenced by animal weight (Supplementary Fig. S6A). B16 tumor growth was reduced by 75% after treatment with anti-CD40, either as monotherapy or in combination with anti-PD1, thus enhancing the response to anti-PD1 alone (Fig. 6A). Such improved response to anti-CD40 involved unique intratumoral re-distribution of F4/80+ TAM, low Ly6Ghi PMN infiltrates, and increased levels of IL-27 and IFNγ, as shown by histology (Fig. 6B; Supplementary Fig. S6B), flow cytometry (Fig. 6C and 6D) and MSD analysis (Fig. 6E). By contrast, non-responsive anti-PD1-treated tumors featured high recruitment of suppressive CD11b+ cells (including Gr1+ and F4/80+ TAM; Fig. 6B and 6C) that locally suppressed IFNγ production from infiltrating CD4/CD8+ T cells (Fig. 6D and 6E). These data demonstrated that agonist CD40 therapy achieve anti-tumor effects and improved response to anti-PD1 in ARE+/− mice.
Figure 6. Agonist CD40 therapy delays melanoma growth in ARE mice.

Seven days post SQ challenge with B16 cells, WT and ARE+/− mice received weekly i.p. injections of anti-CD40, anti-PD1, anti-PD1/CD40 or an isotype control (rat IgG2, 200 ug). (A) Tumor-growth curves. (B) cumulative quantification of F4/80+ IHC staining within whole tumor sections from A (n= 4-5). (C-D) Percentage of tumor immune populations of PMN (C), T cells (CD4+ and CD8+; D left) and monocytes (Ly6Glo; D right) from A, assessed by FACS. (E) Intratumoral levels of IFNγ and IL-27 from A, as determined by MSD (n = 4-5 per group). (F) Immunoblots from spleen lysates showing protein expression of CD40 and indicated IFN signature genes. Side arrows show correct band size. (G) Statistical comparison of serum levels of IFNγ, IL-27, and IL-12p40 in treated groups. (H) HS for nephropathy in ARE+/− mice. (I-J) Spleen size as % body weight (BW) (I) and percentage population of CD11b+Gr1+ MDSC, obtained by FACS (J). Unless indicated, pooled data from two independent experiments are shown as mean ± SEM. Two-way ANOVA: * p <0.05, ** p < 0.01, *** p < 0.001.
Once the tumor response was defined, we examined the systemic impact of immunotherapy. Surprisingly, immunoblotting showed most therapies increased CD40 expression and activated pSTAT1 in ARE+/− splenocytes compared to isotype controls, except for anti-CD40 in WT mice (Fig. 6F). Although IFNγ expression indicates a good therapeutic response in cancer (24), the high circulating levels of IFNγ and IL-27 induced by anti-PD1/CD40 (Fig. 6G) caused systemic inflammation that worsened nephritis in ARE+/− mice, as evidenced by high HS (Fig. 6H), larger spleen sizes (Fig. 6I) , and high circulating levels of CXCL9, CXCL10, and TNFα (Supplementary Fig. S6C and S6D). Moreover, such a landscape induced tumor progression and therapy resistance, as anti-PD1/CD40 elicited extra-pulmonary metastases and diminished induction of CD40 ligation cytokines (IL-12p40, IL-1a, and IL-2) compared to anti-CD40 alone (Fig. 6G; Table S2; Supplementary Fig. S6C). Apparently, this effect comes from anti-PD-1 induction of intrinsic and extrinsic resistance factors, including global activation of pSTAT3 (Fig. 6F) and strong MDSC expansion (Fig. 6J).
