

Abstract
The nucleus accumbens (NAc) plays an important role in regulating multiple behaviors, and its dysfunction has been linked to many neural disorders. However, the molecular, cellular and anatomic heterogeneity underlying its functional diversity remains incompletely understood. In this study, we generated a cell census of the mouse NAc using single-cell RNA sequencing and multiplexed error-robust fluorescence in situ hybridization, revealing a high level of cell heterogeneity in this brain region. Here we show that the transcriptional and spatial diversity of neuron subtypes underlie the NAc’s anatomic and functional heterogeneity. These findings explain how the seemingly simple neuronal composition of the NAc achieves its highly heterogenous structure and diverse functions. Collectively, our study generates a spatially resolved cell taxonomy for understanding the structure and function of the NAc, which demonstrates the importance of combining molecular and spatial information in revealing the fundamental features of the nervous system.
The NAc is a key component of the basal ganglion circuitry, which integrates information from cortical and limbic regions to direct behaviors1. Previous research revealed that the NAc is involved in regulating diverse behaviors, including appetitive and aversive responses, feeding, social interaction, reinforcement and instrumental learning2–6. In addition, clinical and pre-clinical studies have linked NAc dysfunction to multiple neuropsychiatric disorders, including depression, anxiety, schizophrenia and substance abuse7–10. Consistent with its functional diversity, the NAc establishes complex neural connections with different brain regions11–14. Accumulating evidence suggests that specific input/output circuits of the NAc might underlie its different functions2,15–18.
Considering the functional and anatomic complexity of the NAc, understanding of the cellular composition and architecture remains limited. In general, over 90% of NAc neurons are GABAergic medium spiny neurons (MSNs), with a small percentage of interneurons (INs)19,20. The conventional direct/indirect pathway model divides the MSNs into D1 dopamine receptor-expressing and D2 dopamine receptor-expressing subtypes (D1 and D2 MSNs), based on their gene expression, connectivity and function20–23. Although this model provides a framework for understanding the cellular and circuitry organization of the striatum, it largely ignores intra-striatal heterogeneity and, thus, cannot well explain the anatomic and functional diversity of the NAc. For example, the striosome/matrix compartments in the dorsal striatum (DS) and the core/shell organization in the NAc exhibit discrete molecular and anatomic features24,25. Additionally, the MSNs located in different or even the same subregions of the NAc connect with different upstream and downstream targets14,16,26. Moreover, different neural functions have been attributed to different NAc subregions or even to the same neuron subtype (for example, D1 MSN) within close proximity16,27,28.
To understand the cellular basis underlying the anatomical and functional heterogeneity of the NAc, a molecularly defined and spatially resolved cell taxonomy is required to integrate its structural and functional complexity. Recent studies used single-cell gene expression profiling29–31 to characterize the cell composition of the striatum and the spatial profiles of marker gene expression, which revealed both discrete and continuous transcriptional programs underlying the MSN heterogeneity and suggested a correlation between gene expression and spatial distribution of MSNs29,30. However, how molecularly defined cell types contribute to the anatomic and functional heterogeneity of the NAc remains an open question. In this study, we analyzed the mouse NAc at single-cell resolution by combining single-cell RNA sequencing (scRNA-seq) and multiplexed error-robust fluorescence in situ hybridization (MERFISH)32,33, which not only revealed molecularly distinct neuron subtypes but also resolved how these cell types are spatially organized into NAc subregions. Furthermore, integrative analyses of the molecular and spatial features of NAc neuron subtypes enabled us to link different functions to different neuron substrates, illuminating the underlying molecular-cellular-anatomical-functional relationship of the NAc. The neural taxonomy presented here provides a foundation for the further understanding of how different neuron subtypes of the NAc are involved in various physiological and pathological conditions.
Results
scRNA-seq reveals major NAc cell types.
To systematically characterize the cell types of the mouse NAc, we performed scRNA-seq with cells dissociated from the NAc of adult mice (Fig. 1a and Extended Data Fig. 1a). In total, 11 biological samples were collected (Extended Data Fig. 1b and Supplementary Table 1). After quality control, we obtained 37,011 single cells. Our initial ‘low-resolution’ clustering analysis revealed nine major cell populations with distinct gene expression patterns, including four neuronal populations (Snap25+) and five non-neuronal populations (Snap25−) (Fig. 1b–d and Supplementary Table 2), which are present in all samples (Extended Data Fig. 1c). The five non-neuronal populations of the NAc are similar to those identified from other brain regions34–36 (Fig. 1b–d), consistent with their broad distribution in the brain. Because the numbers of unique molecular identifier (UMIs) and genes detected in non-neuronal cells are lower than those in neurons (Extended Data Fig. 1d), we further assigned non-neuronal cells with 800–1,500 genes detected in each cell to corresponding cell populations (Extended Data Fig. 1e,f). For neuronal cells, we identified D1 and D2 MSNs as the major neuron populations in the NAc, which constitute more than 90% of neurons (20,029 of 21,842 neurons), as well as an IN population (Fig. 1b–d). Another neuronal population represents neural stem cells and neuroblasts in the ventral wall of the lateral ventricle (Extended Data Fig. 1g). To characterize the neuronal heterogeneity of the NAc, we focused our further analysis on INs and MSNs, respectively.
Fig. 1 |. scRNA-seq reveals major cell populations in the NAc.
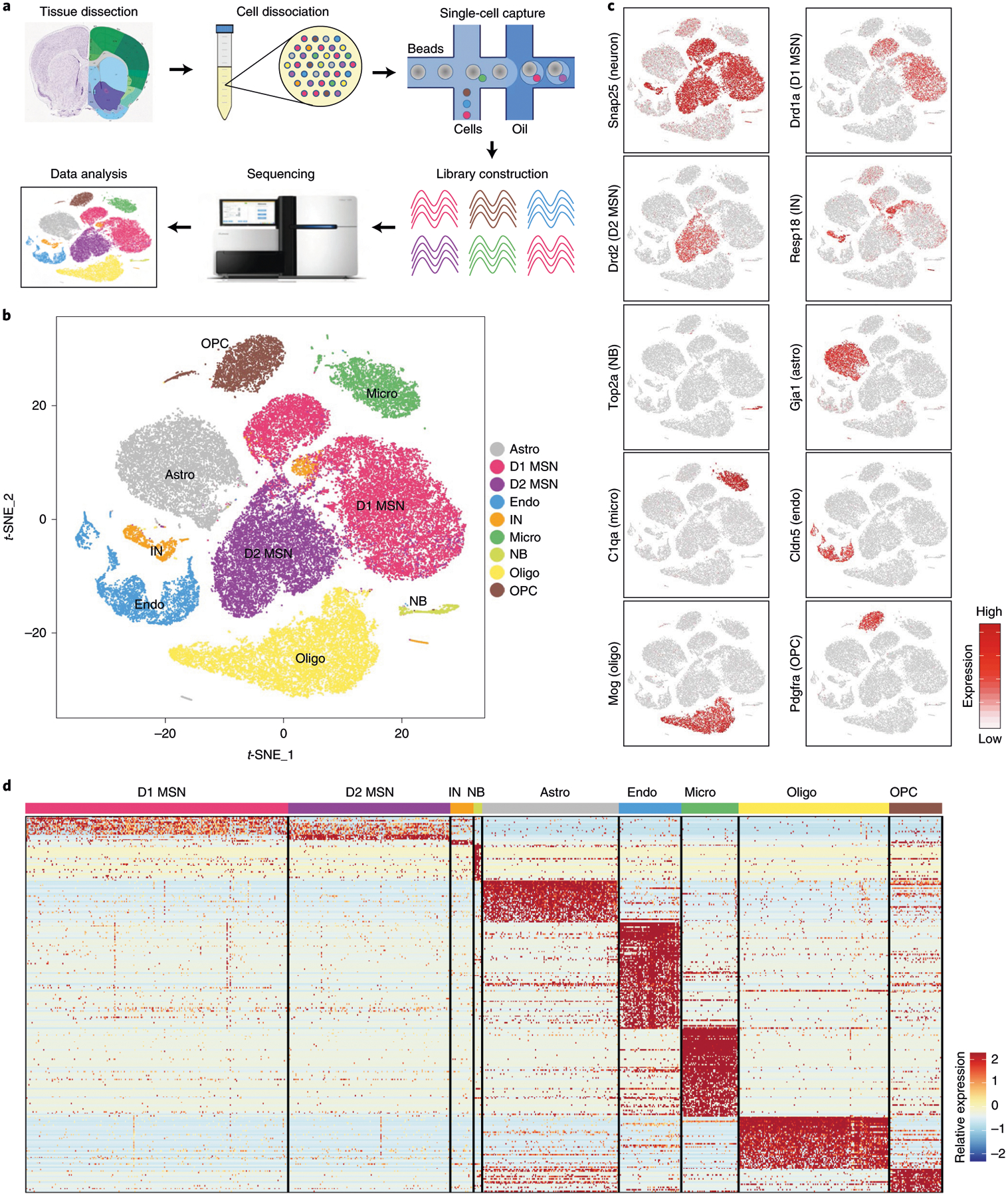
a, Workflow of scRNA-seq of mouse NAc. NAc tissues were dissected from adult mouse brain and dissociated into single-cell suspension. Single cells were captured into droplets with the 10x platform, followed by cDNA synthesis, amplification and library construction. After sequencing, cells were classified by their transcriptomes. b, t-SNE plot showing the different major cell types in the NAc. Different cell clusters are color-coded. c, t-SNE plots showing expression of cell-type-specific markers across different cell subtypes. The gene expression level is color-coded. d, Heat map showing that the cell-type-specific markers are differentially expressed across the nine NAc cell populations. DEGs with power >0.4 and fold change >2 among the nine cell clusters were used to generate the heat map. Columns represent individual cells, and rows represent individual genes. The gene expression level is color-coded. Astro, astrocyte; Endo, endothelial cell; Micro, microglia; NB, neural stem cells and neuroblasts; Oligo, oligodendrocyte; t-SNE, t-distributed stochastic neighbor embedding.
IN subtypes and their distribution in the NAc.
Although INs represent less than 10% of the striatal neurons, they exhibit diverse molecular, morphological and electrophysiological properties19. However, a detailed molecular classification of NAc INs has not been achieved. Further clustering of the initial IN population identified 13 subtypes (Fig. 2a,b and Extended Data Fig. 2a,b), and the marker genes for different IN subtypes were confirmed by in situ hybridization (ISH) (Extended Data Fig. 2c). Examination of the expression of conventional striatal IN markers in our IN subtypes revealed that some of these markers labeled single IN subtypes, such as Chat and Pvalb (Fig. 2b and Extended Data Fig. 2b, d). However, scRNA-seq also allowed us to classify conventional IN populations into different subtypes. For example, the Sst+ INs could be divided into Hhip+ and Hhip− subtypes (Fig. 2b and Extended Data Fig. 2b,d), whereas the Th+ INs can be classified into Trh+ and Calb2+ subtypes (Fig. 2b and Extended Data Fig. 2b,d). These IN subtypes might underlie the morphological and electrophysiological heterogeneity observed in Sst+ and Th+ INs19. Interestingly, we found that Pthlh is a pan-marker for three NAc IN subtypes exhibiting similar transcriptomes (Pthlh+/Pvalb+, Pthlh+/Crhbp+ and Pthlh +/Pvalb−/Crhbp− subtypes) (Fig. 2b and Extended Data Fig. 2b,d). In addition, Afap1 marks an IN subtype expressing Npy but not Sst (subtype 8) (Fig. 2b and Extended Data Fig. 2b), which likely represents the Npy+ neurogliaform cells37. The IN subtypes identified here are similar to those reported in the DS38.
Fig. 2 |. Gene expression and spatial pattern of NAc IN subtypes.
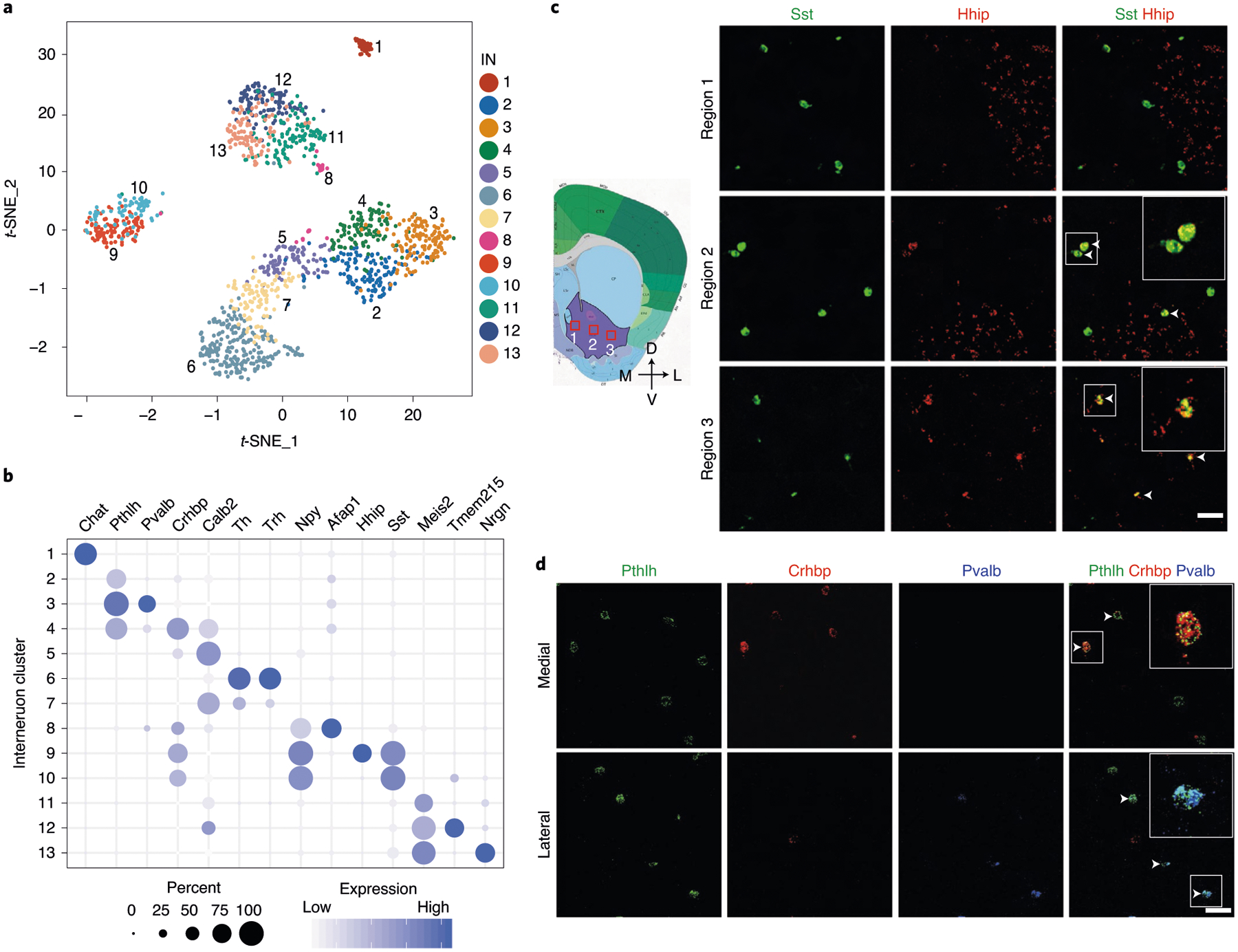
a, t-SNE plot showing the 13 IN subtypes identified in the NAc. DEGs among all subtypes are used for dimension reduction. Different neuron subtypes are color-coded. b, Dot plot showing the expression of selective markers in different NAc IN subtypes. The diameters of the dots represent the percentage of cells within a cluster expressing that gene. The gene expression level is color-coded. c, FISH showing the distribution of Sst+/Hhip+ and Sst+/Hhip− IN subtypes in different subregions of the NAc. The box regions labeled with 1–3 in the left panel indicate different subregions of the NAc (from medial to lateral), which are analyzed in the right panels (from upper to lower). The right panels show the FISH of NAc slice with Sst and Hhip probes. Arrowheads indicate cells co-expressing Sst and Hhip. Three independent experiments were performed with similar results. Scale bar, 100 μm. d, FISH showing the enrichment of distinct Pthlh+ IN subpopulations in different subregions of the NAc. The upper and lower panels represent the medial and lateral regions of the NAc. Triple-color FISH was performed with probes targeting Pthlh, Crhbp and Pvalb. Arrowheads indicate cells co-expressing Pthlh and Crbhp (upper panels) or cells co-expressing Pthlh and Pvalb (lower panels). Three independent experiments were performed with similar results. Scale bar, 50 μm. t-SNE, t-distributed stochastic neighbor embedding.
By performing FISH, we confirmed the identity of Th+/Calb2+, Th+/Trh+, Sst+/Hhip+, Sst+/Hhip−, Pthlh+/Pvalb+, Pthlh+/Crhbp+ and Pthlh+/Pvalb−/Crhbp− IN subtypes in the NAc (Fig. 2c,d and Extended Data Fig. 2e). Notably, different IN subtypes exhibit unique subregion-specific distribution in the NAc. For example, the Sst+/Hhip− and Sst+/Hhip+ INs are preferentially located in the medial and lateral NAc, respectively (Fig. 2c). Similarly, the Pthlh+/Pvalb+ INs are located mainly in the lateral NAc (Fig. 2d), whereas the Pthlh+/Crhbp+ neurons are mainly located in the medial NAc (Fig. 2d). Collectively, our analysis revealed diverse IN subtypes with distinct transcriptional and spatial features in the NAc.
MSN subtypes exhibit distinct spatial patterns in the NAc.
Based on the canonical MSN marker Ppp1r1b39, most neuronal cells captured in scRNA-seq belong to MSNs (Fig. 3a). Consistent with the direct/indirect pathway model20, the entire MSN population can be divided into discrete D1 and D2 MSNs based on established markers, such as Drd1, Pdyn, Drd2 and Adora2a (Fig. 3a). To further understand the heterogeneity within the D1 and D2 MSNs, we separately classified the D1 and D2 populations into eight subtypes each (Fig. 3b and Extended Data Fig. 3a). Consistent with a previous report30, most D1 and D2 MSN subtypes exhibit continuous transcriptional features, as most of the adjacent subtypes were not separated by clear gaps (Fig. 3b). Thus, although different MSN subtypes could be distinguished by the enriched expression of marker genes (Fig. 3c and Supplementary Table 3), D1 and D2 MSNs could each represent a largely continuous spectrum of cell populations.
Fig. 3 |. Transcriptional features of MSN subtypes correlate with their spatial distribution in the NAc.
