

Abstract
Low tidal volume ventilation protects the lung in mechanically ventilated patients. The impact of the accompanying permissive hypoxemia and hypercapnia on endothelial cell recovery from injury is poorly understood. CA (carbonic anhydrase) IX is expressed in pulmonary microvascular endothelial cells (PMVECs), where it contributes to CO2 and pH homeostasis, bioenergetics, and angiogenesis. We hypothesized that CA IX is important for PMVEC survival and that CA IX expression and release from PMVECs are increased during infection. Although the plasma concentration of CA IX was unchanged in human and rat pneumonia, there was a trend toward increasing CA IX in the bronchoalveolar fluid of mechanically ventilated critically ill patients with pneumonia and a significant increase in CA IX in the lung tissue lysates of pneumonia rats. To investigate the functional implications of the lung CA IX increase, we generated PMVEC cell lines harboring domain-specific CA IX mutations. By using these cells, we found that infection promotes intracellular (IC) expression, release, and MMP (metalloproteinase)-mediated extracellular cleavage of CA IX in PMVECs. IC domain deletion uniquely impaired CA IX membrane localization. Loss of the CA IX IC domain promoted cell death after infection, suggesting that the IC domain has an important role in PMVEC survival. We also found that hypoxia improves survival, whereas hypercapnia reverses the protective effect of hypoxia, during infection. Thus, we report 1) that CA IX increases in the lungs of pneumonia rats and 2) that the CA IX IC domain and hypoxia promote PMVEC survival during infection.
Keywords: pneumonia, acidosis, hypercapnia, ultrasound, angiopoietin-2
Clinical Relevance
Lung-protective ventilatory strategies significantly improve acute respiratory distress syndrome survival. However, implications of the accompanying permissive hypoxemia and hypercapnia are poorly understood. Herein, we report that CA (carbonic anhydrase) IX, a hypoxia-responsive protein involved in CO2 homeostasis, is a potential mediator of pulmonary microvascular endothelial cell survival during infection.
The ARDSNet (Acute Respiratory Distress Syndrome [ARDS] Network) trial led to successful implementation of a lung-protective ventilatory strategy in the care of patients with ARDS (1, 2). One of the major findings from the ARDSNet trial was that gas exchange parameters do not correlate with important clinical outcomes (3). This result challenged the conventional target goals for the partial pressure of arterial oxygen (PaO2) and partial pressure of arterial carbon dioxide (PaCO2) in ARDS management, which were mostly based on consensus expert opinion rather than direct scientific evidence. Thus, permissive hypoxia and hypercapnia are presently acceptable, in favor of limiting mechanical injury to the lung (4, 5). Although “suboptimal” gas exchange from low tidal volume ventilation is considered a relatively benign and necessary expense of protecting the lung, little is known about how such blood gas derangements impact clinical outcomes (6, 7).
Recent randomized controlled trials compared conservative with liberal oxygen therapy in critically ill patients, mainly to examine how the current PaO2 target goal may affect cellular physiology. A recent study found that an increase in the fraction of inspired oxygen increases the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) co-receptor expression in lung epithelial cells, providing an example of an unknown but potentially harmful effect of hyperoxia (8). The CLOSE (Conservative [arterial oxyhemoglobin saturation (SpO2) between 88 and 92%] versus Liberal [SpO2 equal or greater than 96%] Oxygenation Targets for Mechanically Ventilated Patients) (9) and the LOCO2 (Liberal [PaO2 between 90 and 105 mm Hg and SpO2 equal or greater than 96%] Oxygenation versus Conservative [PaO2 between 55 and 70 mm Hg and SpO2 between 88 and 92%] Oxygenation in ARDS) (10) trials showed no increase in major adverse outcomes with conservative oxygen therapy. In the OXYGEN-ICU (Effect of Conservative [PaO2 between 70 and 100 mm Hg or SpO2 between 94 and 98%] versus Conventional [PaO2 up to 150 mm Hg or SpO2 between 97 and 100%] Oxygen Therapy on Mortality among Patients in an ICU) trial, mortality was lower in the conservative oxygen therapy group (11). However, the mechanisms involved in these findings were unclear, and the results were not analyzed in the context of accompanying dynamic CO2 changes, despite increasing evidence that hypercapnia may confer detrimental cellular and systemic effects in populations with moderate to severe ARDS (12). This collection of studies supports the current recommendation for lower PaO2 target values in mechanically ventilated patients.
Studies unanimously indicate that CO2 is an important mediator of inflammation during infection (13–15). However, whether this regulatory role is beneficial or harmful is unclear, as the evidence seems to be largely context dependent. Some evidence supporting a beneficial effect of CO2 includes the concept of a “pH paradox,” which involves a protective effect of acidosis during ischemia-reperfusion injury (16). Ongoing studies also seek to determine whether hypercapnia in patients with cardiac arrest has a therapeutic effect by protecting cerebral blood flow (clinicaltrials.gov identifier NCT 03114033). Whether the results from these studies on sterile inflammation can be applied to patients with ARDS remains to be determined. Only a limited number of clinical trials investigated a direct relationship between hypercapnia and mortality in populations with ARDS (12). Among three landmark studies, one supported that the effects of CO2 on mechanically ventilated patients with ARDS were beneficial (17), whereas two supported that the effects were harmful (18, 19). These studies were all retrospective analyses without a specific intervention to control for PaCO2; therefore, a causal relationship between PaCO2 and patient outcomes could not be determined. The complex nature of ARDS, including heterogeneous areas of involvement with dynamic variations in ventilation-to-perfusion matching, underscores the importance of developing a better understanding of the cellular responses to shifts in CO2.