Next, we asked whether pre-existent autoimmunity could drive resistance to PD1-blockade in PD-1 sensitive melanomas, such as the B2905 model featuring high T cell infiltration and expression of PD-L1 (20, 39). To this end, B2905 melanoma cells were injected SQ in ARE+/− mice and received the anti-PD1 protocol. Although B2905-tumors variably responded to PD1 blockade in WT mice (Supplementary Fig. S6E and S6F), anti-PD1-treated B2905 tumors in ARE+/− mice were even more resistant than isotype-treated counterparts due to high infiltration of myeloid cells (Ly6Chi monocytes, F4/80+ TAM and Ly6G+ PMN-MDSC) amid continuous recruitment of CD4+ and CD8+T cells (Supplementary Fig. S6H). Moreover, like B16-resistant tumors, resistance to anti-PD1 featured systemic inflammation evidenced by larger spleen sizes than isotype-treated counterparts (Supplementary Fig. S6G). As constitutive myeloid infiltration and IFNγ activation are known sources of PD-1 resistance (12), these data provide proof of principle that therapeutic targeting of CD40/IL-27 with anti-CD40 mAb could achieved tumor responses with limited nephrotoxicity in an autoimmune landscape. Moreover, our data underscore the importance of alternative approaches to ICB therapies in the context of pre-existent autoimmunity.
Conserved intratumoral CD40/IL-27 signaling relates to ICB response in human melanoma
Next, we analyzed the translational potential of IFNγ/IL-27/CD40 signature in tumor transcriptomic data from melanoma patients with metastatic melanoma prior and after treatment with anti-PD1 or anti-CTLA4 (22–24). Although the mechanisms of innate response to anti-PD1 and anti-CTLA4 are not necessarily similar (22), the expression of IL27 and CD40 was unchanged among anti-PD1 treated tumors or anti-CTLA4-treated human melanoma cells lacking IFNγ signatures (Supplementary Fig. 7A and 7B). As these data and our data indicated IL-27-CD40 acts a tumor cell extrinsic feature, we next employed CytoSig to calculate the “influence score” of IFNγ, IL-27, and CD40 alone or combined, on the expression pattern of melanoma-intrinsic genes (known as melanocytic plasticity signature) reported to predict response to anti-CTLA4 (20). The signaling score can therefore be regarded as the “cytokine” effect over tumors. This time, melanoma samples with high IFNγ scores also exhibited high IL-27 signaling scores prior to treatment (Fig. 7A). As the IL-27-CD40 signature related to macrophages, we employed CIBERSORT to identify putative macrophage subtypes per tumor via deconvolution of transcriptomic data. This method calculated the ratio of one macrophage subset, such as non-activated (M0), pro-inflammatory (M1), or anti-inflammatory (M2), over the global macrophage signature. Examination of the relation between the individual influence scores of IFNγ, IL-27, and CD40 over macrophage subsets in melanoma samples found high signaling scores inversely correlated with the ratio of intratumoral M2 macrophages prior to treatment (Fig. 7B). To further investigate what this association means for therapeutic response, we broke down the melanoma patient cohort into two subsets, non-responders (NR) and better responders (R+LS), which included samples from responders (R) and patients with long term survival (LS). Although subgroups showed no significant individual difference in pre-treatment scores of IFNγ, IL-27 or IFNγ/IL-27 (Fig. 7C), high IL-27 signaling correlated with high CD40L signaling scores among better responders (R+LS) (Fig. 7D), indicating that the IL-27/CD40 signature identified melanoma patients that most likely benefit from anti-CTLA4 or could be potential candidates for agonist therapy.
Figure. 7. The CD40/IL27 signature associates with better response to ICB in human melanoma.