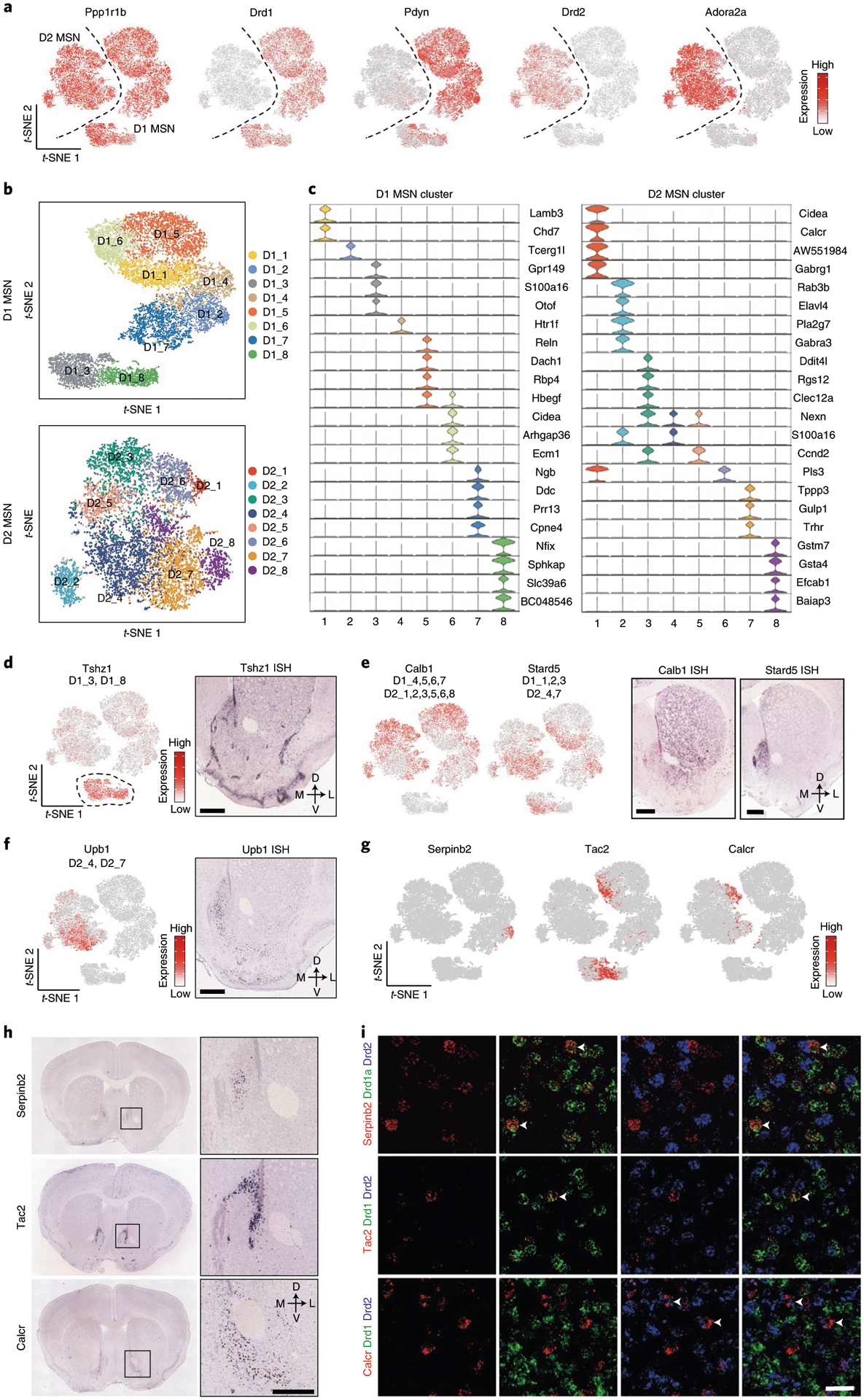
a, t-SNE plots showing the expression pattern of Ppp1r1b, Drd1, Pdyn, Drd2 and Adora2a in different MSN populations. The expression level is color-coded. b, t-SNE plots showing the eight D1 MSN subtypes (upper panel) and eight D2 MSN subtypes (lower panel); different subtypes are color-coded. c, Violin plots showing the expression pattern of MSN subtype-specific gene markers across the D1 MSNs (left) and D2 MSNs (right). Different MSN subtypes are color-coded. The mRNA level is presented on a log scale and adjusted for different genes. d, Tshz1 is enriched in a subpopulation of D1 MSNs. Left, t-SNE plots showing the expression pattern of Tshz1 across MSN populations. The expression level is color-coded. The MSN subtypes labeled by Tshz1 are indicated. Right, ISH of Tshz1 showing the distribution of Tshz1+ cells in the NAc. Coronal section of mouse brain including NAc is shown. Data were obtained from the Allen Mouse Brain Atlas. Scale bar, 500 μm. e, Calb1 and Stard5 showing anti-correlated expression pattern. Left two panels, t-SNE plots showing the expression of Calb1 and Stard5 as detected by scRNA-seq across D1 and D2 MSNs. The gene expression level is color-coded. The MSN subtypes labeled by Calb1 and Stard5 are indicated. Right two panels, ISH of Calb1 and Stard5 showing their anti-correlated expression pattern in the NAc. The ISH data are from the Allen Brain Atlas. Scale bars, 500 μm. f, Upb1+ D2 MSNs are distributed in the medial NAc. Left panel, t-SNE plot showing that Upb1 expression is restricted in certain D2 MSN populations. Right panel, ISH image showing the distribution of Upb1+ cells in the NAc. Data were obtained from the Allen Mouse Brain Atlas. Scale bar, 500 μm. g, t-SNE plots showing the expression of selected MSN subtype markers across MSNs. Transcriptional levels are color-coded. h, ISH images showing the spatial distribution of Serpinb2, Tac2 and Calcr, markers of certain D1/D2 MSN subtypes, in mouse NAc. Boxed regions in left panels are enlarged and shown in the right panels. The data are from the Allen Mouse Brain Atlas. Scale bars, 500 μm. i, Three-color FISH confirms the expression of MSN subtype-specific markers in the NAc. The arrowheads indicate cells that co-express selected MSN subtype markers with Drd1 or Drd2 in mouse NAc. Three independent experiments were performed with similar results. Scale bar, 50 μm. t-SNE, t-distributed stochastic neighbor embedding.
ISH of marker genes revealed that different MSN subtypes tend to locate in different NAc subregions. For instance, a subgroup of D1 MSNs (D1_3 and D1_8) expresses high levels of Tshz1, Lrpprc and Foxp2 but low levels of Foxp1 and Isl1 (Fig. 3d and Extended Data Fig. 3b). ISH indicated that these Tshz1-high cells mainly distribute along the boundary of NAc and form clusters within the NAc (Fig. 3d and Extended Data Fig. 3c). On the other hand, Ddit4l, which labels D1_4, D1_5 and D2_3, is selectively expressed in the dorso-lateral part of the NAc (Extended Data Fig. 3d), whereas Trhr, which mainly marks D1_1, D1_6 and D2_7, is selectively expressed in the medial part of the NAc (Extended Data Fig. 3e).
To assess the relationship between transcriptional feature and spatial distribution of MSN subtypes, we grouped the highly variable genes among MSN clusters based on their co-expression (Extended Data Fig. 3f). We found that different groups of variable genes exhibit different spatial patterns in the NAc, suggesting that the transcriptional profiles of MSN subtypes correlate with their anatomical distribution. Notably, many spatially related genes are broadly expressed in both D1 and D2 subtypes, indicating that a similar transcriptional program underlies the distribution patterns in both D1 and D2 MSNs (such as dorsal-ventral and medial-lateral patterns)30. For example, Calb1 and Stard5 belong to two anti-correlated gene groups, which are expressed in multiple D1 and D2 MSN subtypes (Fig. 3e). ISH demonstrated a clear bias of Calb1 in the lateral NAc and Stard5 in the medial NAc (Fig. 3e)30. On the other hand, some spatially related genes are restricted to either D1 or D2 subtypes, which might underlie the spatial pattern of certain D1/D2 MSN subtypes. For example, Upb1 is selectively expressed in a few D2 MSN subtypes located in the medial NAc (Fig. 3f).
Taking advantage of the large number of MSNs in our dataset, we further classified the D1 and D2 MSNs into 30 D1 and 27 D2 subtypes. These ‘fine’ D1 and D2 MSN subtypes could be distinguished in a few instances by unique markers but, otherwise, mostly by a combination of markers (Extended Data Fig. 3g,h). We analyzed the expression of some genes that uniquely mark MSN subtypes and found that these ‘fine’ MSN subtypes also exhibit distinct spatial patterns, even in restricted NAc subregions. For example, Serpineb2, which selectively marks D1 MSN subtype 14 (Fig. 3g and Extended Data Fig. 3g), is specifically expressed in a small cell population in the medial NAc (Fig. 3h)30. Another D1 MSN subtype (subtype 13), marked by Tac2 (Fig. 3g and Extended Data Fig. 3g), is also restricted to the medial NAc (Fig. 3h). However, the spatial patterns of these two subtypes are clearly different (Fig. 3h). On the other hand, Calcr, a marker for D2 MSN subtype 21 (Fig. 3g and Extended Data Fig. 3h), is expressed selectively in cells around the anterior commissure (AC) (Fig. 3h). Using FISH, we confirmed that Serpinb2 and Tac2 are selectively expressed in the Drd1+ neurons, whereas Calcr is expressed only in the Drd2+ neurons (Fig. 3i), confirming that they belong to distinct MSN subtypes. Collectively, these results reveal a tremendous MSN heterogeneity within the NAc and suggest a relationship between gene expression and spatial features of certain neuron subtypes.
Mapping distinct cell types of the striatum with MERFISH.
The scRNA-seq and ISH analyses indicate a correlation between transcriptional and spatial features of different neuron subtypes in the NAc. However, a comprehensive spatial analysis of all the cell subtypes requires analysis of sufficient genes at single-cell resolution in situ, which is not achievable with conventional ISH. To this end, we performed MERFISH, a single-cell transcriptome imaging method32,33, to systematically map the spatial patterns of different cell subtypes across the NAc.
For MERFISH experiments, we selected 253 gene targets (Supplementary Table 4), including marker genes for major cell populations and neuron subtypes from scRNA-seq, as well as genes with strong functional implications (for example, channels, neuropeptides and receptors). To cover the whole NAc region, we analyzed coronal sections between bregma 1.94 mm and 0.74 mm (100-μm interval between consecutive slices) of the adult mouse brain (Fig. 4a and Extended Data Fig. 4a), with the imaging area spanning the entire striatum.
Fig. 4 |. Mapping transcriptionally distinct cell types in mouse striatum with MERFISH.
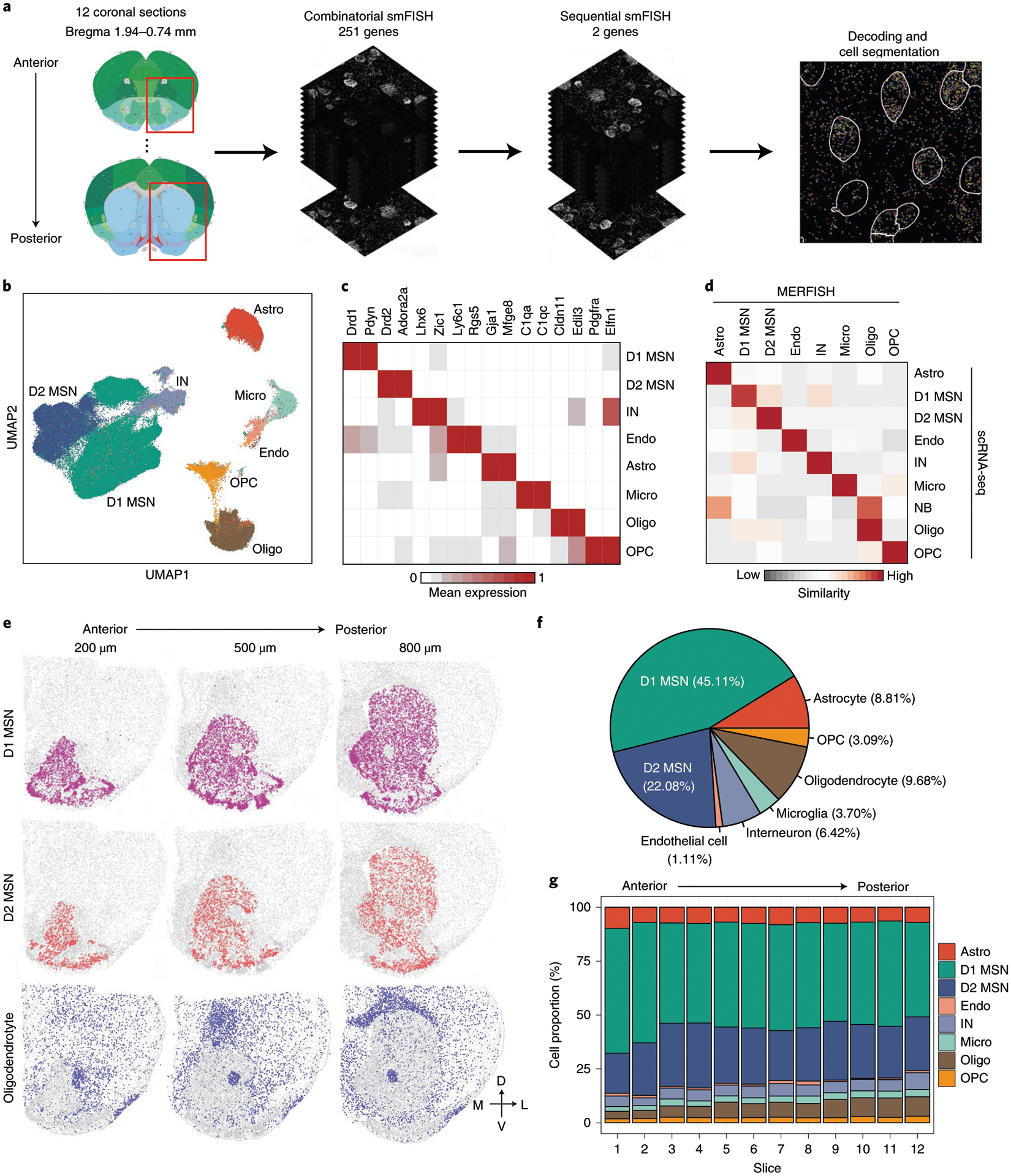
a, The workflow of MERFISH profiling of striatum tissue. b, UMAP plot showing the major striatal cell populations identified by MERFISH. The different cell populations are color-coded. c, Heat map showing the expression pattern of selected marker genes across the major striatal cell populations. The expression level is color-coded. d, Heat map showing the correspondence between scRNA-seq and MERFISH on classification of major striatal cell types. The similarity between scRNA-seq and MERFISH cell clusters is defined as the proportion of scRNA-seq cells that could be matched to each MERFISH cluster for each scRNA-seq cluster (see Methods for details). e, Spatial distribution of D1 MSNs, D2 MSNs and oligodendrocytes in brain slices at different AP positions. Three of the 12 slices from a male mouse are shown. Colored dots are cells belonging to the specified cell populations, whereas gray dots indicate all other cells. The 200-, 500- and 800-μm labels indicate the distance from the anterior position (bregma 1.94 mm). The dorsal-ventral (DV) and medial-lateral (ML) axes are illustrated. f, Pie chart showing the percentages of the major striatal cell populations detected by MERFISH. g, Bar graph showing the percentage of the major cell populations across the 12 brain slices along the AP axis. Astro, astrocyte; Endo, endothelial cell; Micro, microglia; NB, neural stem cells and neuroblasts; Oligo, oligodendrocyte.
Individual mRNA molecules were readily detected, decoded to determine their gene identities and assigned to individual cells based on DAPI and poly(A) staining (Extended Data Fig. 4b,c), thus generating the expression profile of target genes across all analyzed cells (Fig. 4a). Of the two replicates, MERFISH showed high consistency in detecting the mean number of different mRNA molecules (Extended Data Fig. 4d), and the gene expression level determined by MERFISH correlated well with that of bulk RNA-seq (Extended Data Fig. 4e). In addition, the spatial patterns of certain genes determined by MERFISH are consistent with that revealed by ISH (Extended Data Fig. 4f), validating the reliability of the analyses.
In total, we profiled over 700,000 cells in two biological replicates. Clustering analysis with the MERFISH data identified major cell populations representing D1 and D2 MSN, IN, astrocyte, endothelial cell, oligodendrocyte progenitor cell (OPC), oligodendrocyte and microglia (Fig. 4b) based on the expression of known markers (Fig. 4c). These major cell populations and their relative abundance exhibited little bias between the two replicates (Extended Data Fig. 4g,h), suggesting no significant batch effect. In addition, the cell types revealed by MERFISH are consistent with those determined by scRNA-seq (Fig. 4d), suggesting that MERFISH reliably captured molecularly distinct cell populations. Notably, the spatial patterns of major cell types revealed from MERFISH are well in line with known anatomic features: (1) the D1 and D2 MSNs span the entire striatum, including the DS, the NAc and the olfactory tubercle (OT) (Fig. 4e); (2) the oligodendrocytes are highly enriched in the AC and corpus callosum, with others dispersed across the striatum (Fig. 4e); and (3) INs, astrocytes, OPCs, endothelial cells and microglia are dispersed across the entire striatum (Extended Data Fig. 4i). These results demonstrate the capacity of MERFISH in resolving spatial patterns of molecularly defined cells in situ. Based on the MERFISH result, we estimated the cellular composition of the striatum (Fig. 4f) and found that the relative abundance of major neuronal and non-neuronal populations is largely stable across different coronal sections along the anterior–posterior (AP) axis of striatum (Fig. 4g).
Consistency of MERFISH and scRNA-seq results.
To reveal the spatial patterns of different neuronal subtypes, we classified the D1 and D2 MSN population into 15 D1 and nine D2 subtypes based on the MERFISH dataset (Fig. 5a, b). Similarly to scRNA-seq, many D1 and D2 MSN subtypes form continuous transcriptional spectra (Fig. 5a, b), consistent with a previous report30. These subtypes were named based on their spatial patterns (DS, dorsal striatum; NAc, nucleus accumbens; OT, olfactory tubercle; IC, islands of Calleja; AT, atypical). The similar uniform manifold approximation and projection (UMAP) distribution and relatively stable ratio for each of the D1 and D2 subtypes between the two MERFISH replicates suggest no obvious batch effect (Extended Data Fig. 5a–c). Different MSN subtypes showed enriched expression of different gene sets, but most of them do not have a single distinguishable marker (Fig. 5c,d, Extended Data Fig. 5d,e and Supplementary Table 5), suggesting that a complex transcriptional program underlies these subtypes. Integrative analysis comparing the neuron subtypes identified by scRNA-seq and MERFISH (Fig. 5e and Extended Data Fig. 5f–h) revealed that most D1 and D2 subtypes identified by the two methods have good correspondence (Fig. 5f,g). Furthermore, corresponding MSN subtypes from scRNA-seq and MERFISH show highly consistent expression patterns of subtype-enriched genes (Extended Data Fig. 5i).
Fig. 5 |. Identification of striatal D1 and D2 MSN subtypes by MERFISH.
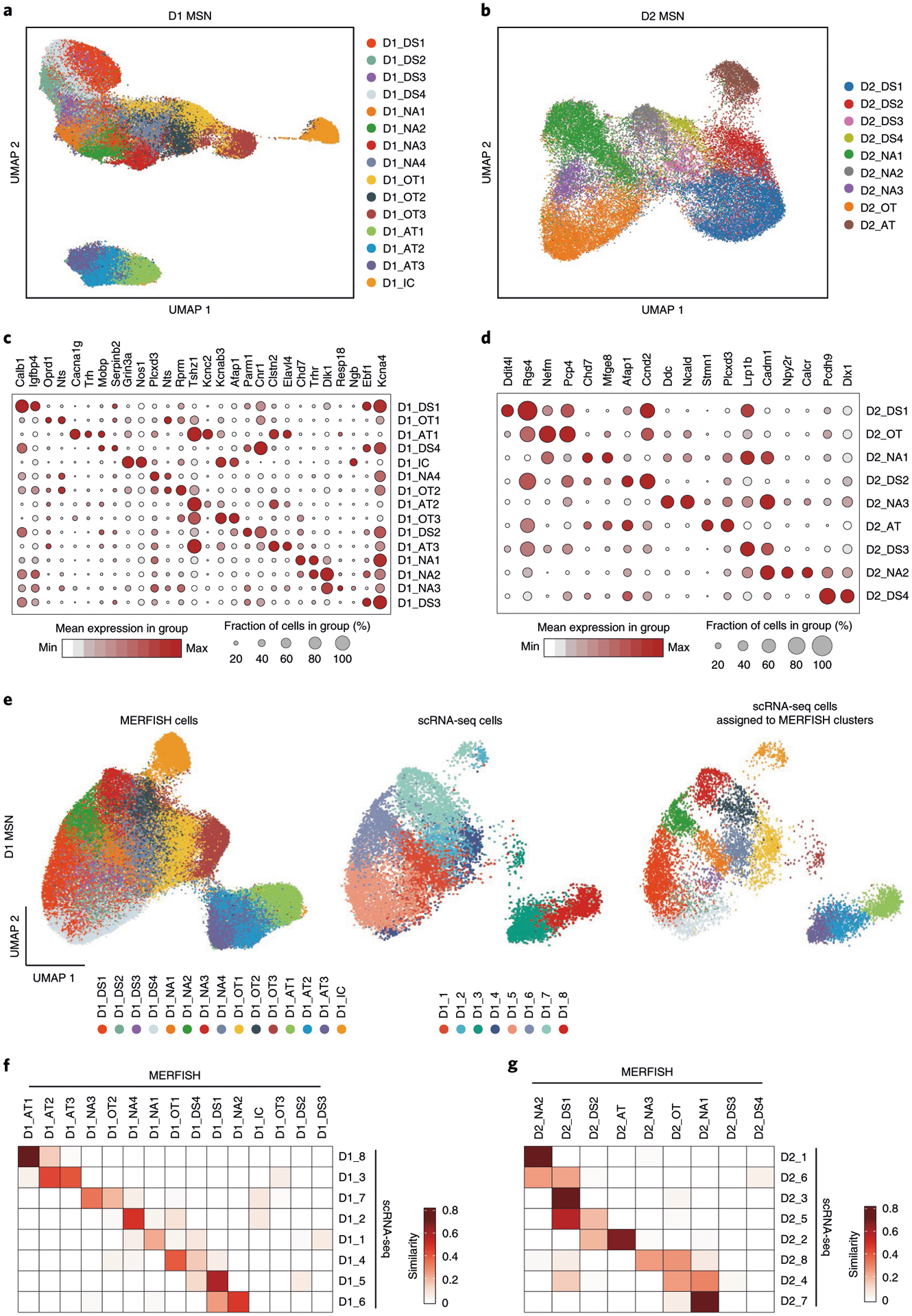
a, b, UMAP plot showing the 15 D1 MSN (a) and nine D2 MSN (b) subtypes based on MERFISH data. Different MSN subtypes are presented in different colors in the UMAP plot. The subtypes were named based on their spatial distribution: DS, dorsal striatum; NAc, nucleus accumbens; OT, olfactory tubercle; AT, atypical; IC, island of Calleja. c, d, Dot plot showing the expression pattern of selected genes across the 15 D1 (c) and nine D2 (d) MSN subtypes. The expression level is color-coded. Dot size represents the fraction of cells expressing the gene in each D1 or D2 subtype. e, Integrative analysis of D1 MSNs from MERFISH and scRNA-seq experiments. The D1 MSNs from MERFISH (left) and scRNA-seq (middle) experiments were integrated into the same UMAP space. The identity of each cell is color-coded. Based on the nearest neighbors from the MERFISH experiments, the cells from scRNA-seq were assigned to one of the MERFISH D1 MSN subtypes and are shown in the right panel. f, g, Heat map showing the correspondence between D1 (f) and D2 (g) MSN subtypes revealed by MERFISH and scRNA-seq. The degree of similarity between scRNA-seq and MERFISH cell clusters is defined as the proportion of scRNA-seq cells that could be matched to each MERFISH cluster for each scRNA-seq cluster. The MERFISH subtypes without corresponding scRNA-seq subtypes were largely distributed outside of the NAc.