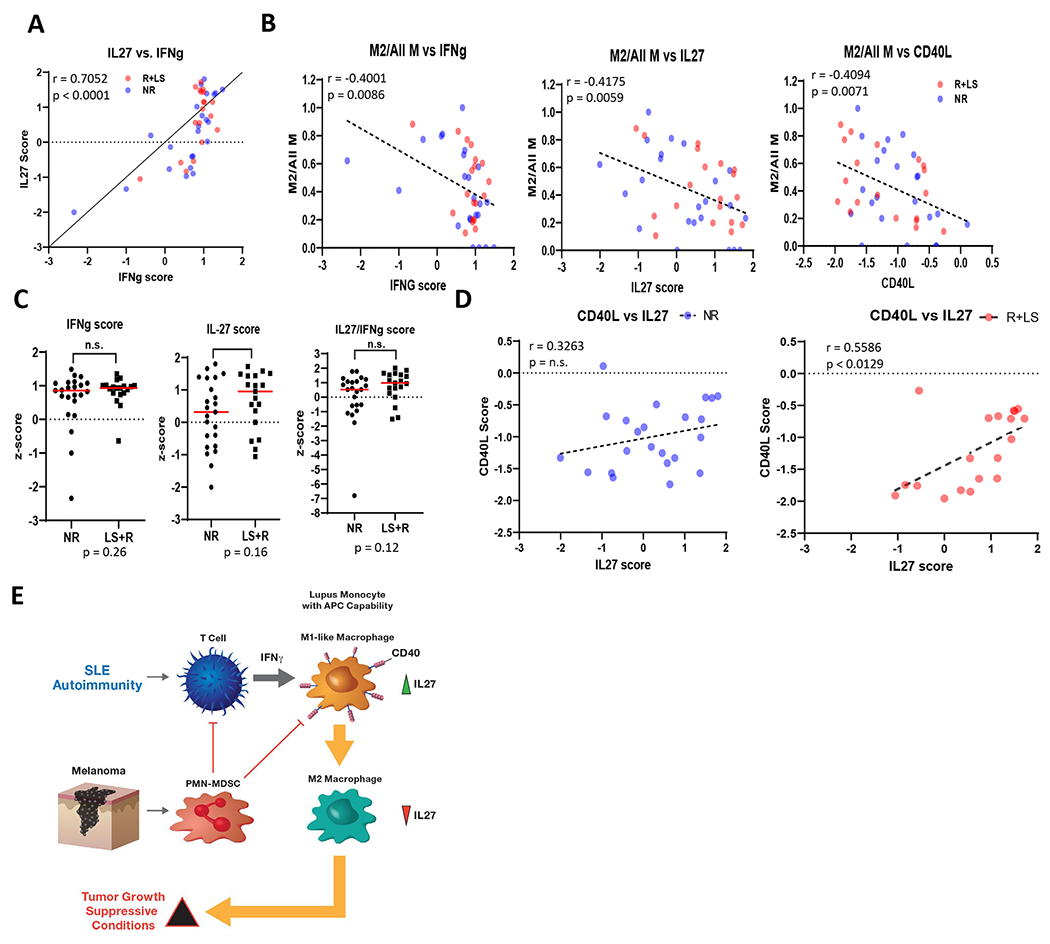
Transcriptomic data of melanoma tumors from a clinical study of CTLA4 were analyzed to generate signaling scores as described in Methods. (A), Correlation of IL27 scores (vertical axis) with IFNγ scores (horizontal axis) prior to treatment. (B) Correlation of the ratio of M2 macrophage subtype signature, as determined by CIBERTSORT. Data shown as the M2 macrophage signature over total tumor-associated macrophage (M2/All M) signatures. (C) Following the result in A, comparison of signaling scores (z-score) in non-responder patients (NR) and in responders (R) plus long-survival (LS) patients (R+LS). (D) Following the result in C, calculation of scores for CD40L signaling, which includes CD40, significantly improved correlation with IL27 scores only in the responsive (R+LS) group (right panel), but not in non-responders (NR) (left panel). (E) Cartoon details how PMN-MDSC inhibits IL-27 production and CD40 expression in ARE monocytes to induce M2-polarization and TME suppression.
DISCUSSION
Different from single disease models, the concurrent existence of cancer and autoimmunity in patients challenge the role of autoimmune features and IFN signaling in promoting systemic inflammation and suppressing tumor development. As tumor growth leans on both tumor-cell intrinsic features (e.g., IFNγ sensitivity), and manipulation of external settings (e.g., infiltrating cells), we report here that melanoma expansion of SLE-generated PMN-MDSC establishes immune evasion in a SLE-prone model of chronic autoimmunity (ARE mice). Thus, PMN-MDSC lessened CD40 expression and suppress IL-27 signaling in Ly6Chi monocytes, two factors associated with dysfunctional myeloid responses in SLE (28, 37). Blockade studies of IL-27p28 or depletion of Ly6G cells in ARE mice demonstrated such inactivation leads to global expansion and subsequent infiltration of F4/80 macrophages into tissues and tumors, which co-establish suppressive TME conditions and worsens tubular renal damage in tumor bearing ARE mice. Although administration of exogenous IL-27 counteracted those outcomes for melanomas sensitive to IFNγ, the known risk of T cell cytotoxicity compromised OS gains in the context of autoimmunity (40). Thus, we believe that manipulation of IL-27 by melanomas with a high density of tumor infiltrating lymphocytes, weakens tumor malignancy both in tumor-bearing WT mice or SLE-prone mice, respectively (41, 42).
Using side by side comparison of immune landscapes in type 1 IFNAR-deficient and -sufficient ARE mice challenged with and without pigmented or non-pigmented melanoma models, we identified that IL-27 levels act as an external indicator of monocyte function in autoimmune landscapes. Although IL-27 is a cytokine with context-defined inflammatory properties, its role is generally unnoticed in host autoimmune responses due to overlying functions with IFNs or the use of suppressive immunotherapy (43). The fact that glucocorticoids decrease IL-27 levels and induced M2-macrophage polarization is consistent with our proposal that IL-27 levels reflect the polarization status of macrophages and the status of autoimmunity. As loss of IL-27 signaling can reactivate dendritic cells and enhance peripheral pathogenic T cell responses (40), our finding that loss of type 1 IFN signaling increased IL-27 levels and lessened suppression capacity of PMN-MDSC in type 1 ifnar-deficient ARE mice (Fig. 4J) underscores the importance of modulating CD40/IL-27 signaling to limit side effects and moderate T cell responses in autoimmunity. Additional support for such an outcome originates from studies where loss of IL-27 had deleterious effects in human and mouse models of rheumatoid arthritis (RA), Sjogren’s syndrome, and SLE (42). The latter observations underscore the importance of assessing cytokine networks when immune-modulating comorbidities, such as melanoma, arise in patients with AID.
CD40 is an IFNγ-regulated molecule essential to tolerogenic immunity that is aberrantly expressed in SLE monocytes (37). Although the regulatory role of IL-27 over co-stimulatory molecules has been reported in dendritic cells from hosts without autoimmunity (40), we found that IL-27 also supports IFNγ regulation of CD40 in the pro-inflammatory phenotype of SLE monocytes. As melanoma inhibits CD40 signaling in monocytes, macrophages acquire a pro-tumoral phenotype by losing the reported ability to regulate secretion of IL-27 and other pro-inflammatory cytokines (44, 45). Such pro-tumoral function of macrophages was observed when tumors grew in CD40 (TNFRSF5), CD40L (CD154), or IL-27ra (WSX-1) knockout mice (7, 46). Importantly, this mechanism of innate dysfunction is not unique to melanoma, as intracellular pathogens reaching chronic phases such as Mycobacteria tuberculosis or Plasmodium falciparum, also expand MDSC and target CD40 or IL-27, respectively (47–49). Thus, our study identifies CD40/IL-27 signaling as an indicator of macrophage polarization status within co-existent autoimmune landscapes and cancer.
Our finding that metastatic melanoma exacerbates nephritis and limits survival gains in tumor-bearing ARE−/− mice, has also provided insights on the reported high risk association between cancer and membranous nephropathy (50). Although aberrant organ infiltration of macrophages and Ly6G+ granulocytic cells has been reported separately in lupus-prone mice and murine melanoma models (8, 17), our study is the first to define how macrophages commonly shape co-existent kidney and tumor immune landscapes. For example, we report here that melanoma-associated macrophages significantly infiltrate kidneys and tumors in ARE−/− mice. Therapeutically, instead of electing direct administration of IL-27 and risk the reported enhancement of CD8+ T cell responses and cytotoxicity (40), we opted for limiting melanoma-associated monocyte deactivation using agonist CD40 abs by levering the fact that IFNγ facilitates binding of agonist CD40 mAb to CD40 (51). We report that targeting CD40 in monocytes, rather than T cell exhaustion markers, prevented the decline of IL-27 and the inactivation of TAM precursors, an effect that had limited toxicity on lupus nephritis. Although anti-CD40 treated ARE mice were not tumor free, the anti-tumor activity of anti-CD40 outperformed that of anti-PD1 as monotherapy in the autoimmune landscapes of ARE mice. Nevertheless, we found that the enhanced potency of anti-PD1/CD40 exacerbated systemic inflammation and induced therapy resistance, via strong co-induction of pSTAT1, pSTAT3 and MDSC expansion (52).