Nevertheless, some differences between scRNA-seq and MERFISH subtypes were noted, especially that some neuron subtypes revealed by MERFISH lack corresponding subtypes in the scRNA-seq dataset (Fig. 5f,g). These differences were caused mainly by the different coverage of the two datasets, as scRNA-seq focused mainly on the NAc, whereas MERFISH analyzed the entire striatum. Because most neuronal subtypes are enriched in certain striatal subregions (see below), the scRNA-seq data are depleted of the neuron subtypes largely located outside the NAc (Extended Data Fig. 5h). For example, the D1_DS2, D1_DS3, D2_DS3 and D2_DS4 subtypes from MERFISH are largely restricted to the DS; thus, the corresponding subtypes were not identified by scRNA-seq (Fig. 5f). Similarly, the MERFISH subtype representing IC cells (D1_IC) and OT ruffle cells (D1_OT3) were absent in scRNA-seq (Fig. 5f), because they were largely removed during tissue dissection for scRNA-seq. Overall, despite the differences caused by experimental coverage, the neuron subtypes identified by scRNA-seq and MERFISH are largely consistent, which laid a solid foundation for understanding the spatial features of these neuron subtypes.
MSN subtypes underlie the striatal structural heterogeneity.
By plotting MSN subtypes identified by MERFISH in serial striatal sections, we found that different MSN subtypes exhibit distinct spatial patterns in individual sections (Fig. 6a,b). Additionally, most MSN subtypes exhibit biased distribution along the AP axis (Fig. 6c,d), which is in sharp contrast to the largely even distribution of the whole D1/D2 populations along the AP axis (Fig. 4g), suggesting that the anatomic heterogeneity of striatum is at least partially caused by the differential spatial distribution of MSN subtypes.
Fig. 6 |. The molecular and spatial features of MSN subtypes underlie anatomic organization of the striatum.
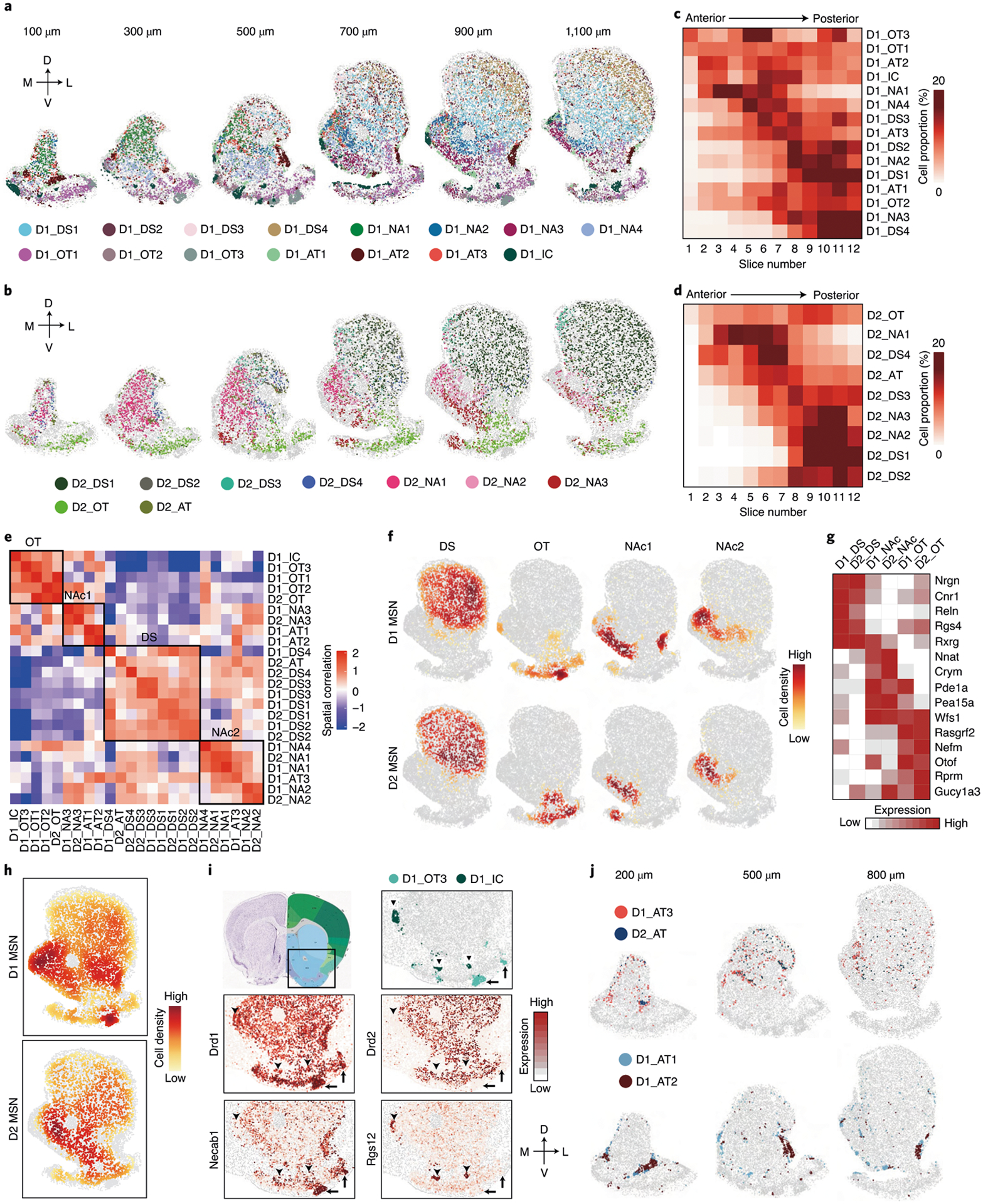
a, b, Spatial patterns of different D1 MSN subtypes (a) and D2 MSN subtypes (b) in coronal sections at different AP positions. Six of the 12 slices from a male mouse are shown. Different subtypes are presented by different colors. The 100-, 300-, 500-, 700-, 900- and 1,100-μm labels indicate the distance from the anterior position (bregma 1.94 mm). The dorsal-ventral (DV) and medial-lateral (ML) axes are illustrated. c, d, Heat maps showing the proportion of each D1 MSN (c) and D2 MSN (d) subtype across the 12 coronal sections along the AP axis. e, Heat map showing the spatial correlation across different D1 and D2 MSN subtypes. The black boxes illustrate cell types located in different major striatal divisions, including DS, OT and NAc. f, Density maps showing spatial enrichment of different subtype groups of D1 and D2 MSNs in striatal subregions. The MSN subtype groups (DS, OT, NAc1 and NAc2) are the same as in e. The patterns of D1 and D2 MSNs are shown in the upper and lower panels, respectively. g, Heat map showing the expression patterns of selected genes in different subtype groups of D1 and D2 MSNs located in major striatal divisions. The subtype groups corresponding to different anatomic regions were determined according to e, but the two NAc groups were combined. h, Density maps showing overall spatial distribution of D1 and D2 MSNs across the striatum. The patterns of D1 and D2 MSNs are shown in the upper and lower panels, respectively. i, Specific D1 MSN subtypes represent OT ruffle and IC. The boxed region in the diagram indicates the regions shown in other panels. The OT ruffle and IC are indicated by arrows and arrowheads, respectively. The spatial patterns of D1_OT3 and D1_IC are shown in the upper right panel. The heat maps in the lower panels show the expression of Drd1, Drd2, Necab1 (marker for D1_OT3) and Rgs12 (marker for D1_IC) in the corresponding region. j, Spatial patterns of three atypical D1 MSN subtypes and one atypical D2 MSN subtype in the striatum at different AP positions. Different subtypes are presented by different colors. MSN subtypes with higher spatial correlation are shown together. The 200-, 500- and 800-μm labels indicate the distance from the anterior position (bregma 1.94 mm).
To understand the relationship between MSN subtype distribution and the anatomic organization of striatum, we assessed the spatial co-localization of MSN subtypes. We found that different MSN subtypes are grouped into blocks; the subtypes within the same blocks are spatially closer to each other, whereas subtypes from different blocks are spatially separated (Fig. 6e). Analyzing the spatial patterns of MSN subtypes in different blocks indicated that they represent major striatal anatomic divisions (Fig. 6f and Extended Data Fig. 6a). Specifically, D1_DS1–D1_DS4, D2_DS1–D2_DS4 and D2_AT form the ‘DS’ block (Fig. 6e), representing the DS region (Fig. 6f and Extended Data Fig. 6a); the ‘OT’ block containing D1_OT1–D1_OT3, D1_IC and D2_OT (Fig. 6e) corresponds to the OT region (Fig. 6f and Extended Data Fig. 6a). Interestingly, the other MSN subtypes separated into two blocks, with the ‘NAc1’ block (D1_NA1, D1_NA2, D1_NA4, D1_AT3, D2_NA1 and D2_NA2) closer to the ‘OT’ block and the ‘NAc2’ block (D1_NA3, D1_AT1, D1_AT2 and D2_NA3) closer to the ‘DS’ block (Fig. 6e). They represent NAc MSN subtypes enriched in different subregions (Fig. 6f and Extended Data Fig. 6a). These results demonstrate that the spatial patterns of MSN subtype groups resemble major striatal subregions. By further comparing the gene expression of MSNs belonging to different blocks, we identified genes enriched in the three major subregions: DS, NAc and OT (Extended Data Fig. 6b,c and Supplementary Table 6). Many of these genes are shared between D1 and D2 MSNs (Fig. 6g), suggesting a global molecular program underlying major striatal anatomic divisions.
In addition to resolving DS, NAc and OT, the MSN subtypes further illustrate fine anatomic features within these subregions. For instance, the spatial and molecular features discriminate D1 MSN subtypes that represent the striosome (D1_DS2) and matrix (D1_DS1, D1_DS3 and D1_DS4) compartments25 in the DS (Extended Data Fig. 6d,e). Additionally, the D1_DS4, D1_DS1 and D1_DS3 form a medial-to-lateral pattern within the DS (Extended Data Fig. 6a), consistent with its anatomic and functional distinction along the medial-lateral axis40,41. Similarly, we found D2 MSN subtypes representing matrix (D2_DS1, D2_DS3)/striosome (D2_DS2) structure (Extended Data Fig. 6e) and medial (D2_DS3)/lateral (D2_DS1) striatum (Extended Data Fig. 6a). We noted that most D1 MSN subtypes have corresponding D2 subtypes that exhibit similar spatial patterns (Fig. 6e and Extended Data Fig. 6a). However, some D1 MSN subtypes lack parallel D2 subtypes, which form specific anatomic structures with high D1/D2 ratio (Fig. 6h). For instance, the D1_OT1 and D1_OT2 are distributed in the OT flat, with D2_OT occupying a similar region (Extended Data Fig. 6a). However, the D1_OT3 represents the OT ruffle structure, which contains only D1 MSNs, without a corresponding D2 subtype (Fig. 6i, arrows). Similarly, the IC is exclusively composed of D1_IC, which also lacks a parallel D2 subtype (Fig. 6i, arrowhead).
Consistent with scRNA-seq results (Fig. 3d and Extended Data Fig. 3b), MERFISH also revealed atypical MSN subtypes (D1_AT1, D1_AT2, D1_AT3 and D2_AT) with distinct transcriptional and spatial features (Fig. 5a,b). Comparing to other MSNs, Tshz1 and Foxp2 were enriched in the three D1_ATs, whereas Th and Tac1 were enriched in D2_AT (Extended Data Fig. 6f). Based on their gene expression, these atypical MSN subtypes correspond to the recently reported eSPN/D1-H SPN (D1_ATs) and patch-like D2H SPN (D2_AT)29–31. Our analysis resolved three D1_ATs with distinct transcriptional and spatial features (Fig. 6j, Extended Data Fig. 6g and Supplementary Table 7). Specifically, the D1_AT3 (Tshz1+/Spon1+) aggregates into a patch-like structure along the border of the anterior striatum, which gradually decreases and shifts to the dorso-medial NAc along the AP axis, with increasing non-aggregating D1_AT3 cells scattered in both the DS and NAc (Fig. 6j). On the other hand, D1_AT2 (Tshz1+/Pdyn+/Penk+) forms a dense patch structure at the ventro-lateral corner of the NAc, whereas D1_AT1 (Tshz1+/Cacna1g+) forms cell clusters along the border of the NAc (Fig. 6j). Interestingly, D2_AT and D1_AT3 show similar distribution patterns (Fig. 6j), suggesting that they are corresponding MSN subtypes. On the other hand, D1_AT1 and D1_AT2 exhibit high spatial correlation (Fig. 6e,i) but lack corresponding D2 subtypes. The transcriptional and spatial features of these atypical MSN subtypes suggest that they might be involved in distinct functions42. Collectively, these results suggest that the spatial distribution of MSN subtypes underlie the anatomic complexity of the striatum.
Substantial MSN heterogeneity in the NAc along the AP axis.
The MSN subtypes in the NAc exhibit diverse distribution patterns along the AP axis. First, the MSN subtype composition in the NAc varies from anterior to posterior, as several D1/D2 subtypes exhibit strong bias along the AP axis. For example, D1_NA1, D1_NA4 and D2_NA1 are enriched in the anterior NAc, whereas D1_NA2, D1_NA3, D2_NA2 and D2_NA3 are enriched in the posterior NAc (Fig. 7a,b and Extended Data Fig. 7a,b). The sharp contrast of MSN subtype composition between the anterior and posterior NAc aligns with the anatomic and functional heterogeneity along the AP axis6. Second, the spatial pattern of MSN subtypes also varies from the anterior to posterior NAc. In the anterior NAc, the MSN subtypes form either a dorsal-ventral pattern (D1_NA1 and D1_NA4) (Fig. 7a) or a largely homogenous distribution (D2_NA1) (Extended Data Fig. 7a), whereas, in the posterior NAc, the MSN subtypes form a core/shell-like structure. Currently, a core/shell organization of the rodent NAc has been widely accepted, but the underlying cellular basis remains unclear. Our data revealed that, in the posterior NAc (700–1,100-μm slices), four major MSN subtypes (D1_DS1, D1_NA2, D2_DS1 and D2_NA2) occupy the region surrounding the AC, which is considered as the NAc core (Fig. 7c and Extended Data Fig. 7c). Other MSN subtypes are located largely surrounding this region, which is considered as the NAc shell (Fig. 7d and Extended Data Fig. 7c). The density map of different MSN groups clearly illustrates the core/shell subregions (Fig. 7e). Notably, this MERFISH-defined core/shell pattern could not be simply recapitulated by the expression pattern of any single gene, although some genes (for example, Calb1 and Gucy1a3) are highly enriched in MSN subtypes located in the core/shell region (Extended Data Fig. 7d), suggesting that complex transcriptional programs underlie the core/shell organization. On the other hand, although most MSN subtypes are largely restricted to either the core or the shell, they do occupy overlapping regions (Extended Data Fig. 7c). This might explain why previous studies using limited marker genes could not identify a definitive border of the core/shell subregions12,24. Together, our analyses reveal substantial neural heterogeneity along the AP axis of the NAc and suggest that spatial enrichment of specific MSN subtypes contributes to the anatomic features of the NAc, including the core/shell division.
Fig. 7 |. The molecular and spatial features of MSN subtypes underlie anatomic heterogeneity of the NAc.
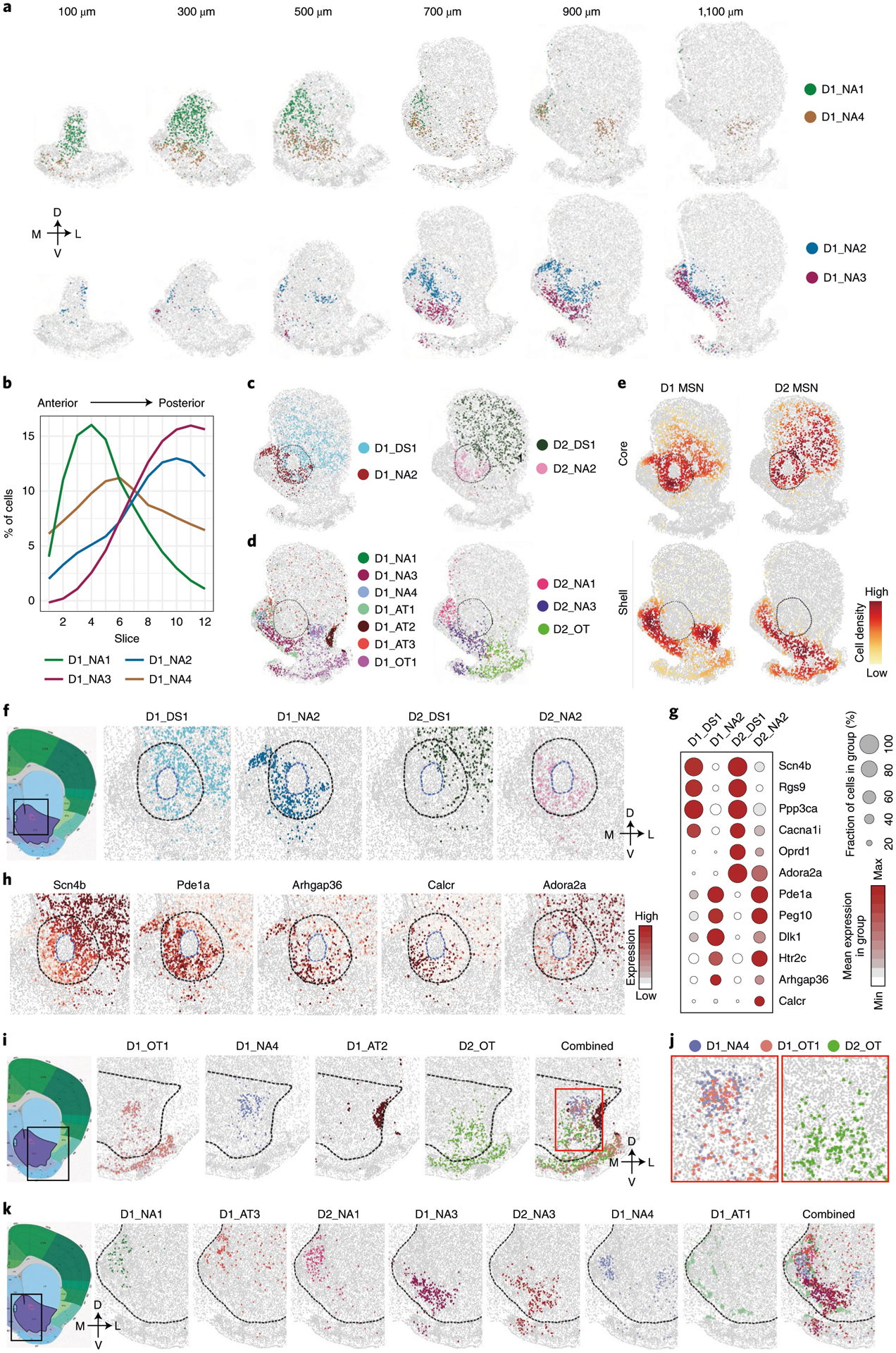
a, Spatial patterns of selected D1 MSN subtypes in coronal sections of different AP positions. The subtypes enriched in the anterior and posterior NAc are shown in the upper and lower panels, respectively. Different subtypes are presented by different colors. The 100-, 300-, 500-, 700-, 900- and 1,100-μm labels indicate the distance from the anterior position (bregma 1.94 mm). The dorsal-ventral (DV) and medial-lateral (ML) axes are illustrated. b, The distribution of the four D1 MSN subtypes shown in a along the AP axis. c, The spatial pattern of D1 (left) and D2 (right) MSN subtypes enriched in the NAc core. Different subtypes are shown in different colors. The dashed line indicates the NAc core. d, The spatial pattern of D1 (left) and D2 (right) MSN subtypes enriched in the NAc shell. Different subtypes are shown in different colors. e, Density maps showing the spatial pattern of D1 and D2 MSN subtypes enriched in either core or shell subregion of the NAc. The subtypes enriched in core and shell subregions of the NAc are listed in c and d, respectively. The core region is labeled with a dashed line. f, Spatial pattern of different MSN subtypes in the NAc core. The boxed region in the diagram indicates the regions analyzed in the other panels. The NAc core and AC structure are indicated with black and blue dashed lines, respectively. The DV and ML axes are indicated. g, Dot plot showing the expression of selected genes across D1 and D2 MSN subtypes enriched in different subregions of the NAc core. The expression level is color-coded. Dot size represents the fraction of cells expressing the gene in each subtype. h, Heat map showing the differential expression of Scn4b, Pde1a, Arhgap36, Calcr and Adora2a between the dorso-lateral and ventro-medial part of the NAc core. The same region as in f is shown. The gene expression level is color-coded. i, Spatial patterns of different MSN subtypes in the lateral shell of the NAc. The boxed region in the diagram is shown in the other panels. Different MSN subtypes are color-coded. The dashed line represents the border of the NAc. The DV and ML axes are indicated. j, The red box region in i is enlarged to show the spatial distribution of D1_NA4, D1_OT1 and D2_OT in the dorsal part of the NAc lateral shell. k, Spatial patterns of different MSN subtypes in the medial and ventral shell of the NAc. The boxed region in the left panel was analyzed in the other panels. Different MSN subtypes are color-coded. The dashed line represents the border of the NAc. AC, anterior commissure.
MSN heterogeneity in the core/shell subregions of the NAc.
In addition to the differences between the NAc core and shell described above, our data uncovered previously unrecognized heterogeneity within the core and shell subregions. In the NAc core, MERFISH revealed a ventro-medial-to-dorso-lateral patten: the D1_NA2 and D2_NA2 subtypes are preferentially located in the ventro-medial part of the core, whereas D1_DS1 and D2_DS1 subtypes are enriched in the dorso-lateral part of the core (Fig. 7f and Extended Data Fig. 7e). This pattern suggests that the dorso-lateral part of the NAc core possesses DS features12, as D1_DS1 and D2_DS1 are two of the major MSN subtypes in the DS (Fig. 7c and Extended Data Fig. 7c). To gain molecular insights into the ventro-medial–dorso-lateral pattern of the NAc core, we identified differentially expressed genes (DEGs) between MSN subtypes enriched in the two subregions (Fig. 7g, Extended Data Fig. 7f and Supplementary Table 8). As expected, these DEGs exhibited substantial bias in corresponding subregions (Fig. 7h and Extended Data Fig. 7g), and many DEGs are shared between D1 and D2 subtypes (Fig. 7g), suggesting a common transcriptional program underlying the cellular heterogeneity of the NAc core. Interestingly, some DEGs are D1 or D2 MSN specific, such as Arhgap36 (D1) and Calcr and Adora2a (D2) (Fig. 7g,h), suggesting that a cell-type-specific mechanism is also involved. Because quite a few DEGs are pertinent to neuronal function, including genes encoding receptors (Adora2a, Htr2c, Calcr and Oprd1), channels (Cacna1i and Scn4b) and enzymes (Ppp3ca and Pde1a) (Fig. 7g,h), MSN subtypes enriched in different NAc core subregions can differentially respond to upstream signals and might have distinct neuronal functions.
Previous studies revealed prominent anatomic and functional heterogeneity within the NAc shell6,16,24,27,43. Consistently, we found that MSN subtypes within this region form a complex topographic map. First, at least seven D1 (D1_NA1, D1_NA3, D1_NA4, D1_AT1, D1_AT2, D1_AT3 and D1_OT1) and three D2 (D2_NA2, D2_NA3 and D2_OT) MSN subtypes have considerable distribution within the NAc shell (Fig. 7d and Extended Data Fig. 7c), whereas only two D1 and two D2 subtypes are enriched in the NAc core (Fig. 7c and Extended Data Fig. 7c). In addition, different MSN subtypes exhibit distinct spatial patterns, leading to different neuronal composition in the lateral and medial part of the NAc shell. In the lateral shell, the D1_NA4 and D1_OT1 are located in its medial part (adjacent to the NAc core), whereas D1_AT2 forms a dense patch structure in the most lateral region (adjacent to cortex) (Fig. 7i). On the other hand, D2_OT is the dominant D2 subtype in the lateral shell (Fig. 7i). Interestingly, the D2_OT is enriched in the ventral part but depleted from the dorsal part of the lateral shell; thus, the dorso-lateral shell is largely occupied by D1 MSNs (Fig. 7j). This observation was confirmed by MERFISH and ISH detection of Drd2 (Extended Data Fig. 7h), suggesting that the dopamine signaling in this region is predominantly mediated by D1 MSNs (D1_NA4 and D1_OT1).
Compared to the lateral shell, the medial shell exhibits even higher cellular and spatial heterogeneity. At least five D1 (D1_NA1, D1_NA3, D1_NA4, D1_AT1 and D1_AT3) and two D2 (D2_NA1 and D2_NA3) MSN subtypes are enriched in the medial shell (Fig. 7k). Among these subtypes, D1_NA1, D1_AT3 and D2_NA1 occupy the dorsal part of the medial shell (Fig. 7k and Extended Data Fig. 7i), whereas D1_NA3 and D2_NA3 mainly occupy the ventro-medial and ventral parts of the shell (Fig. 7k and Extended Data Fig. 7i). In addition, D1_NA4 mainly occupies the middle between D1_NA2 and D1_NA3 (Fig. 7k and Extended Data Fig. 7i), whereas D1_AT1 forms small cell clusters along the border of the medial shell (Fig. 7k). Despite their distinct spatial patterns, multiple MSN subtypes always co-occupy any given subregion in the medial shell (Fig. 7k).
Notably, the topological organization of the core/shell subregions described here are observed mainly in the posterior NAc (700–1,100-μm slices in our MERFISH dataset), whereas MSN subtypes and their distribution patterns are substantially different in the anterior NAc (100–500-μm slices; see above), which adds another layer of anatomic complexity to the NAc. Overall, our results reveal highly heterogeneous molecular and spatial features of NAc neuron subtypes, supporting the notion that neuronal diversity underlies the anatomic heterogeneity of this region.
Link MSN subtypes to potential specific neuronal functions.
Having established that the spatial pattern of MSN subtypes underlies the anatomic heterogeneity of the NAc, we wondered whether these subtypes are related to the functional complexity of the NAc. Indeed, the spatial distribution of MSN subtypes is also well aligned with functional heterogeneity observed in the NAc. For example, previous studies revealed functional differences between the NAc core and shell44–46. These core- or shell-specific functions could be linked to MSN subgroups located in corresponding regions (Fig. 7c–e). As another example, applying opioid receptor ligands to the medial NAc along the AP axis has been shown to have a different effect on hedonic response (‘hot spot’ and ‘cold spot’)47. This functional change correlates with the change of MSN subtypes along the AP axis (Fig. 7a,b and Extended Data Fig. 7a,b). Similarly, the functional difference of Pdyn+ (a pan-D1 MSN marker) MSNs that reside in the dorso-medial and ventro-medial NAc27 could also be explained by the enrichment of distinct Pdyn+ (D1 MSN) subtypes in these subregions (Fig. 7k and Extended Data Fig. 7i). Furthermore, a recent study showed that dopamine signaling in the ventro-medial NAc specifically encodes aversive stimuli17, and this same region is occupied by D1_NA3 and D2_NA3 (Fig. 7k and Extended Data Fig. 7i), suggesting that these two MSN subtypes might be the neuronal substrates of this region-specific dopamine signal. Notably, another recent study showed a specific role of the Tshz1+/Pdyn− D1 MSNs (corresponding to the D1_AT3) in aversive coding and learning42, providing strong evidence that molecularly defined MSN subtypes are functionally distinct.
Expression of functional relevant genes in MSN subtypes.
To gain molecular insights into the functional complexity of the striatum, we assessed the expression of genes relevant to neural function (neural peptide, receptors and ion channels) among different MSN subtypes and found that these genes exhibit diverse expression patterns (Fig. 8a). For example, Trhr (Trh receptor) is expressed mainly in the D1_NA1, D1_NA2, D2_NA1 and D2_NA2 in the NAc (Fig. 8a), suggesting that these MSN subtypes might mediate the intra-NAc function of Trh in feeding regulation48. On the other hand, Oxtr (oxytocin receptor), Drd3 (dopamine receptor 3) and Npy1r (NPY receptor 1) are highly enriched in D1_IC (Fig. 8a), suggesting that the IC might integrate divergent upstream signals to regulate behavior.
Fig. 8 |. The molecular and spatial features of neuron subtypes underlie functional complexity of the NAc.
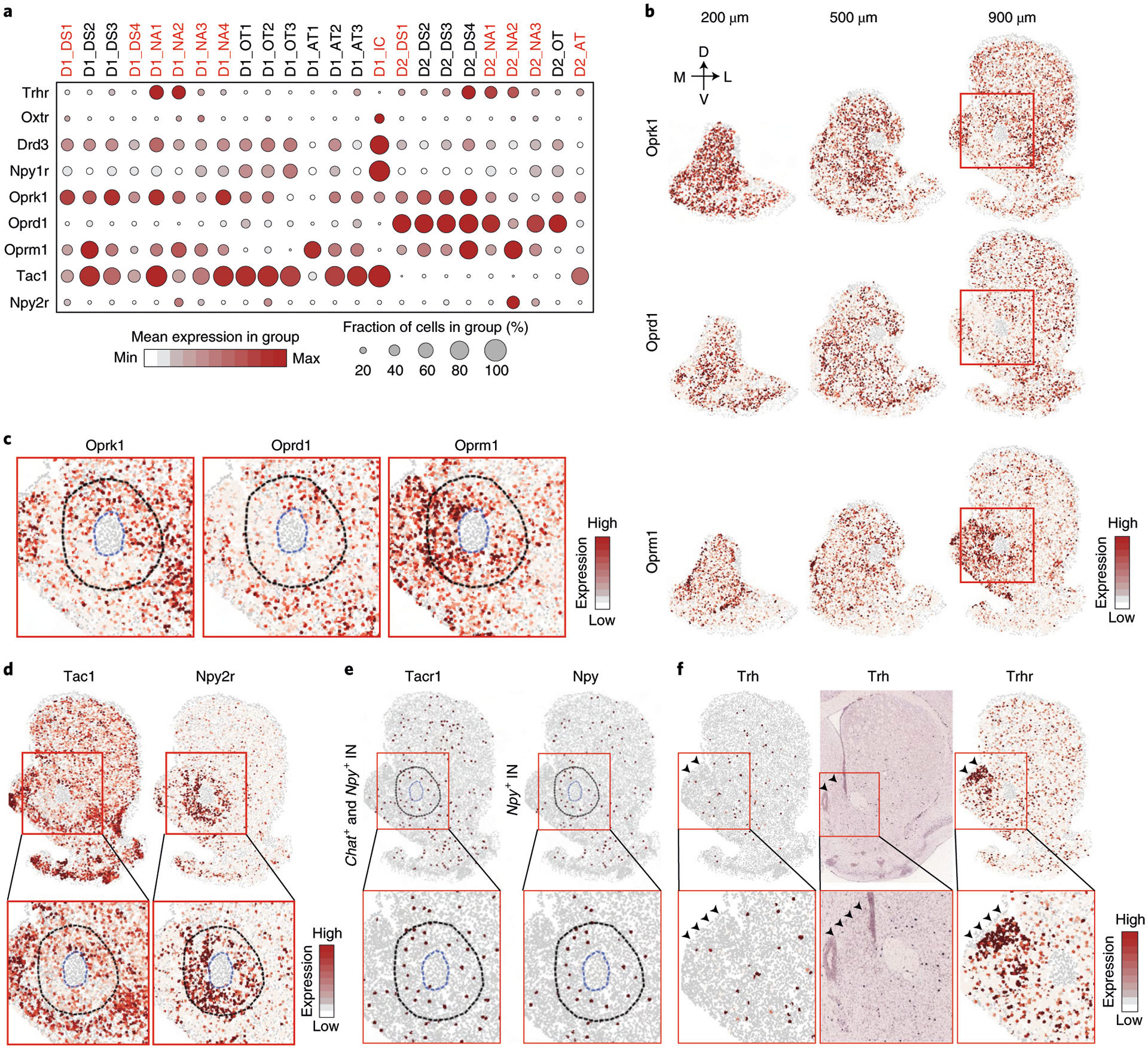
a, Dot plot showing the expression patterns of selected functional relevant genes across all the D1 and D2 MSN subtypes. The expression level is color-coded. Dot size represents the fraction of cells expressing the gene in each subtype. The MSN subtypes mentioned in the text are labeled in red. b, Heat map showing the expression of Oprk1, Oprd1 and Oprm1 in coronal sections at different AP positions. The expression level is color-coded. The 200-, 500- and 900-μm labels indicate the distance from the anterior position (bregma 1.94 mm). The dorsal-ventral (DV) and medial-lateral (ML) axes are indicated. c, Heat map showing the expression patterns of Oprk1, Oprd1 and Oprm1 in the NAc core. The boxed regions in b are enlarged and shown. The NAc core and AC structure are labeled with dashed lines. The gene expression level is color-coded. d, Heat map showing the expression patterns of Tac1 and Npy2r in the striatum. The boxed regions in the upper panels are enlarged and shown in the lower panels. The NAc core and AC structure are labeled with blue and black dashed lines, respectively. The gene expression level is color-coded. e, Heat map showing the expression of Tacr1 in Chat+ and Npy+ INs (left) and Npy in Npy+ INs (right). The boxed regions in the upper panels are enlarged and shown in the lower panels. The NAc core and AC structures are labeled with dashed lines. f, Heat map and ISH showing the expression pattern of Trh and Trhr in the striatum. Arrowheads indicate the dorso-medial NAc with enriched Trhr expression. The boxed regions in the upper panels are enlarged and shown in the lower panels. ACC, anterior cingulate cortex.
We paid special attention to opioid receptors genes as their functions are complex in the striatum49,50. Consistently, different opioid receptors exhibit distinct expression patterns (Fig. 8a–c). For example, Oprk1 (κ-opioid receptor) is expressed in most D1 and D2 MSN subtypes but with significant variation along the AP axis. Specifically, Oprk1 is enriched in D1_NA1 and D1_NA2 but depleted in D1_NA3, D2_NA2 and D2_NA3 (Fig. 8a). Because these two groups of MSNs are respectively biased toward the anterior and posterior parts of the NAc (Fig. 6c,d), Oprk1 exhibits a high-to-low expression pattern along the AP axis in the ventro-medial NAc (Fig. 8b). In contrast, Oprd1 (δ-opioid receptor) is restricted to D2 MSNs and expressed in most D2 subtypes (Fig. 8a). However, two D2 subtypes are significantly depleted in Oprd1 expression, including the D2_AT dispersed across the striatum (Fig. 6j) and the D2_NA2 located in the ventro-medial part of the NAc core (Fig. 7f). Because Oprd1 is highly expressed in the D2_DS1 that occupies the dorso-lateral core (Fig. 7f), we see substantial differential expression of Oprd1 in the NAc core along the ventro-medial–dorso-lateral axis (Fig. 8b,c). Interestingly, Oprm1 (μ-opioid receptor) is enriched in D1_NA2 and D2_NA2 but shows relatively low expression in the D1_DS1, D2_DS1, D1_NA1 and D2_NA1 (Fig. 8a), thus forming complementary spatial patterns to that of Oprk1 and Oprd1. Specifically, along the AP axis, Oprm1 exhibits a low-to-high pattern in the medial NAc, which is complementary to Oprk1 (Fig. 8b), whereas, in the NAc core, Oprm1 is enriched in the ventro-medial part, forming a complementary pattern to both Oprk1 and Oprd1 (Fig. 8c). The cell-type-specific and region-specific enrichment of different opioid receptors might underlie the complex interaction of different MSN subtypes51,52 and contribute to functional heterogeneity of NAc subregions6,47.
Potential region-specific and cell-type-specific interaction in the NAc.
In addition to inferring the functional properties of individual neuronal subtypes, our data could also imply potential cell–cell interactions53. To illustrate this, we focused on the neuropeptide–receptor pairs suggesting potential region-specific and cell-type-specific regulatory relationships between MSN and IN subtypes. For example, because Tac1 is expressed in most D1 MSN subtypes (Fig. 8a,d), and its receptor gene, Tacr1, is expressed in both Chat+ (cholinergic) and Npy+ INs (Fig. 8e), a widespread interaction between D1 MSN and these two IN subtypes through a Tac1–Tacr1 pathway is expected54. Interestingly, three D1 MSN subtypes (D1_NA2, D1_DS1 and D1_DS4) showed a significantly lower Tac1 level (Fig. 8a,d), suggesting that this interaction is attenuated in the subregions harboring these subtypes, including the NAc core (Fig. 8d) and the matrix in the DS. On the other hand, Npy+ IN (Fig. 8e) might exert a region-specific regulation on the NAc core through the Npy–Npy2r pathway, as Npy2r is enriched in the MSN subtypes (D1_NA2 and D2_NA2) located in this region (Fig. 8a,d). Notably, with spatial information, the cell–cell communication implied from gene expression could be evaluated more accurately. For instance, Trh is expressed in Th+/Trh+ INs (Fig. 8f), and Trhr is expressed in certain MSN subtypes (Fig. 8a,f). However, few Trh+ INs are distributed in the Trhr+ MSN-enriched region (Fig. 8f, arrowhead), suggesting that an additional Trh source, such as Trh+ neurons in the hypothalamus55, might act on these Trhr+ MSNs. Although our analysis focused only on genes directly detected by MERFISH, the results indicate that highly divergent cell–cell interactions might also contribute to the functional complexity of striatum, which depend on both transcriptional and spatial features of different neuronal subtypes.
Discussion
Although previous studies illustrated the complexity of striatal cell composition correlated with spatial organizions29–31, a systematic and high-resolution characterization of the spatial organization of striatal neuronal subtypes remains highly desirable to bridge the molecular and spatial features of cell types with the anatomic and functional heterogeneity of this brain region. By combining scRNA-seq and MERFISH, we generated a NAc taxonomy with transcriptional and spatial information at single-cell resolution. Our study not only revealed diverse neuronal subtypes with distinct molecular and spatial features but also linked the gene expression and spatial distribution of these neuron subtypes to the anatomic and functional complexity of the NAc, thus providing insights into how the NAc, which has a seemingly simple neuronal composition (D1 and D2 MSNs), accomplishes its observed structural and functional diversity.
The anatomic heterogeneity of the NAc, especially the core/shell division, has been recognized for decades24. However, the basis of these anatomic structures remains unclear, and definitive criteria distinguishing the core and shell are lacking. By assessing the spatial organization of MSN subtypes, we identified a cellular basis for the core/shell subregions. Furthermore, with the power of detecting hundreds of genes at single-cell resolution in situ, we uncovered novel anatomic complexity beyond the core/shell subregions. All these findings support the notion that molecularly defined neuronal subtypes and their spatial distribution is a fundamental factor underlying the anatomic organization of the NAc.