Unlike prior cancer models, our model predicts that tumor progression and choice of therapy depends on how solid tumors impact pre-existent or newly diagnosed autoimmune conditions (Fig. 7E), such as those reported after ICB immunotherapy (53). Translational support for this model comes from transcriptomic data of melanoma tumors with IFNγ signatures in patients with better response to anti-CTLA4 (20, 24). The latter finding supports the relevance of CD40/IL-27 as an external modulatory factor complementing the predictive power for tumor cell intrinsic (MPS) factors (20). Recent clinical studies have shown that high circulation and infiltration of PMN-MDSC in melanoma patients was associated with a worst response and shorter survival to ICB (54, 55). Collectively, the combination of histologic, molecular, and functional data provides a detailed overview of how melanoma growth impacts nephritis in SLE-prone ARE mice. These data not only link the loss of CD40 regulation and IL-27 signaling to a poor therapeutic prognosis, but also sets a proof of principle for use of agonistic CD40 therapy to counteract the global immunosuppression generated by melanoma in hosts with previous or newly diagnosis AD.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Myeloid-derived suppressor cells induce macrophage reprogramming by suppressing CD40/IL-27 signaling to drive melanoma progression, simultaneously affecting underlying autoimmune disease and facilitating resistance to immunotherapy within pre-existing autoimmune landscapes.
ACKNOWLEDGEMENTS
This project has been funded in whole or in part with Federal funds directly from the intramural research programs of the National Cancer Institute, CCR, LCIM under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
The authors declare no competing financial interests.
REFERENCES
- 1.Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrand-Rosenberg S, Sinha P, Beury DW, and Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Seminars in Cancer Biology. 2012;22(4):275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ji J, Li P, Shen C, Dou H, Wang T, Shi L, et al. MDSCs: friend or foe in systemic lupus erythematosus. Cell Mol Immunol. 2019;16(12):937–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu H, Zhen Y, Ma Z, Li H, Yu J, Xu ZG, et al. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci Transl Med. 2016;8(331):331ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan P-Y, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 Is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Research. 2010;70(1):99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu C, Savage N, Singh N, and Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. The Journal of Immunology. 2017;198(1 Supplement):124.9–.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha P, Clements VK, Bunt SK, Albelda SM, and Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179(2):977–83. [DOI] [PubMed] [Google Scholar]
- 8.Iwata Y, Boström EA, Menke J, Rabacal WA, Morel L, Wada T, et al. Aberrant macrophages mediate defective kidney repair that triggers nephritis in lupus-susceptible mice. The Journal of Immunology. 2012;188(9):4568–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Q, Shilkrut M, Zhao Z, Li M, Batty N, and Barber B. Autoimmune comorbidities in patients with metastatic melanoma: a retrospective analysis of us claims data. BMC Cancer. 2018;18:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson DB, Sullivan RJ, Ott PA, Carlino MS, Khushalani NI, Ye F, et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncolog. 2016;2(2):234–40. [DOI] [PubMed] [Google Scholar]
- 11.Criscitiello C, Bagnardi V, Esposito A, Gelao L, Santillo B, Viale G, et al. Impact of autoimmune diseases on outcome of patients with early breast cancer. Oncotarget. 2016;7(32):51184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menzies AM, Johnson DB, Ramanujam S, Atkinson VG, Wong AN, Park JJ, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol. 2017;28(2):368–76. [DOI] [PubMed] [Google Scholar]
- 13.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167(6):1540–54 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M, et al. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell. 2020;38(4):500–15.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tocut M, Brenner R, and Zandman-Goddard G. Autoimmune phenomena and disease in cancer patients treated with immune checkpoint inhibitors. Autoimmunity Reviews. 2018;17(6):610–6. [DOI] [PubMed] [Google Scholar]