A large body of work has revealed the functional diversity of the NAc in health and disease1,7–10, but the general mechanism underlying these diverse functions is unclear. Our work helped fill in this knowledge gap by linking the functional diversity of the NAc to its different neuronal subtypes and their distinct molecular and spatial features. Notably, our data are consistent with the conventional dichotomy model, as most NAc subregions were well covered by both D1 and D2 MSNs, and most D1 MSN subtypes have corresponding D2 subtypes that show similar spatial distribution patterns. These data suggest that the D1/D2 MSN subtypes work in concert as fundamental components of computation across different NAc subregions.
Our transcriptomic-based NAc cell taxonomy can serve as a framework to help further understand the structure and function of the NAc. First, identification of transcriptionally different neuron subtypes in the NAc will facilitate the development of new genetic tools for monitoring and manipulating specific neuronal types. Second, the cell taxonomy could be used to annotate and infer neuronal substrates if genetic/molecular information (for example, transgenic line or marker gene staining) is available, making direct comparison of different studies possible. Third, the MERFISH and scRNA-seq approaches used in this study can be combined with neural tracing and activity mapping33,56 to integrate additional anatomic and functional information. Fourth, in addition to linking known anatomic and functional heterogeneity to different neuronal subtypes, our transcriptional and spatial analyses also revealed previously uncharacterized molecular and cellular heterogeneity in the NAc, which can serve as the basis for further studies.
Methods
Mice.
All experiments were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees of Boston Children’s Hospital and Harvard Medical School. For scRNA-seq, we used 10-week-old young adult male C57BL/6N mice (cat. no. 000664, Jackson Laboratory), and the NAc tissues were collected for scRNA-seq. For FISH and MERFISH assays, we used 8–10-week-old male C57BL/6N mice. The mice were housed in groups (3–5 mice per cage) at 22–24 °C and 20–30% humidity on a 12-h light/dark cycle, with food and water ad libitum.
Tissue dissection and dissociation.
Dissection and cell dissociation were performed in 11 separate experiments, with brain tissues from two mice pooled for cell dissociation and library preparation in each experiment. Each experiment was regarded as one biological replicate. No randomization or blinding were performed during sample collection as no comparisons of different conditions were performed. For single-cell dissociation of the NAc, the mice were anesthetized with isoflurane, and the brain was quickly removed and transferred into ice-cold Hibernate A/B27 medium (60 ml of Hibernate A medium with 1 ml of B27 and 0.15 ml of GlutaMAX). Coronal sections containing NAc were cut using a brain matrix and sliced into 0.5-mm slices in ice-cold Hibernate A/B27 medium. NAc tissue was then removed from each slice under a dissection microscope and subjected to tissue dissociation. NAc tissues were further dissociated into single-cell suspension using a papain-based dissociation protocol57 with some modifications. Briefly, the tissues from two animals were cut into small pieces and incubated in dissociation medium (Hibernate A-Ca medium with 2 mg ml−1 of papain and 2× GlutaMAX) at 30 °C for 35–40 min with constant agitation. After washing with 5 ml of Hibernate A/B27 medium, the tissues were triturated with fire-polished glass Pasteur pipettes ten times in 2 ml of Hibernate A/B27 medium to generate single-cell suspension, which was repeated three times. To remove debris, the 6 ml of single-cell suspension was loaded on a four-layer OptiPrep gradient57 and centrifuged at 800g for 15 min at 4 °C. Fractions 2–4 were then collected and washed with 5 ml of Hibernate A/B27 medium and 5 ml of DPBS with 0.01% BSA. The cells were spun down at 200g for 3 min and re-suspended in 0.2 ml of DPBS with 0.01% BSA. A 10-μl cell suspension was stained with trypan blue, and the live cells were counted. During the entire procedure, the tissues and cells were kept in ice-cold solutions, except for the papain digestion.
Single-cell capture, library preparation and sequencing.
The cell suspension was diluted with DPBS containing 0.01% BSA to 300–330 cells per microliter for single-cell capture. Single cells and barcoded beads were captured into droplets with the 10x Chromium platform (10x Genomics) according to the manufacturer’s protocol58. After cell capture, reverse transcription, cDNA amplification and sequencing library preparation were performed as described previously58. The libraries were sequenced on an Illumina HiSeq 2500 sequencer with paired-end sequencing (Read1: 26 bp; Index: 8 bp; Read2: 98 bp).
FISH (RNAscope) and imaging.
For sample preparation, young adult male (8–10-week-old) C57B6 mice (Jackson Laboratory) were anesthetized and perfused with PBS followed by 4% paraformaldehyde (PFA) in PBS. The whole brain was dissected and post-fixed in 4% PFA overnight, followed by dehydration in PBS containing 30% sucrose at 4 °C until the tissues sank to the bottom of the tube. The brains were frozen in optimal cutting temperature (OCT) embedding media, and 20-μm (for FISH) or 40-μm (for immunostaining) coronal sections were cut with cryostat. For FISH, the slices were mounted on Superfrost Plus slides, air dried and stored at −80 °C until use. For immunostaining, the slices were stored in PBS at 4 °C until use. The multi-color FISH experiments were performed following the manual of ACD RNAscope Fluorescent Multiplex Assay. For imaging, brain sections were imaged on a Zeiss confocal microscope (LSM800) with a ×10 (0.3 NA) or ×20 (0.8 NA) objective. z stacks were taken with 1-μm optical sectioning. For some sections, tiled images were acquired, which covered the whole NAc region.
Generation of single-cell gene expression matrix.
Raw sequencing data were processed with Cell Ranger (v1.3.1)58 for sample demultiplexing, cell barcode detection and single-cell expression matrix generation. Briefly, the ‘cellranger mkfastq’ command was used to demultiplex the different samples, extract the UMI barcodes and generate the FASTQ files. The gene–cell expression matrices for each sample were generated separately using the ‘cellranger count’ command by aligning the reads to the mm9 genome. In total, 11 single-cell gene expression matrices corresponding to the 11 biological replicates were generated.
Single-cell transcriptomic data filtering and quality control.
The R package Seurat (v2.1.0)59 and some customized R scripts were used for data analyses. First, the expression matrices of all the 11 samples were pooled together into a global Seurat object using the ‘MergeSeurat’ function. Genes expressed in fewer than three cells were excluded. The cells with more than 10% of their total transcripts from mitochondrial transcriptome and cells with a large number of uniquely expressed genes (more than the 99th percentile, 4,766 genes) were removed. For the initial analysis, all single cells expressing ≤1,500 genes were also excluded. In total, an expression matrix including 19,458 genes and 37,011 cells was obtained. The gene expression profile of each single cell was then normalized to count per million (cpm) and natural log-transformed. To further exclude potential experimental variations, a linear model was generated by using the ScaleData function in Seurat to regress out the effect of (1) the number of detected UMIs in each cell, (2) the percent of mitochondrial genes and (3) sample-to-sample variation.
Separation of cells into neuronal and non-neuronal populations and detection of major cell populations.
To detect the major neuronal and non-neuronal populations, 2,104 genes with cpm >1.5 and dispersion (variance/mean expression) larger than one standard deviation away from the expected dispersion were selected as variable genes using the Seurat function ‘FindVariableGenes’. The variable genes were used to calculate the top 30 principal components (PCs), and the significant PCs were selected using the ‘JackStraw’ method in the Seurat package (P < 1 × 10−3). The cells were then classified into 25 clusters with the Seurat ‘FindClusters’ function. Then, the 25 initial clusters were merged using a random forest classifier with ten-fold cross-validation and 500 trees built with the caret R package60, until the markers of each cluster had a strong predictive power (>80%), which led to 13 cell clusters. Based on the expression of neuron-specific gene Snap25, the 13 cell clusters were identified as neuronal or non-neuronal clusters. In total, five neuronal clusters with 26,429 cells and eight non-neuronal clusters with 10,582 cells were detected. These 13 major cell clusters were further aggregated into five non-neuronal populations (astrocytes, endothelial cells, microglia, oligodendrocytes and OPCs) and four neuronal populations (D1 MSNs, D2 MSNs, INs and new-born neurons) based on the expression of established marker genes, which is presented in Fig. 1.
Inclusion of non-neuronal cells with more than 800 genes detected.
Previous studies showed that neuronal cells tend to have a higher number of mRNAs (UMI) and genes than non-neuronal cells34,35, which is also confirmed in our dataset (Extended Data Fig. 1d). Thus, the criteria used above (>1,500 genes detected) excluded many non-neuronal cells. Hence, we decided to include non-neuronal cells with more than 800 detected genes, which was regarded as high-quality cells in previous high-throughput scRNA-seq studies61,62. To this end, the cells expressing ≥1,500 genes were used to train a random forest classifier (500 trees and ten-fold cross-validation; Extended Data Fig. 1e) to predict the identity of the cells expressing 800–1,500 genes (Extended Data Fig. 1f). Only cells predicted as non-neuronal populations were included. This led to a dataset containing 26,429 neuronal cells and 26,913 non-neuronal cells.
Clustering analysis of each major clusters.
To further cluster the major clusters, the variable genes for each major cluster were identified with the ‘FindVariableGenes’ function in Seurat. In this function, genes were placed into 20 non-overlapping bins according to their average expression, and then a z-score between their dispersion (variance/mean expression) and the mean dispersion of the bin was calculated. The genes with the expression cpm >1.5 and dispersion ≥1 standard deviations away from the expected dispersion in the corresponding bin were selected as variable genes. In some cases, the threshold of dispersion was set to a higher value (up to 3 standard deviations) if the number of variable genes was greater than 2,500. Then, the variable genes of each major cluster were used to calculate the top 30 PCs. Significant PCs with a P value of 1 × 10−3 were selected with the JackStraw method in the Seurat package. If too many PCs were significant, the top 10 PCs were selected. After principal component analysis (PCA), the coordinates of the cells in the significant PCs were used as input for cell clustering. For each major cluster, we first forced the ‘FindClusters’ function to generate a large number of clusters by setting the resolution parameter to 3 or 4. Then, the marker genes of these clusters were calculated, and any pair of clusters with fewer than five DEGs (fold change >2 and P < 0.01) were merged. Finally, we manually filtered out the cell clusters with substantial expression of (1) neuronal and non-neuronal marker or (2) established non-neuronal markers of different subtypes, because they potentially represent double droplets. We also excluded the cell clusters expressing a high level of glutamatergic neuron markers (Slc17a6 and Slc17a7), because NAc does not contain glutamatergic neuron. In addition, cell clusters contributed by cells from fewer than three samples were also excluded. After these processes, our pruned dataset included 25,734 non-neuronal cells and 21,842 neuronal cells.
Identification of cluster markers.
The cluster-specific markers were identified by detecting the DEGs in the saline-treated samples by comparing their expression in the given cluster versus the other clusters. Specifically, the cluster markers were calculated by using a likelihood ratio test with an underlying negative binomial distribution (FindAllMarkers function of the Seurat package) while controlling for (1) the number of UMIs in each cell, (2) the percent of mitochondrial transcripts and (3) sample-to-sample variation. False discovery rates were corrected using the Benjamini–Hochberg method. The gene expression counts were assumed to follow a negative binomial distribution, but this was not formally tested. For MERFISH data, the two-sided, non-parametric Mann–Whitney test with Benjamini–Hochberg multiple testing correction was used (FindAllMarkers function of the Seurat package).
Cell clustering of scRNA-seq data for MERFISH gene selection.
Before selection of the MERFISH gene panel, the scRNA-seq data were clustered in three successive rounds: (1) all cells classified into major cell types (neuronal and non-neuronal); (2) neuronal cells classified into major neuronal cell types (D1, D2 and IN); and (3) each neuronal cell type classified separately into cell subtypes. For each round of clustering, the raw counts for each cell were normalized to the total counts per cell and then logged (natural logarithm). Then, a group of highly variable genes was identified using the ‘highly_variable_genes’ function in Scanpy63, an implementation of the z-scored normalized dispersion (variance/mean) method described previously64, applying cutoffs for the minimum dispersion and minimum and maximum mean of 0.5, 0.025 and 4.0, respectively. Next, a simple linear regression was used to remove potential biases due to the total gene count or percentage of mitochondrial genes per cell, as implemented in the Scanpy ‘regress_out’ function. The data were then scaled to have a mean of zero and unit variance. Then, a linear dimensionality reduction was performed using PCA on the identified highly variable genes and used the first 50, 44, 38, 28 and 26 PCs for the all-cells, the neuronal cells and the D1, D2 and IN clustering, respectively. To determine the number of PCs to keep, we calculated the largest eigenvalue after randomly shuffling the gene values of the cell-by-gene matrix and then kept all the PCs that had an eigenvalue larger than the median of the largest eigen values across ten such shuffling iterations33. Finally, cells were embedded in a k-nearest neighbor (kNN) graph in PC space using Scanpy63 for a range of k sizes before performing Louvain community detection65 on each, as implemented in the Python Louvain-igraph package. To select an optimal k value, we used a bootstrapping analysis to evaluate cluster stability, as described previously33, and selected a k value of 6, 30, 8, 8 and 8, for the all-cells, neuronal cells and D1, D2, and IN clustering, respectively. After the first round of clustering (all-cells), Snap25 and Gad1 were used to identify neuronal clusters to use in the second round of clustering. After the second round of clustering (neurons), expression of Ppp1r1b and Drd1 was used to identify D1 neurons, Ppp1r1b and Drd2 to identify D2 neurons and Resp18 to identify INs, which were each used in a third round of clustering to identify subtypes of each.
Gene selection for MERFISH.
To transcriptionally profile distinct cell populations with MERFISH, we designed a panel of 253 genes. Of these, 133 were chosen manually based on established markers for known cell types (excitatory and inhibitory neurons, oligodendrocytes, oligodendrocyte precursors, microglia, endothelial cells, ependymal, new-born neurons) or specifically pertinent to NAc biology (such as ion channels, ligands and receptors). To discriminate the subclasses of the major neuronal cell types of the NAc (D1, D2 and IN; see next section), we selected the remaining genes using a combination of two complimentary approaches: (1) mutual information (MI) analysis and (2) DEG analysis. We selected the top 50 genes with most MI for each of the three groups of neuronal subclusters, as described previously56. Because of overlap among the three groups, this yielded a total of 114 genes. For DEGs, we included the top 2 DEGs for each subcluster when compared to the remaining cells in its respective group, as determined by the ‘rank_genes_group’ function of the Scanpy package, which employs a t-test to determine differential significance. This yielded 88 DEGs.
The combination of the genes selected manually and those selected by the data-driven approaches above resulted in a final gene panel of 253 genes. After screening the gene list to identify any genes that have a relatively high expression level, which are potentially challenging for MERFISH imaging33, we selected two genes (Penk and Sst) to be imaged separately in a single two-color FISH imaging round, after the MERFISH run that imaged the remaining 251 genes.
Design and construction of encoding probes.
MERFISH encoding probes for the 251 genes were designed as described previously32,33. Each of the 251 genes was assigned to a unique binary barcode drawn from a 20-bit, Hamming distance 4, Hamming weight 4 encoding scheme. We included 34 extra ‘blank’ barcodes that were not assigned to any genes to provide a measure of the false-positive rate, as described previously32,33. As previously described66, we first identified all possible 30-mer targeting regions within each selected gene transcript. For each gene, we then randomly selected 60 of the 30-mer target sequences to comprise 60 encoding probes. For transcripts that were too short and had fewer than 60 targeting regions, we allowed the 30-mers to overlap by as much as 20 nucleotides (nt) to increase the total number of probes targeting that transcript. We then assigned two readout sequences to each of the encoding probes associated with each gene. Each bit in the 20-bit code was associated with a unique readout sequence, and, for each gene, the readouts corresponding to the four ‘on-bits’ of the gene’s assigned barcode were evenly distributed over the entire set of its encoding probes. Finally, each encoding probe was flanked by the sequence of two PCR primers, the first comprising the T7 promoter and the second being a random 20-mer with homology to the encoding probes66. Template DNA for encoding probes used for the 251 multiplexed genes was synthesized as a complex oligo pool (Twist Biosciences) and used to construct the final MERFISH probe set as described previously33. Encoding probes for the two genes measured in sequential two-color FISH rounds were designed in a similar fashion as described above, except: (1) 48 30-mer targeting sequences were chosen for each gene; (2) one unique readout sequence was used for each gene; and (3) PCR primer sequences were omitted. Encoding probes were then synthesized in a 96-well plate format (Integrated DNA Technologies) and mixed to a suitable final concentration.
Design and construction of readout probes.
Twenty-two readout probes were designed to uniquely complement each of the 22 readout sequences used in the set of encoding probes. Twenty of the 22 readouts correspond to the 20-bit barcode used for MERFISH imaging, and the remaining two readout probes correspond to each of the two genes imaged in the sequential two-color FISH rounds. Each of the readout probes was conjugated to one of two dye molecules (Cy5 or Alexa750) via a disulfide linkage, as described previously66. Readout probes were synthesized and purified by Bio-Synthesis, resuspended in Tris-EDTA (TE) buffer, pH8 (Thermo Fisher Scientific) to a concentration of 100 μM and stored at −20 °C.
Tissue slides preparation for MERFISH imaging.
Wild-type male C57BL/6N (8–10-week-old) mice were used for preparing striatal slices for MERFISH experiments. No randomization or blinding was performed during sample collection as no comparison of different conditions was performed. The animals were euthanized with isoflurane, and the brains were quickly harvested and rinsed with ice-cold PBS. Each brain was cut into two hemispheres along the middle line and frozen immediately with dry ice in OCT compound (Tissue-Tek; VWR, 25608–930) and stored at −80 °C until slice cutting. The brains were cut into 10-μm-thick coronal sections with a cryostat (Leica, CM3050S). Before slice cutting, the frozen brains embedded in OCT were put in the cryostat (−20 °C) for at least 30 min to stabilize the temperature. Slices were discarded until the NAc region was reached. Because there is no clear border between the NAc and the4 DS, the brain tissues were trimmed to include the whole striatum. To cover the entire NAc from anterior to posterior, a total of 14 10-μm-thick slices were taken at approximately 100-μm intervals.
The first 12 slices were distributed over three coverslips such that each of the three coverslips contained four slices spanning the anterior, mid and posterior sections of the NAc (for example, coverslip 1 received slices numbered 1, 4, 7 and 10; coverslip 2 received slices numbered 2, 5, 8 and 11). The fourth coverslip received slices numbered 13 and 14. Before slicing, the coverslips were prepared as described previously33. Once placed on the coverslips, tissue slices were immediately fixed by incubating in 4% PFA in 1× PBS for 15 min at room temperature, which was followed by three successive washes with 1× PBS. Samples were then stored in 70% ethanol at 4 °C for at least 18 h to permeabilize the cell membranes. Under these conditions, the four coverslips could be stably stored for up to 2 weeks, with no appreciable degradation observed, while they were further processed in series for MERFISH imaging.
Next, the tissue slices were stained with the MERFISH probe set as described previously33. Briefly, samples were washed three times with 2× saline sodium citrate (2× SSC, Thermo Fisher Scientific, AM9765) and equilibrated with encoding probe wash buffer (30% formamide (Thermo Fisher Scientific, AM9342) in 2× SSC) for 5 min at room temperature. Wash buffer was then aspirated, and the coverslip was inverted onto a 50-μl droplet of an encoding probe mixture on a parafilm-coated petri dish. The encoding probe mixture comprised ~0.1 nM of each encoding probe used in the MERFISH rounds, ~0.3 nM for each encoding probe used in the sequential two-color FISH round and 1 μM of a poly(A) anchor probe (Integrated DNA Technologies) in 2× SSC with 30% vol/vol formamide, 0.1% wt/vol yeast tRNA (Thermo Fisher Scientific, AM7119) and 10% vol/vol dextran sulfate (Millipore, S4030). Samples were stained for ~48 h at 37 °C. The poly(A) anchor probe contained a mixture of both DNA and locked nucleic acid (LNA) nucleotides (/5Acryd/ TTGAGTGGATGGAGTGTAAT T + TT + TT + TT + TT + TT + TT + TT + TT + TT + T, where ‘T + ‘ denotes a thymidine LNA nucleotide and ‘/5Acryd/’ denotes a 5′ acrydite modification) and was used to hybridize to the poly(A) sequence of polyadenylated mRNAs and anchor these RNAs to a polyacrylamide gel, as described below. After staining, samples were incubated twice with encodingprobe wash buffer for 30 min at 47 °C to remove excess and non-specifically bound probes. Samples were then cleared to remove background fluorescence as previously described33. Briefly, samples were embedded in a thin 4% polyacrylamide gel and then incubated for 48 h at 37 °C in digestion buffer (50 mM Tris, pH 8.0 (Thermo Fisher Scientific, 15568025), 1 mM EDTA (Thermo Fisher Scientific, 15575020), 2% SDS (Thermo Fisher Scientific, AM9823), 0.5% Triton X-100 (Sigma-Aldrich T9284) and 1:100 proteinase K (New England Biolabs, P8107S)), refreshing the buffer once after the first 24 h. After digestion, samples were washed four times with 2× SSC and stored at 4 °C in 2× SSC supplemented with 1:1,000 murine RNase inhibitor (New England Biolabs, M0314L) before imaging.