- 16.Mamlouk O, Selamet U, Machado S, Abdelrahim M, Glass WF, Tchakarov A, et al. Nephrotoxicity of immune checkpoint inhibitors beyond tubulointerstitial nephritis: single-center experience. Journal for ImmunoTherapy of Cancer. 2019;7(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark MR, Trotter K, and Chang A. The pathogenesis and therapeutic implications of tubulointerstitial Inflammation in human lupus nephritis. Seminars in nephrology. 2015;35(5):455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bae HR, Hodge DL, Yang GX, Leung PSC, Chodisetti SB, Valencia JC, et al. The interplay of type I and type II interferons in murine autoimmune cholangitis as a basis for sex-biased autoimmunity. Hepatology. 2018;67(4):1408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodge DL, Berthet C, Coppola V, Kastenmuller W, Buschman MD, Schaughency PM, et al. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice. J Autoimmun. 2014;53:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pérez-Guijarro E, Yang HH, Araya RE, El Meskini R, Michael HT, Vodnala SK, et al. Multimodel preclinical platform predicts clinical response of melanoma to immunotherapy. Nature Medicine. 2020;26(5):781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blasi E, Radzioch D, Merletti L, and Varesio L. Generation of macrophage cell line from fresh bone marrow cells with a myc/raf recombinant retrovirus. Cancer Biochem Biophys. 1989;10(4):303–17. [PubMed] [Google Scholar]
- 22.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165(1):35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167(2):397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bauer JW, Baechler EC, Petri M, Batliwalla FM, Crawford D, Ortmann WA, et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006;3(12):e491–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proceedings of the National Academy of Sciences. 1994;91(14):6458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jablonska J, Leschner S, Westphal K, Lienenklaus S, and Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest. 2010;120(4):1151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MH, Gallo PM, Hooper KM, Corradetti C, Ganea D, Caricchio R, et al. The cytokine network type I IFN-IL-27-IL-10 is augmented in murine and human lupus. Journal of Leukocyte Biology. 2019;106(4):967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarting A, Tesch G, Kinoshita K, Maron R, Weiner HL, and Kelley VR. IL-12 drives IFN-gamma-dependent autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol. 1999;163(12):6884–91. [PubMed] [Google Scholar]
- 30.Murugaiyan G, Mittal A, and Weiner HL. Identification of an IL-27/osteopontin axis in dendritic cells and its modulation by IFN-gamma limits IL-17-mediated autoimmune inflammation. Proc Natl Acad Sci U S A. 2010;107(25):11495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boswell JM, Yui MA, Burt DW, and Kelley VE. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. The Journal of Immunology. 1988;141(9):3050–4. [PubMed] [Google Scholar]
- 32.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7(9):929–36. [DOI] [PubMed] [Google Scholar]
- 33.Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. 2002;16(6):779–90. [DOI] [PubMed] [Google Scholar]
- 34.Zhao X, Ting SM, Liu CH, Sun G, Kruzel M, Roy-O’Reilly M, et al. Neutrophil polarization by IL-27 as a therapeutic target for intracerebral hemorrhage. Nat Commun. 2017;8(1):602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alicea-Torres K, Sanseviero E, Gui J, Chen J, Veglia F, Yu Q, et al. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat Commun. 2021;12(1):1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson APS, et al. Functional Crosstalk between Type I and II Interferon through the Regulated Expression of STAT1. PLOS Biology. 2010;8(4):e1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katsiari CG, Liossis S-NC, Souliotis VL, Dimopoulos AM, Manoussakis MN, and Sfikakis PP. Aberrant expression of the costimulatory molecule CD40 ligand on monocytes from patients with systemic lupus erythematosus. Clinical Immunology. 2002;103(1):54–62. [DOI] [PubMed] [Google Scholar]
- 38.Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, and Rakhmilevich AL. CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol. 2005;174(10):6013–22. [DOI] [PubMed] [Google Scholar]
- 39.Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells. Cancer Immunol Res. 2016;4(10):845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nature Immunology. 2013;14:1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimizu S, Sugiyama N, Masutani K, Sadanaga A, Miyazaki Y, Inoue Y, et al. Membranous glomerulonephritis development with Th2-type immune deviations in MRL/lpr mice deficient for IL-27 receptor (WSX-1). J Immunol. 2005;175(11):7185–92. [DOI] [PubMed] [Google Scholar]
- 42.Yoshida H, and Hunter CA. The immunobiology of interleukin-27. Annu Rev Immunol. 2015;33:417–43. [DOI] [PubMed] [Google Scholar]
- 43.Qiu F, Song L, Yang N, and Li X. Glucocorticoid downregulates expression of IL-12 family cytokines in systemic lupus erythematosus patients. Lupus. 2013;22(10):1011–6. [DOI] [PubMed] [Google Scholar]
- 44.Blahoianu MA, Rahimi AAR, Kozlowski M, Angel JB, and Kumar A. IFN-γ-induced IL-27 and IL-27p28 expression are differentially regulated through JNK MAPK and PI3K pathways independent of Jak/STAT in human monocytic cells. Immunobiology. 2014;219(1):1–8. [DOI] [PubMed] [Google Scholar]
- 45.Dibra D, Cutrera JJ, and Li S. Coordination between TLR9 signaling in macrophages and CD3 signaling in T cells induces robust expression of IL-30. J Immunol. 2012;188(8):3709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ford ML, Adams AB, and Pearson TC. Targeting co-stimulatory pathways: transplantation and autoimmunity. Nature Reviews Nephrology. 2014;10(1):14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samten B, Thomas EK, Gong J, and Barnes PF. Depressed CD40 ligand expression contributes to reduced gamma interferon production in human tuberculosis. Infect Immun. 2000;68(5):3002–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ayimba E, Hegewald J, Segbena AY, Gantin RG, Lechner CJ, Agosssou A, et al. Proinflammatory and regulatory cytokines and chemokines in infants with uncomplicated and severe Plasmodium falciparum malaria. Clin Exp Immunol. 2011;166(2):218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El Daker S, Sacchi A, Tempestilli M, Carducci C, Goletti D, Vanini V, et al. Granulocytic Myeloid Derived Suppressor Cells Expansion during Active Pulmonary Tuberculosis Is Associated with High Nitric Oxide Plasma Level. PLOS ONE. 2015;10(4):e0123772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lefaucheur C, Stengel B, Nochy D, Martel P, Hill GS, Jacquot C, et al. Membranous nephropathy and cancer: Epidemiologic evidence and determinants of high-risk cancer association. Kidney Int. 2006;70(8):1510–7. [DOI] [PubMed] [Google Scholar]
- 51.Singh M, Vianden C, Cantwell MJ, Dai Z, Xiao Z, Sharma M, et al. Intratumoral CD40 activation and checkpoint blockade induces T cell-mediated eradication of melanoma in the brain. Nat Commun. 2017;8(1):1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen PA, Ko JS, Storkus WJ, Spencer CD, Bradley JM, Gorman JE, et al. Myeloid-derived suppressor cells adhere to physiologic STAT3-vs STAT5-dependent hematopoietic programming, establishing diverse tumor-mediated mechanisms of immunologic escape. Immunological Investigations. 2012;41(6–7):680–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amos SM, Duong CP, Westwood JA, Ritchie DS, Junghans RP, Darcy PK, et al. Autoimmunity associated with immunotherapy of cancer. Blood. 2011;118(3):499–509. [DOI] [PubMed] [Google Scholar]
- 54.Krebs FK, Trzeciak ER, Zimmer S, Ozistanbullu D, Mitzel-Rink H, Meissner M, et al. Immune signature as predictive marker for response to checkpoint inhibitor immunotherapy and overall survival in melanoma. Cancer Med. 2021;10(5):1562–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gondois-Rey F, Paul M, Alcaraz F, Bourass S, Monnier J, Malissen N, et al. Identification of an Immature Subset of PMN-MDSC Correlated to Response to Checkpoint Inhibitor Therapy in Patients with Metastatic Melanoma. Cancers (Basel). 2021;13(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang P, Zhang Y, Ru B et al. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nat Methods 2021; 18: 1181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.