MERFISH Imaging.
The homemade imaging platform was described previously67. Before imaging, samples were stained with a hybridization mixture containing the readout probes associated with the first round of imaging, and an Alexa 488-conjugated readout probe complementary to the poly(A) anchor probe. The hybridization mixture was comprised of readout probes at a concentration of 3 nM in hybridization buffer: 2× SSC, 10% (vol/vol) ethylene carbonate (Sigma-Aldrich, E26258),and 0.1% Triton X-100 (Sigma-Aldrich, T9284). Staining incubated for 15 min at room temperature, and samples were washed in hybridization buffer for 10 min at room temperature. Samples were then incubated with a solution of 10 μg ml−1 of DAPI (Thermo Fisher Scientific, D1306) in 2× SSC for 5 min at room temperature, washed briefly in 2× SSC and finally imaged.
For imaging, the sample coverslip was held inside a flow chamber (Bioptechs, FCS2) with a 0.75-mm-thick gasket (Bioptechs DIE no. P47132), and buffer exchange within the chamber was directed using a custom-built automated fluidics system composed of three 12-port valves (IDEX, EZ1213-820-4) and a peristaltic pump (Gilson, MP3), configured as described previously32. Imaging buffer comprised 2× SSC, 2 mM trolox (Sigma-Aldrich, 238813), 50 μM trolox-quinone, 5 mM protocatechuic acid (Sigma-Aldrich, 37580), 1:500 recombinant protocatechuate oxidase 3,4-dioxygenases (rPCO 46852004, OYC Americas), 1:500 murine RNase inhibitor (New England Biolabs, M0314L) and 5 mM NaOH (VWR 0583) to adjust final pH to 7.0. Cleavage buffer comprised 50 mM TCEP (Gold Bio, TCEP50) in 2× SSC.
Each imaging round consisted of readout probe hybridization (10 min), washing with hybridization buffer (5 min), flowing on imaging buffer (defined below), imaging of each field of view (FOV) (220 μm × 220 μm per FOV), readout fluorophore cleavage by cleavage buffer (15 min) and washing with 2× SSC (5 min). In the first round, we imaged the cell nucleus stained with DAPI and poly(A) anchors stained with an Alexa 488-labeled readout probe at seven focal planes separated by 1.5 μm in z. We then performed 12 rounds of two-color imaging, wherein the first ten rounds imaged the barcode-encoded RNA species (combinatorial single molecule FISH (smFISH) rounds); the 11th round imaged the individually labeled RNA species (sequential smFISH rounds); and the final 12th round imaged the sample unlabeled to serve as a measure of background fluorescence. For each combinatorial round, images were acquired with 750-nm and 650-nm illumination at seven focal planes separated by 1.5 μm z to image the readout probes. For the sequential and background rounds, images were acquired with 750-nm and 650-nm illumination at a single focal plane 3.5 μm above the glass surface. In addition, every imaging round described above also included a single z-plane image with 546-nm illumination to image the fiducial beads on the glass surface for image registration. Finally, the number of FOVs imaged varied for each sample base on the size and number of tissues present on the coverslip.
Image analysis and cell segmentation.
All MERFISH image analysis was performed using the MERlin (https://github.com/emanuega/MERlin) Python package, which, in turn, uses algorithms similar to what has been described previously33,67. First, for each FOV, the images from each imaging round were aligned using their respective fiducial bead image to correct for x–y drift in the stage position relative to the first round. For MERFISH rounds, images stacks for each FOV were then high-pass filtered to remove background fluorescence, deconvolved using ten rounds of Lucy–Richardson deconvolution and finally low-pass filtered to account for small movements in the centroid of RNA spots across the imaging rounds. Individual RNA molecules were then identified using pixel-based decoding, as described previously66. After decoding, adjacent pixels that were assigned the same barcode were aggregated as putative RNA molecules, and then the list of putative RNA molecules with an area greater than 1 pixel was filtered, as described previously67, to enrich for correctly identified transcripts with a gross misidentification rate of 5%. Single pixel spots were excluded because they are disproportionally prone to be spurious barcodes generated by random fluorescent fluctuations.
Next, we identified cell segmentation boundaries for each FOV using a seeded watershed approach, as described previously33. The DAPI images were used as the seeds, and the poly(A) signals were used to identify segmentation boundaries. After segmentation, individual RNA molecules were assigned to individual cells based on if they fell within the segmented boundaries. For the sequential two-color FISH rounds, images were high-pass filtered, and the expression level of each gene in each cell was calculated as the sum of the fluorescence intensity of all pixels within the segmentation boundary of the central z plane of each cell. Finally, the signals from the two genes from the sequential two-color FISH round were merged with the RNA counts matrix from the 251 genes measured in the MERFISH run and used for cell clustering analysis.
Cell clustering analysis of MERFISH data.
The cell-by-gene matrix as obtained above was pre-processed with several steps. First, to remove spurious segmentation artifacts, we removed segmented ‘cells’ that had a volume less than 100 μm3 or greater than 4,000 μm3, which was chosen empirically to ensure retention of some larger inhibitory cell subtypes that were observed in subsequent analysis. Because the expression units of the 251 genes measured in the MERFISH rounds was fundamentally different from that of the two genes measured in sequential FISH rounds (integer counts per cell versus total fluorescence intensity per cell, respectively), pre-processing of these two types of measurements required a different set of steps. For the counts matrix from the 251 genes the steps were as follows: (1) to reduce potential noise introduced by cells with few measured RNA molecules, we removed cells with fewer than five total RNA counts; (2) to normalize for variation in cell size, owing to either true biological differences between cell types or states, or to the fact that the entire soma of every cell did not occupy the 10-μm-thick tissue slice, we divided the RNA counts per cell by the imaged volume of each cell; and (3) to correct for minor batch fluctuations in measured RNA counts across slices, we normalized the total counts per cell to the same value (100 in this case). For the intensity matrix from the two sequential round genes, we applied two steps to remove background fluorescence. (1) To equalize the baseline fluorescence for each gene across all slices, within each slice, the median intensity per gene was subtracted from each cell. (2) The final two-color imaging round was ‘blank’ in that no readout probes were added, providing an autofluorescence background measurement for each cell in each imaging channel. This background was subtracted for each gene in each cell according to its imaging color channel.
After the pre-processing described above, we applied three successive rounds of clustering analysis to identify cell types and subtypes. The first round was applied to all cells to identify major non-neuronal, neuronal and IN populations. In the second round, we selected only the MSNs identified in the first round to further discriminate Drd1-positive (D1) and Drd2-positive (D2) neurons. In the first two rounds of clustering, established marker genes were used to identify cell populations: Ppp1r1b (MSN), Resp18 (IN), Snap25 (other-neuronal), Aldoc (astrocytes), C1qa (microglia), Mobp (oligodendrocytes), Pdgfra (oligodendrocyte precursors), Rgs5 (endothelial), Ccdc153 (ependymal), Drd1 (D1 MSN) and Drd2 (D2 MSN). In the third round, the D1, D2 and IN (identified in the first round) populations were each clustered separately into the final reported subtypes. Each round of clustering followed the same basic workflow, similarly to what was done for the clustering of the scRNA-seq data before MERFISH gene panel selection described above. Briefly, the cell-by-gene expression matrix was log-transformed, and each gene was scaled to have unit variance and a mean of zero. Next, PCA was applied, using all genes, to reduce dimensionality, and the number of PCs to keep was selected in each instance (26, 21, 22, 25 and 28 for all-cells, MSN, D1, D2, and IN clustering, respectively), as described above and previously33. Then, as described above, cells were embedded in a kNN graph in PC space and clustered using Leiden community detection68 as implemented in the Leidenalg Python package. To remove an apparent batch effect between the datasets from each of the two mouse brains used, we employed the Harmony algorithm69, as implemented in the Harmonypy Python package, to correct PCs in the MSN, D1, D2 and IN analysis before clustering. Optimal k values of 25, 12, 10, 25 and 45 were chosen for the all-cells, MSN, D1, D2, and IN clustering, respectively, using the bootstrapping method described above. Finally, we manually filtered the clustering results by checking the gene expression and spatial distribution of each cell type (subtype), to remove cell clusters representing doublets (based on co-expression of established makers of multiple cell types) or located out of striatum.
Integration of scRNA-seq and MERFISH data.
This analysis was done using the Seurat R package (v3.2.0)70 in combination with the R version of the Harmony algorithm69 (v1.0, https://github.com/immunogenomics/harmony) and some customized R scripts. We used the list of MERFISH genes to build a combined count matrix between the scRNA-seq counts and the normalized MERFISH RNA counts. Next, the counts were normalized to the library size and log-scaled, and their batch-un-corrected PCAs were calculated using the Seurat functions ‘NormalizeData’, ‘FindVariableFeatures’, ‘ScaleData’ and ‘RunPCA’, respectively. The ‘RunHarmony’ function was then used with default parameters for batch correction in the PCA space. Next, the top 20 batch-corrected PCs were used to generate the UMAP embeddings. The same approach was followed to respectively integrate D1 and D2 MSN data between scRNA-seq and MERFISH.
Estimation of the similarity between scRNA-seq and MERFISH clusters.
As MERFISH clusters capture both the transcriptional and spatial distribution, we used the MERFISH-identified clusters as our reference. The degree of correspondence between scRNA-seq and MERFISH clusters was calculated based on the kNN classification. Given the top 20 batch-corrected PCs from Harmony, we identified for each scRNA-seq cell its 30 nearest MERFISH cells using the ‘Seurat:::FindNN’ function with parameters ‘cells1 = scRNACells, cells2 = merFISHCells, internal.neighbors = NULL, dims = 1:20, reduction=‘harmony’, nn.reduction=‘harmony’, k = 30, nn.method=‘rann’ and eps = 0, and then we used the function Seurat::GetIntegrationData to get the results. An scRNA-seq cell was assigned to a MERFISH cluster if more than 70% of its kNN MERFISH neighbors belong to the same MERFISH cluster; otherwise, the scRNA-seq cell was considered as ambiguous. The degree of similarity between scRNA-seq transcriptional clusters and MERFISH cell clusters was calculated as the proportion scRNA-seq cells that could be matched to each MERFISH cluster for each scRNA-seq cluster. From the total scRNA-seq data, only 3.04% of scRNA-seq cells could not be confidently matched to a MERFISH cluster, whereas 24.97% and 21.45% of scRNA-seq D1 and D2 MSN cells, respectively, could not be confidently matched to a corresponding D1 or D2 MERFISH cluster.
Estimation of cluster density in a MERFISH slice.
To visualize the spatial density distribution of a MERFISH cluster or group of clusters in a MERFISH slice, we used the Gaussian kernel density estimator implemented in the function embedding_density from the Python module Scanpy (v1.6.0)63.
Generation of plots and heat maps.
R and Python were used interchangeably to generate the different plots and heat maps. Heat maps were generated using the R/Bioconductor package ComplexHeatmap71 or the pheatmap R package72. All the other plots were generated using the ggplot2 package73. Dimensional embedding plots were mainly generated using the Seurat R package (v3.9.9.9008)70. Scanpy (v1.6.0)63 was also used to generate some gene expression heat maps, dot plots and the density plots.
Interactive visualization of MERFISH data.
To enable the interactive exploration of our scRNA-seq and MERFISH data, we built upon the open-source version of cellxgene v0.15.0 (https://github.com/chanzuckerberg/cellxgene). We did some additional customization to the color schemes and selection behavior to fit our purpose. The actual version can be accessed at http://35.184.4.122:5050/ (MERFISH data: https://www.zhanglab.tch.harvard.edu/neuro-group/nac/merfish) and http://35.184.4.122:9090/(scRNA-seq data: https://www.zhanglab.tch.harvard. edu/neuro-group/nac/scrnaseq).
Statistics and reproducibility.
No statistical method was used to predetermine sample size, but our sample sizes are similar to those reported in previous publications33,74. No randomization or blinding was performed during sample collection and data analysis as no comparisons of different conditions were performed. The criteria for excluding data are detailed in the ‘Single-cell transcriptomic data filtering and quality control’ section.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Identification of the transcriptionally distinct cell types in mouse NAc.
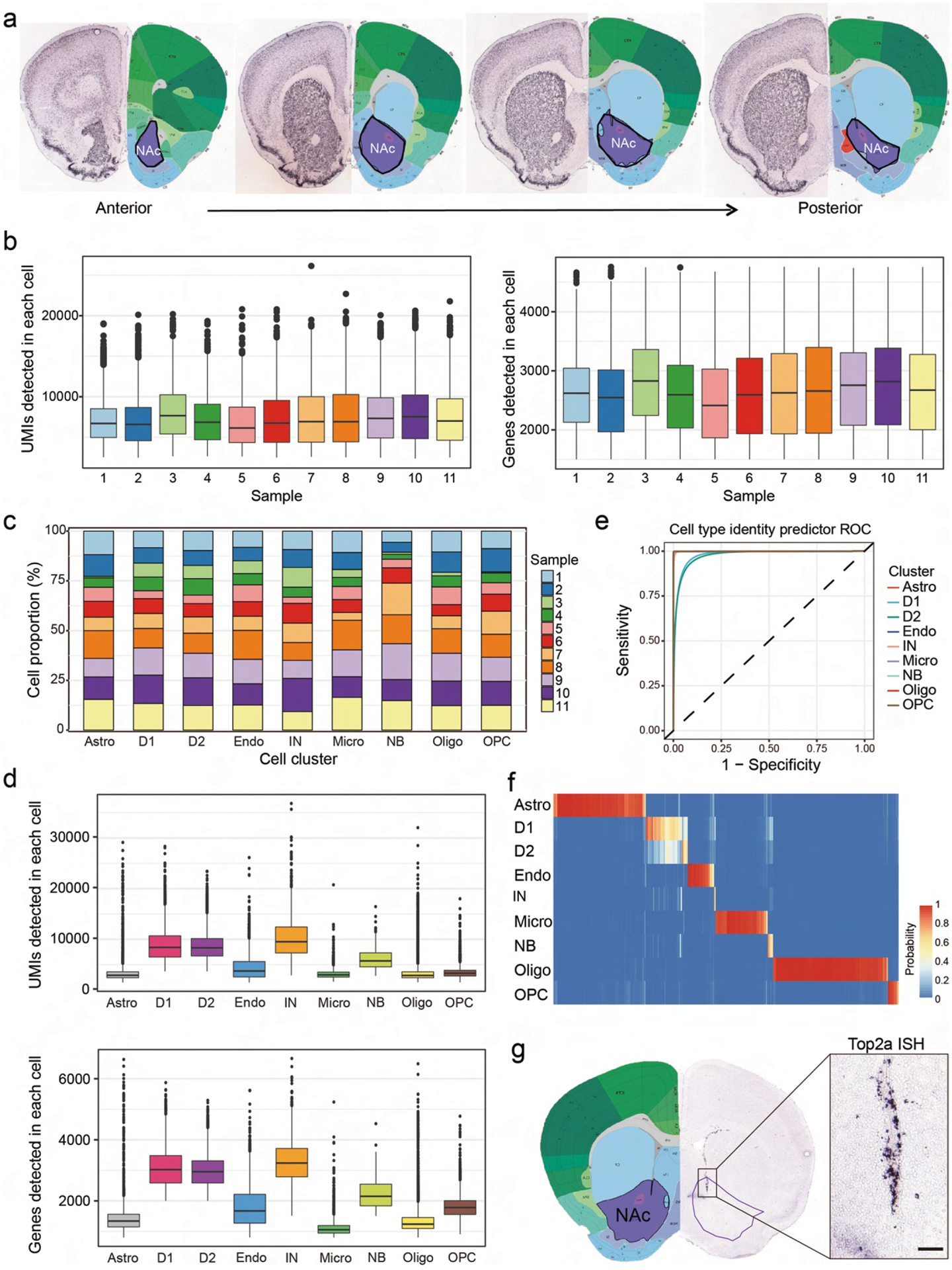
a, Schematic diagram showing the nucleus accumbens region used for single-cell RNA sequencing. Adult mouse brain was first cut into 0.5mm-thick serial coronal sections and then NAc tissues (shown in purple contours) were dissected from successive slices along the rostral-caudal axis. The brain pictures were taken from Allen Mouse Brain Atlas. b, Bargraphs showing the distribution of UMI and gene number detected in each cell across the 11 samples. Different samples are color-coded. The black central line is the median, the box limits indicate the upper and lower quartiles, the whiskers indicate the 1.5 interquartile range and dots represent outliers. c, Histograms showing the percentage of cells from each sample that contribute to the major cell clusters. Different samples are color-coded. d, Bargraphs showing the distribution of UMI and gene number detected in each cell across the 9 major cell clusters. Different cell clusters are color-coded. The black central line is the median, the box limits indicate the upper and lower quartiles, the whiskers indicate the 1.5 interquartile range and dots represent outliers. Astro, astrocyte; D1, medial spiny neuron, D1-receoptor subtype; D2, medial spiny neuron, D2-receptor subtype; Endo, endothelial cell; IN, interneuron; Micro, microglia; NB, neural stem cells and neuroblast; Oligo, oligodendrocyte; OPC, oligodendrocyte progenitor cell. e, ROC curves showing the high accuracy of the cell identity predictor, especially for non-neuronal cells. The curves of different cell types are represented by different colors. f, Heatmap showing the results of predicted identity of cells with 800 to 1,500 genes detected. The prediction probability is color-coded. g, In situ hybridization of the cell-cycle gene, Top2a, showing the distribution of neural stem cells and neuroblasts in the ventral wall of the lateral ventricle. The purple contour indicates the NAc, and the boxed region in the left panel is enlarged and shown on the right. The ISH data was obtained from Allen Mouse Brain Atlas. Scale bar, 100 μm.
Extended Data Fig. 2 |. Gene expression and spatial pattern of NAc interneuron subtypes.
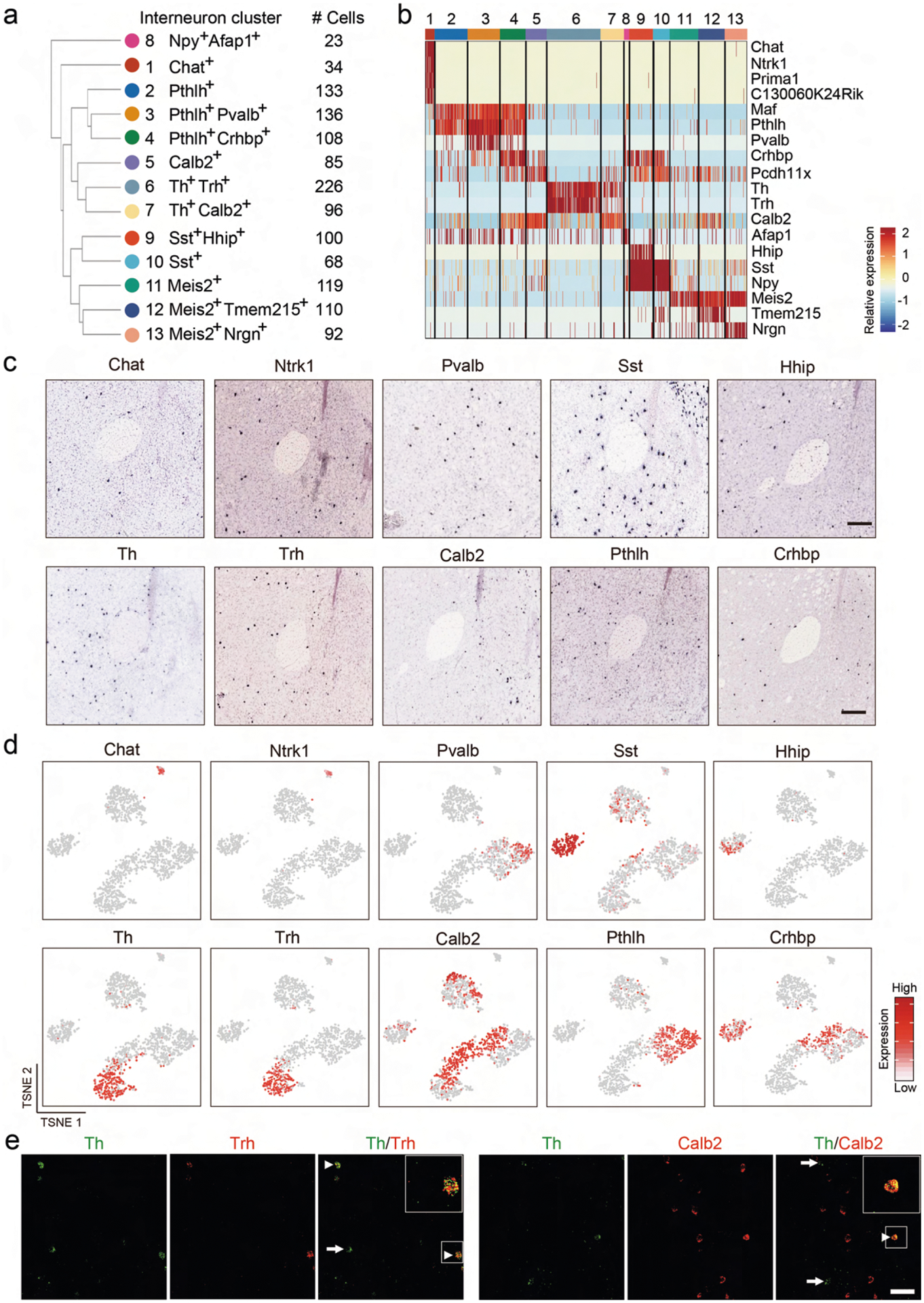
a, Hierarchical relationship of the 13 NAc interneuron subtypes identified from scRNA-seq. The dendrogram indicate the relatedness among interneuron subtypes based on their gene expression. The markers and the number of cells of each subtypes are shown. b, Heatmap showing the expression pattern of interneuron markers across different subtypes. Each column represents a single cell. The gene expression level is color coded. The 13 interneuron subtypes are indicated by different colors on top. c, ISH showing the expression of selected interneuron subtype markers identified by scRNA-seq in NAc. The regions around the anterior commissure (AC) were shown except for Pvalb, which was more enriched in lateral part of NAc. The images were obtained from the Allen Mouse Brain Atlas. d, tSNE plots showing the expression of interneuron subtype markers across NAc interneuron subtypes. The gene expression level is color-coded. e, FISH showing the overlap of selected interneuron markers in NAc. Arrow heads indicate the cells co-express the two genes. Arrows indicate cells expressing one marker gene. Three independent experiments were performed with similar results. Scale bar, 50 μm.
Extended Data Fig. 3 |. Molecularly defined MsN subtypes exhibit distinct spatial distribution in NAc.
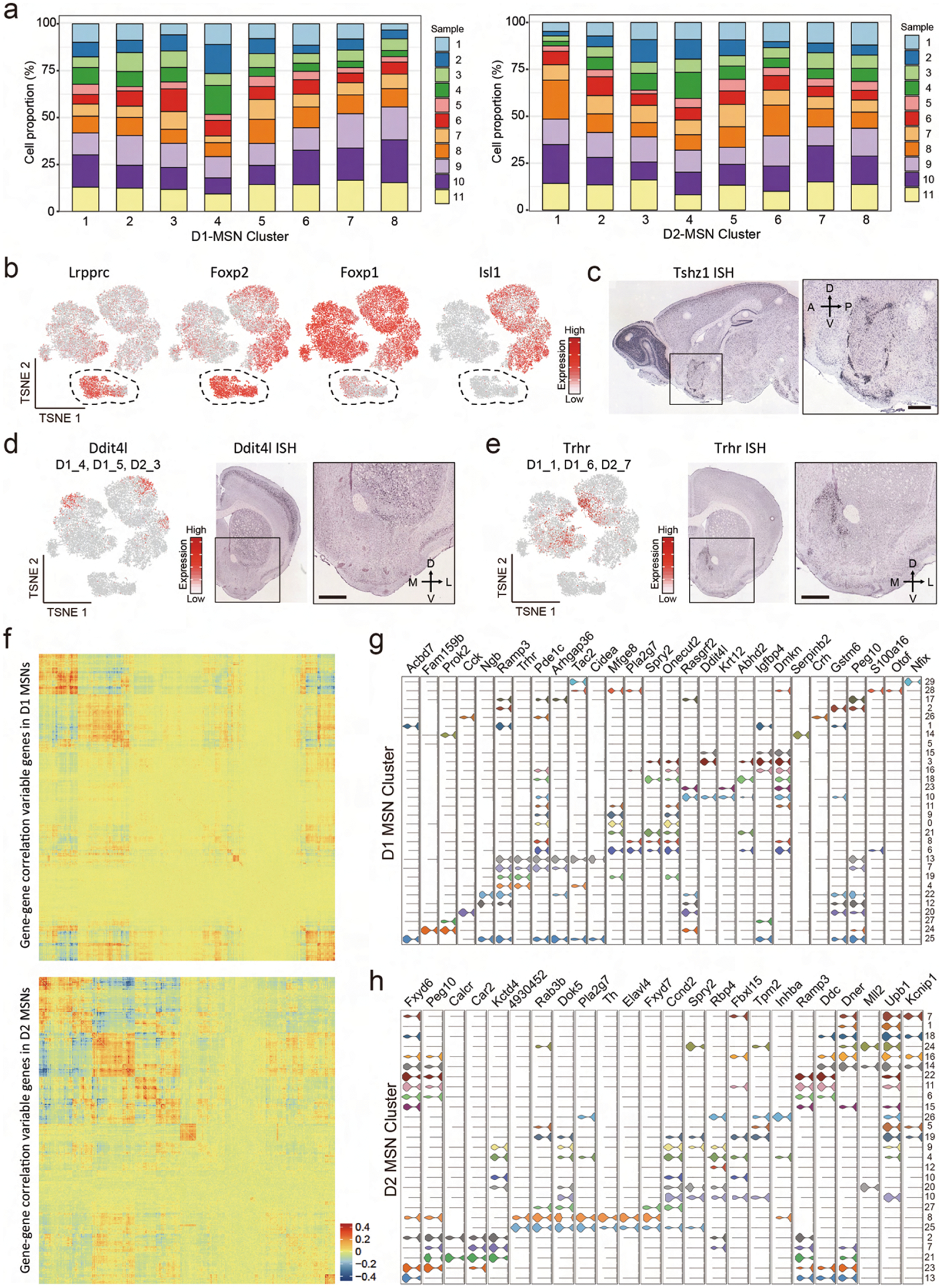
a, Histograms showing the percentage of cells from each of the 11 samples across D1 and D2 MSN subtypes. Different samples are represented in different colors. b, tSNE plots showing the expression pattern of Lrpprc, Foxp2, Foxp1 and Isl1 across MSN populations. The expression level is color-coded. c, ISH of Tshz1 showing the distribution of Tshz1+ cells in NAc. Sagittal section of mouse brain including NAc is shown. The boxed region is enlarged and shown on the right. Data are obtained from Allen Mouse Brain Atlas. Scale bars, 500 μm. d, e, Ddit4l (d) and Trhr (e) are enriched in subpopulation of MSNs. Left panel, tSNE plots showing the expression pattern of Ddit4l (d) and Trhr (e) across MSN populations. The expression level is color-coded. The MSN subtypes labeled by Ddit4l or Trhr were indicated. Right panels, ISH image showing the distribution of Ddit4l+ (d) and Trhr+ (e) cells in NAc. The boxed regions are enlarged and shown on the right. Data is obtained from Allen Mouse Brain Atlas. Scale bar, 500 μm. f, Heatmaps showing the gene expression correlation of differentially expresses genes in D1 MSNs (upper panel) and D2 MSNs (lower panel). The correlation is color-coded, and genes were sorted into groups with higher intra-group correlation. g, h, Violin plots showing the expression of selected markers across high-resolution D1 (g) and D2 (h) MSN subtypes. Different MSN subtypes are color-coded. The mRNA level is presented on a log scale and adjusted for different genes.
Extended Data Fig. 4 |. MERFISH revealed major cell types in striatum.
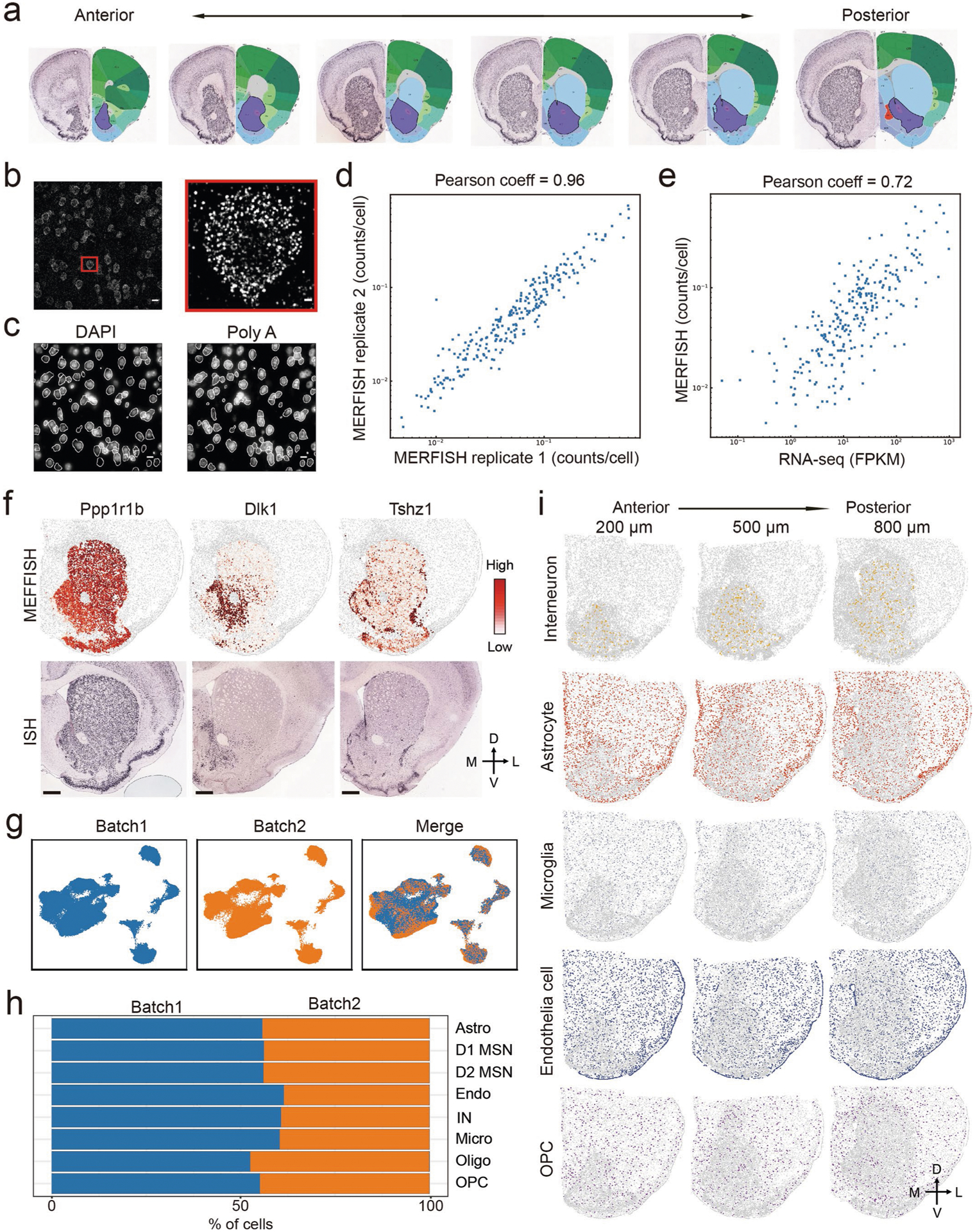
a, Schematic diagram showing the striatal slices used for MERFISH. Adult mouse brain was first cut into 10 μm -thick serial coronal sections. Twelve brain slices with 100 μm interval between adjacent slices were used for MERFISH experiment. The brain pictures were taken from Allen Mouse Brain Atlas, note that only 6 slices were shown. b, One example image showing the maximum projection of images taken in one representative field-of-view (FOV) during MERFISH. The boxed region was enlarged and shown on the right. Individual RNA molecules were detected as single dots. c, DAPI (left) and poly(A) RNA (right) images were used to define the boundaries of each cell in white. The mRNA molecules detected were assigned to different cells based on the cell boundaries. d, Scatterplot showing the average counts of each genes per cell detected by MERFISH in the two biological replicates. e, Scatterplot showing the average copy number of each genes per cell detected by MERFISH and bulk RNA-seq. The Pearson correlation coefficient is 0.72. f, Heatmap showing the expression pattern of selected genes in striatum as determined by MERFISH (upper panels), which are highly similar to the patterns determined by conventional ISH (lower panels). The ISH data are obtained from Allen Mouse Brain Atlas. Scale bars, 500 μm. g, the Harmony algorithm69 based UMAP plots showing the cells from the two replicates of MERFISH experiments with good overlap. h, Bar graph showing the proportion of major cell types from the two batches of MERFISH experiments after the Harmony integration. i, Spatial pattern of interneuron, astrocyte, microglia, endothelial cell and OPC in coronal brain sections at different anterior-posterior positions. Three of the twelve slices from a male mouse were shown. Colored dots were cells belong to the specified cell populations, while gray dots indicate all other cells. The 200, 500 and 800 μm labels indicate the distance from the anterior position (Bregma 1.94 mm). The dorsal-ventral (DV) and medial-lateral (ML) axes are indicated.
Extended Data Fig. 5 |. MERFISH identifies molecularly distinct D1 and D2 MsN subtypes in striatum.
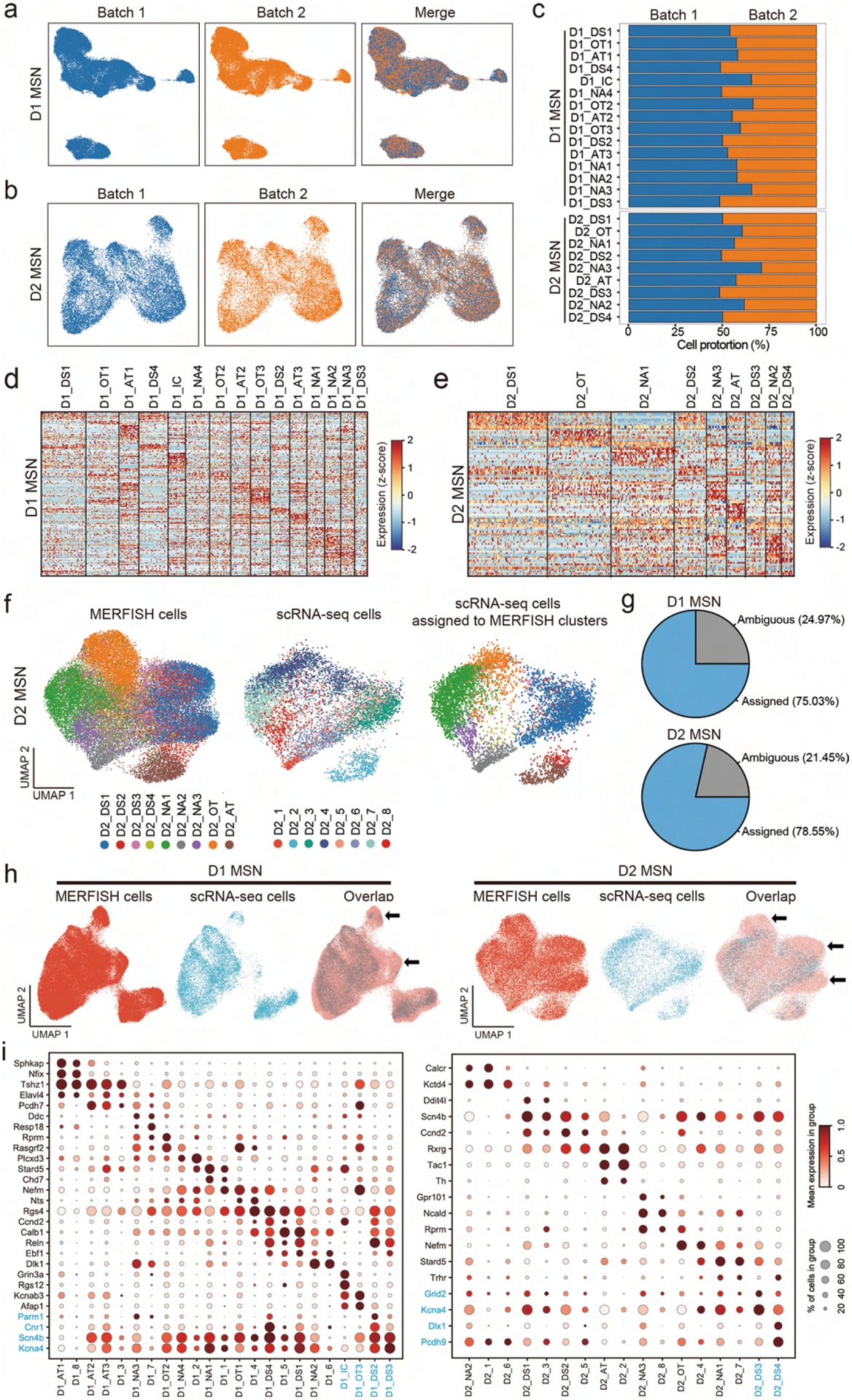
a, b, The Harmony algorithm69 based UMAP plots showing the D1 MSNs (a) and D2 MSNs (b) from the two replicates of MERFISH experiments. c, Bar graph showing the proportion of different D1 and D2 MSN subtypes from the two replicates of MERFISH experiments. d, e, Heatmaps showing the expression pattern of differentially expressed genes detected by MERFISH across D1 (d) and D2 (e) MSN subtypes. The expression level is color coded. The width of each column represents the abundance of each MSN subtype. f, Integrative analysis (Using the Harmony algorithm) of D2 MSNs from MERFISH and scRNA-seq experiments. The D2 MSNs from MERFISH and scRNA-seq experiments were integrated into the same UMAP space. The initial identity of each cell was color coded and shown in the left and middle panels. Based on the nearest neighbors from the MERFISH experiments, the cells from scRNA-seq were assigned to one of the MERFISH D2 MSN subtypes shown on the right panel. g, Pie charts showing the percentage of D1 (upper) and D2 (lower) MSNs from scRNA-seq experiments which could or could not be assigned to a certain MEFFISH identify due to insufficient MERFISH k-NN belonging to the same cluster. h, The Harmony algorithm based UMAP showing the D1 MSNs (upper panels) and D2 MSNs (lower panels) from scRNA-seq and MERFISH experiments. The cells from different experiments were integrated into the same UMAP spaces. The arrows indicated UMAP spaces that are mainly occupied by cells from non-NAc region, thus the scRNA-seq cells were depleted comparing to MERFISH cells. i, Dotplots showing the expression of selected MSN subtype markers in D1 (left) and D2 (ringht) MSN subtypes defined by MERFISH and scRNA-seq. The expression level is color-coded (MERFISH and scRNA-seq data are normalized and scaled separately). Dot size represents the fraction of cells expressing the gene in each subtype. Corresponding MERFISH and scRNA-seq clusters are grouped together. The names and marker genes of MSN subtypes identified by MERFISH but without corresponding scRNA-seq subtypes are marked in blue.
Extended Data Fig. 6 |. Molecular and spatial features of MsN subtypes underlie anatomic organization of striatum.
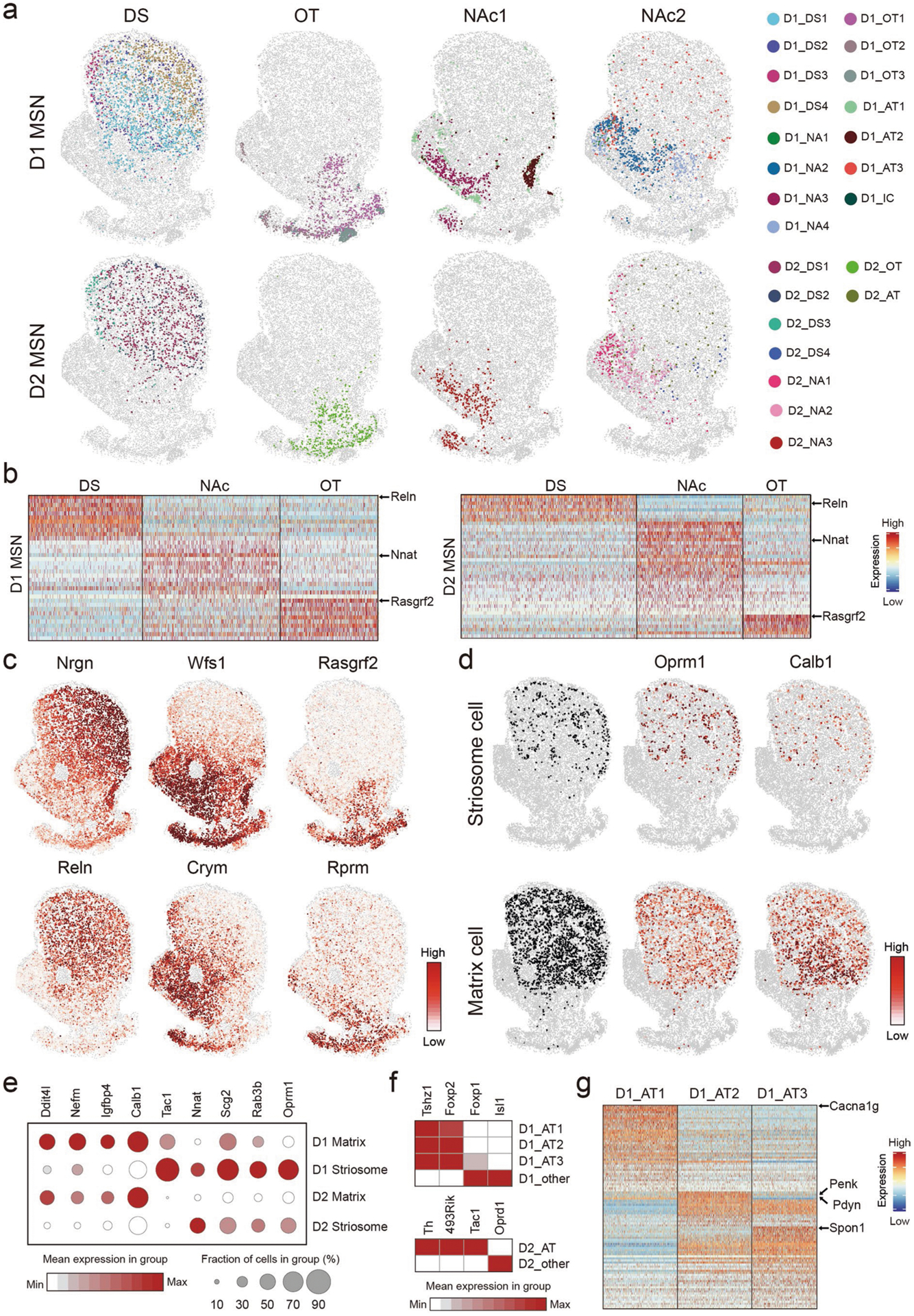
a, Spatial patterns of different MSNs groups in striatal region. The MSN subtype groups are the same as Fig. 6e. Different MSN subtypes are color coded. The D1 and D2 MSNs are shown in upper and lower panels, respectively. b, Heatmap showing the patterns of differentially expressed genes among D1 (left panel) and D2 (right panel) MSN subtype groups located in major striatal divisions. The MSN groups corresponding to different anatomic regions are the same as Fig. 6e, but the two NAc groups are combined. Reln, Nnat and Rasgrf2 are enriched in DS, NAc and OT, respectively, and the patterns are shared in D1 and D2 MSN subtypes. c, The spatial heatmaps showing the expression pattern of Nrgn, Reln, Wfs1, Crym, Rasgrf2, Rprm in coronal sections, which are enriched in different striatal divisions. The expression level is color coded. d, Spatial and gene expression features of D1 MSN subtypes representing striosome (D1_DS2) and matrix (D1_DS1, D1_DS3 and D1_DS4) structure in DS. The upper and lower panels show D1 subtypes representing striosome and matrix, respectively. The left panels show spatial pattern of D1 subtypes corresponding to striosome and matrix. The middle and right panels are heatmaps showing the expression of striosome enriched gene Oprm1 and matrix enriched gene Calb1 in these D1 MSN subtypes. e, Dotplot showing the expression pattern of selected genes in D1 and D2 MSN subtypes representing striosome and matrix. D1 Matrix: D1_DS1, D1_DS3 and D1_DS4; D1 Striosome: D1_DS4; D2 Matrix: D2_DS1 and D2_DS3; D2 Striosome: D2_DS2. The expression level is color coded. Dot size represents the fraction of cells in the subtype. f, Heatmap showing the expression of selected marker genes that distinguish atypical D1 and D2 MSN subtypes from other D1 and D2 MSN subtypes. The expression level is color coded. g, Heatmap showing the pattern of differentially expressed genes among the three atypical D1 MSN subtypes. Selected marker genes enriched in different atypical D1 MSN subtypes are labeled.
Extended Data Fig. 7 |. Molecular and spatial features of MsN subtypes underlie anatomic heterogeneity of NAc.
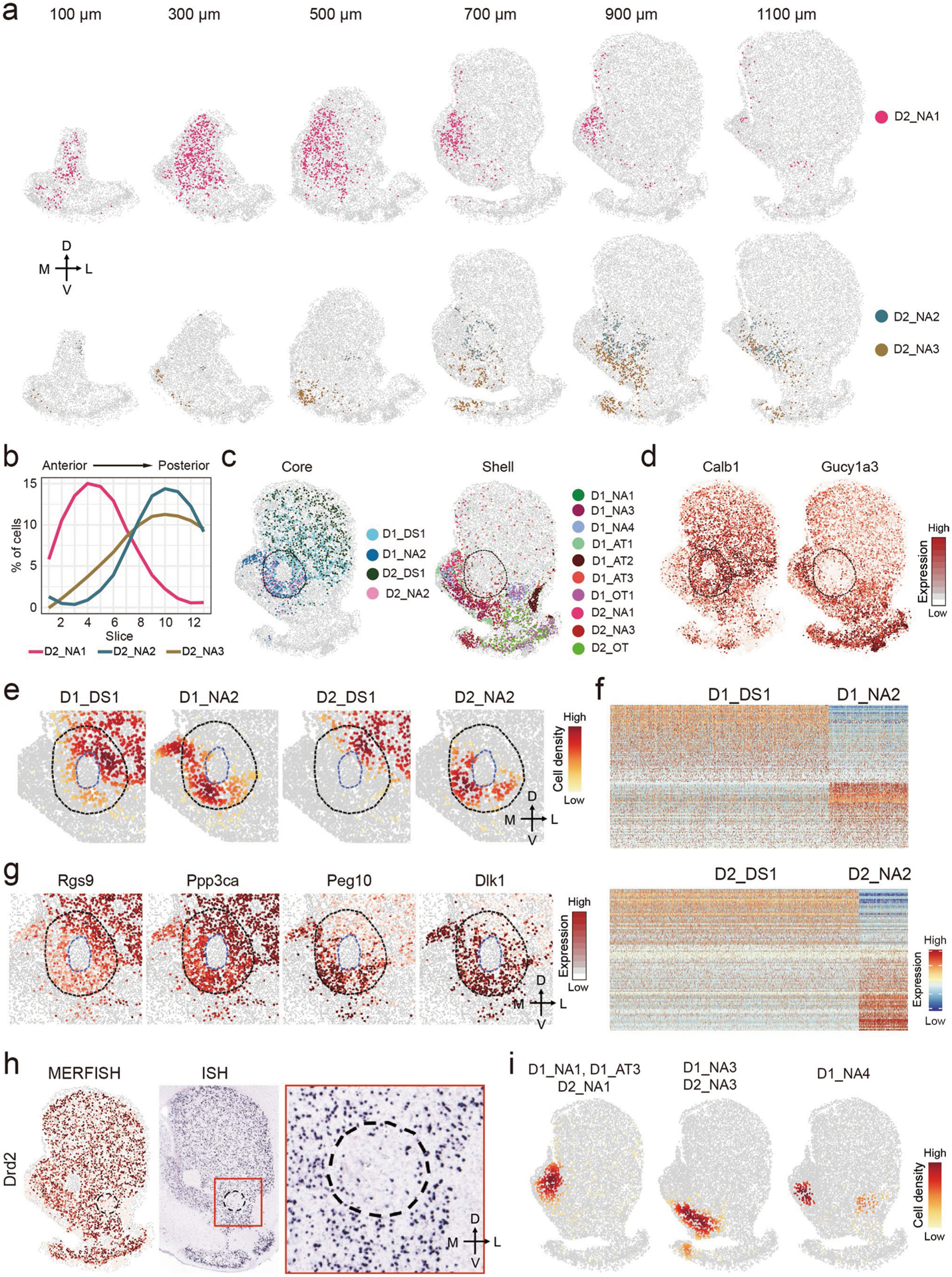
a, Spatial patterns of selected D2 MSN subtypes in coronal sections at different anterior-posterior positions. The subtypes enriched in anterior and posterior NAc are shown in upper and lower panels, respectively. Different subtypes are represented by different colors. The 100, 300, 500, 700, 900 and 1100 μm labels indicate the distance from the anterior position (Bregma 1.94 mm). The dorsal-ventral (DV) and medial-lateral (ML) axes are indicated. b, The distribution of the three D2 MSN subtypes shown in (a) along the AP axis. c, The spatial pattern of MSN subtypes enriched in NAc core (left panel) or shell (right panel). Different subtypes are indicated with different colors. The core region is indicated by dashed line. d, Heatmap showing the expression of core enriched gene Calb1 and shell enriched genes Gucy1a3 in coronal brain sections. The expression level was color coded and the core region was indicated with dashed line. e, Density maps showing the enrichment of different D1 and D2 MSN subtypes in the dorsolateral and ventromedial part of NAc core. The same region as Fig. 7f are shown. The NAc core and AC structure are labeled with dashed lines. The dorsal-ventral (DV) and medial-lateral (ML) axes are indicated. f, Heatmaps showing the differentially expressed genes between D1 (upper panel) and D2 (lower panel) MSN subtypes enriched in different subregions of NAc core. The expression level is color coded. g, Heatmap showing the differential expression of Rgs9, Ppp3ca, Peg10 and Dlk1 between the dorsolateral and ventromedial part of NAc core. The same region as Fig. 7f is presented. The NAc core and AC structure are labeled with dashed lines. The gene expression level is color coded. h, MERFISH and ISH detection of Drd2 expression in striatum. The dorsal part of NAc lateral shell with low Drd2 expression is labeled with dashed line. The boxed region in the ISH image is enlarged and shown on the right. The ISH data is from Allen Brain Atlas. i, Density maps showing the enrichment of different groups of MSN subtypes in distinct subregions in NAc medial shell.
Supplementary Material
Acknowledgements
We thank Z. Chen and Q. Yin for their help with sequencing and B. L. Sabatini for critical reading of the manuscript. We acknowledge the support of the HMS Neurobiology Imaging Facility (supported by NINDS P30 Core Center Grant no. NS072030) and its staff, R. Carelli and M. Ocana, and the Mouse Behavior Core of Harvard Medical School and its director, B. Caldarone. This project was supported by the National Institutes of Health (NIDA R01DA042283 to Y.Z. and NIMH U19MH114821 to X.Z.) and the Howard Hughes Medical Institute. Y.Z. and X.Z. are investigators of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41593-021-00938-x.
Code availability
Code for MERFISH image acquisition is available at https://github.com/ZhuangLab. Code for MERFISH image analysis is available at https://github.com/ZhuangLab/MERlin. Code for MERFISH and scRNA-seq integration is available at https://github.com/YiZhang-lab/NAcMERFISHscRNAseqAnalysis.
Competing interests
X.Z. is a co-founder and consultant of Vizgen. The remaining authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41593-021-00938-x.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41593-021-00938-x.
Data availability
The scRNA-seq data have been deposited at the Gene Expression Omnibus with accession number GSE118020. The MERFISH data are available at the Brain Image Library (https://download.brainimagelibrary.org/fc/4c/fc4c2570c3711952/).
References
- 1.Floresco SB The nucleus accumbens: an interface between cognition, emotion, and action. Annu. Rev. Psychol 66, 25–52 (2015). [DOI] [PubMed] [Google Scholar]
- 2.O’Connor EC et al. Accumbal D1R neurons projecting to lateral hypothalamus authorize feeding. Neuron 88, 553–564 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Dolen G, Darvishzadeh A, Huang KW & Malenka RC Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 501, 179–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith-Roe SL & Kelley AE Coincident activation of NMDA and dopamine D1 receptors within the nucleus accumbens core is required for appetitive instrumental learning. J. Neurosci 20, 7737–7742 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kravitz AV, Tye LD & Kreitzer AC Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat. Neurosci 15, 816–818 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berridge KC & Kringelbach ML Pleasure systems in the brain. Neuron 86, 646–664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pascoli V, Turiault M & Luscher C Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 481, 71–75 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Krishnan V et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131, 391–404 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Bewernick BH et al. Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol. Psychiatry 67, 110–116 (2010). [DOI] [PubMed] [Google Scholar]
- 10.McCollum LA, Walker CK, Roche JK & Roberts RC Elevated excitatory input to the nucleus accumbens in schizophrenia: a postmortem ultrastructural study. Schizophr. Bull 41, 1123–1132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh SW et al. A mesoscale connectome of the mouse brain. Nature 508, 207–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groenewegen HJ, Wright CI, Beijer AV & Voorn P Convergence and segregation of ventral striatal inputs and outputs. Ann. N. Y. Acad. Sci 877, 49–63 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Salgado S & Kaplitt MG The nucleus accumbens: a comprehensive review. Stereotact. Funct. Neurosurg 93, 75–93 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Britt JP et al. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76, 790–803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pascoli V et al. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509, 459–464 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Yang H et al. Nucleus accumbens subnuclei regulate motivated behavior via direct inhibition and disinhibition of VTA dopamine subpopulations. Neuron 97, 434–449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Jong JW et al. A neural circuit mechanism for encoding aversive stimuli in the mesolimbic dopamine system. Neuron 101, 133–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lammel S et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature 491, 212–217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tepper JM, Tecuapetla F, Koos T & Ibanez-Sandoval O Heterogeneity and diversity of striatal GABAergic interneurons. Front. Neuroanat 4, 150 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerfen CR & Surmeier DJ Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci 34, 441–466 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreitzer AC & Malenka RC Striatal plasticity and basal ganglia circuit function. Neuron 60, 543–554 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerfen CR et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432 (1990). [DOI] [PubMed] [Google Scholar]
- 23.Yawata S, Yamaguchi T, Danjo T, Hikida T & Nakanishi S Pathway-specific control of reward learning and its flexibility via selective dopamine receptors in the nucleus accumbens. Proc. Natl Acad. Sci. USA 109, 12764–12769 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voorn P, Gerfen CR & Groenewegen HJ Compartmental organization of the ventral striatum of the rat: immunohistochemical distribution of enkephalin, substance P, dopamine, and calcium-binding protein. J. Comp. Neurol 289, 189–201 (1989). [DOI] [PubMed] [Google Scholar]
- 25.Brimblecombe KR & Cragg SJ The striosome and matrix compartments of the striatum: a path through the labyrinth from neurochemistry toward function. ACS Chem. Neurosci 8, 235–242 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Kupchik YM et al. Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat. Neurosci 18, 1230–1232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Hasani R et al. Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron 87, 1063–1077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gibson GD et al. Distinct accumbens shell output pathways promote versus prevent relapse to alcohol seeking. Neuron 98, 512–520 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Gokce O et al. Cellular taxonomy of the mouse striatum as revealed by single-cell RNA-seq. Cell Rep 16, 1126–1137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stanley G, Gokce O, Malenka RC, Sudhof TC & Quake SR Continuous and discrete neuron types of the adult murine striatum. Neuron 105, 688–699 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Saunders A et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen R, Wu X, Jiang L & Zhang Y Single-cell RNA-seq reveals hypothalamic cell diversity. Cell Rep 18, 3227–3241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeisel A et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Zeisel A et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ibanez-Sandoval O et al. A novel functionally distinct subtype of striatal neuropeptide Y interneuron. J. Neurosci 31, 16757–16769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz-Manchado AB et al. Diversity of interneurons in the dorsal striatum revealed by single-cell RNA sequencing and PatchSeq. Cell Rep 24, 2179–2190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walaas SI, Aswad DW & Greengard P A dopamine- and cyclic AMP-regulated phosphoprotein enriched in dopamine-innervated brain regions. Nature 301, 69–71 (1983). [DOI] [PubMed] [Google Scholar]
- 40.Hintiryan H et al. The mouse cortico-striatal projectome. Nat. Neurosci 19, 1100–1114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J, Wang W & Sabatini BL Anatomically segregated basal ganglia pathways allow parallel behavioral modulation. Nat. Neurosci 23, 1388–1398 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao X et al. A genetically defined compartmentalized striatal direct pathway for negative reinforcement. Cell 183, 211–227 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castro DC & Bruchas MR A motivational and neuropeptidergic hub: anatomical and functional diversity within the nucleus accumbens shell. Neuron 102, 529–552 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaudhri N, Sahuque LL, Schairer WW & Janak PH Separable roles of the nucleus accumbens core and shell in context- and cue-induced alcohol-seeking. Neuropsychopharmacology 35, 783–791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Chiara G Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav. Brain Res 137, 75–114 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Ito R, Robbins TW & Everitt BJ Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat. Neurosci 7, 389–397 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Castro DC & Berridge KC Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness ‘liking’ and ‘wanting’. J. Neurosci 34, 4239–4250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puga L, Alcantara-Alonso V, Coffeen U, Jaimes O & de Gortari P TRH injected into the nucleus accumbens shell releases dopamine and reduces feeding motivation in rats. Behav. Brain Res 306, 128–136 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Darcq E & Kieffer BL Opioid receptors: drivers to addiction? Nat. Rev. Neurosci 19, 499–514 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Le Merrer J, Becker JA, Befort K & Kieffer BL Reward processing by the opioid system in the brain. Physiol. Rev 89, 1379–1412 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banghart MR, Neufeld SQ, Wong NC & Sabatini BL Enkephalin disinhibits mu opioid receptor-rich striatal patches via delta opioid receptors. Neuron 88, 1227–1239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tejeda HA et al. Pathway- and cell-specific kappa-opioid receptor modulation of excitation-inhibition balance differentially gates D1 and D2 accumbens neuron activity. Neuron 93, 147–163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eng CL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Francis TC, Yano H, Demarest TG, Shen H & Bonci A High-frequency activation of nucleus accumbens D1-MSNs drives excitatory potentiation on D2-MSNs. Neuron 103, 432–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lechan RM & Fekete C The TRH neuron: a hypothalamic integrator of energy metabolism. Prog. Brain Res 153, 209–235 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Zhang M et al. Molecular, spatial and projection diversity of neurons in primary motor cortex revealed by in situ single-cell transcriptomics Preprint at https://www.biorxiv.org/content/10.1101/2020.06.04.105700v1 (2020).
- 57.Brewer GJ & Torricelli JR Isolation and culture of adult neurons and neurospheres. Nat. Protoc 2, 1490–1498 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Zheng GX et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuhn M Building predictive models in R using the caret package. J. Stat. Softw 1, 2008 (2008). [Google Scholar]
- 61.Mayer C et al. Developmental diversification of cortical inhibitory interneurons. Nature 555, 457–462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shekhar K et al. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell 166, 1308–1323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blondel VD, Guillaume J-L, Lambiotte R & Lefebvre E Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008 (2008). [Google Scholar]
- 66.Moffitt JR et al. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl Acad. Sci. USA 113, 11046–11051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xia C, Fan J, Emanuel G, Hao J & Zhuang X Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl Acad. Sci. USA 116, 19490–19499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gu Z, Eils R & Schlesner M Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Kolde R Pheatmap: pretty heatmaps [Software] (2015).
- 73.Wickham H ggplot2: Elegant Graphics for Data Analysis (Springer, 2016). [Google Scholar]
- 74.Tasic B et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq data have been deposited at the Gene Expression Omnibus with accession number GSE118020. The MERFISH data are available at the Brain Image Library (https://download.brainimagelibrary.org/fc/4c/fc4c2570c3711952/).