CAs (carbonic anhydrases) are important regulators of CO2. These enzymes are the principal mediators of the bicarbonate buffer system; they are best known for their roles in catalyzing the reversible conversion of CO2 to carbonic acids. Although “buffer system” implies a temporary and finite nature of activity, increasing reports on noncatalytic functions of CA isoforms suggest that they possess broader roles beyond pH regulation (20). In particular, the CA IX isoform has been extensively studied for its noncatalytic activities related to cancer invasion and migration (21), but its role in inflammatory conditions such as infection is not known. We have previously shown that CA IX is abundantly expressed in pulmonary microvascular endothelial cells (PMVECs) (22), which are critical constituents of the alveolar capillary barrier (23, 24). CA IX possesses the traditional pH regulatory role in PMVECs, and its noncatalytic functions extend to the modulation of metabolism, migration, and network formation (25). However, it is unclear whether CA IX plays a role in PMVEC survival during infection, and furthermore, there is uncertainty about which of the protein’s functional domains mediate a cell-survival signal. Here, we sought to determine the roles of CA IX in pneumonia by examining human subjects and rats with pneumonia and by using CA IX functional domain mutants to elucidate domain-specific roles of CA IX in PMVEC survival in the context of hypoxic and hypercapnic environments during infection.
Methods
Human Plasma CA IX and Ang-2
Patients were enrolled from the medical ICU at the University of South Alabama University Hospital (USAH) and the trauma–burn, neurological, surgical, and medical ICUs at the University of Alabama at Birmingham Hospital (UABH). All studies were approved by the institutional review boards of the respective universities (26). Plasma CA IX and Ang-2 (angiopoietin-2) and BAL-fluid (BALF) CA IX were measured by using an ELISA.
Rat Pneumonia Model
All experimental procedures were approved by the U.S. Institutional Animal Care and Use Committee. PBS with or without Pseudomonas aeruginosa was introduced into the tracheas of male Fischer rats. After 24 hours, cardiopulmonary ultrasonography was performed, followed by abdominal aortic blood sampling and tissue harvesting (27).
Isolation of Rat Lung Endothelial Cells
Cell isolation procedures were approved by the U.S. Institutional Animal Care and Use Committee. PMVECs were isolated from male Sprague-Dawley rats and verified as previously described (28). All cells were from one animal. All mutant cell lines reached greater than 10 passages after genetic modification and selection, and they were maintained on bicarbonate-buffered media in Dulbecco’s modified Eagle medium, 10% FCS, and 1% penicillin–streptomycin at 37°C in ambient air (5% CO2).
Generation of CA IX Mutant Cell Lines
CA IX possesses an N-terminal proteoglycan-like (PG) domain, a catalytic CA domain, a transmembrane (TM) domain, and an intracellular (IC) domain (29). To investigate domain-specific CA IX function in PMVECs, we used CRISPR–Cas9 to create CA IX–knockout PMVECs (22). We rescued CA IX–knockout PMVECs with wild-type (WT) CA IX (rescued WT [rWT]) or mutant alleles that lacked the PG (ΔPG), CA (ΔCA), or IC (ΔIC) domains. This was achieved by using a conditional expression system, in which CA IX mutant expression is controlled by doxycycline (tetracycline off [Tet-Off]), as previously described (30, 31). Cell-line establishment was confirmed by using Western blotting and immunocytochemistry, as shown in the data supplement.
Analysis of CA IX Secretory Mechanisms and Infection-induced Cytotoxicity In Vitro
PMVECs were seeded on bicarbonate-buffered media at 37°C in ambient air (5% CO2). Two days later, media were replaced with Hanks’ balanced salt solution with DMSO or an MMP (metalloproteinase) inhibitor (batimastat or marimastat [MM]) with or without P. aeruginosa, and cells were incubated under normoxia (21% O2) or hypoxia (1% O2) with 5% or 10% CO2. Three and 6 hours later, cells were photographed, and LDH (lactate dehydrogenase) activity was measured in the culture media. The CA IX protein abundance was measured in supernatants and lysates by using Western blotting. ATP was measured at 6 hours, and propidium iodide staining was performed at 5.5 hours.
Statistics
Student’s t tests, one-way ANOVA with Bonferroni post hoc tests, chi-square tests (or Fisher exact tests for small sample sizes), and Wilcoxon rank sum tests were used as indicated. Significance was denoted by P < 0.05.
See the data supplement for further details.
Results
CA IX Does Not Change in the Plasma but May Increase in the BALF of Mechanically Ventilated Patients with Pneumonia within 24 Hours of ICU Admission
Elevated circulating concentrations of CA IX have been suggested to portend poor outcomes in patients with circulatory compromise, such as right heart dysfunction during pulmonary embolism (32) and cardiogenic shock requiring extracorporeal support (33). Although infection enhances glycolytic metabolism and causes systemic acidosis, whether the plasma concentrations of CA IX, a TM molecule with a powerful extracellular catalytic domain, change during pneumonia is not known. To address this issue, we measured CA IX in the plasma of noninfectious control subjects and patients with pneumonia on the first day of ICU admission in the USAH medical ICU and the UABH trauma–burn, neurological, surgical, and medical ICUs.
For USAH patients, stroke was more common in the control group, and the majority of patients with pneumonia were septic (Table 1). ICU and hospital lengths of stay were significantly longer in the pneumonia group. Plasma CA IX was not different between groups within 24 hours of the ICU admission (Figure 1A; USAH control group vs. USAH pneumonia group, P = not significant [ns]). We also measured plasma Ang-2, a known endothelium-derived marker of lung permeability and clinical outcomes during pneumonia (34), for comparison. The plasma Ang-2 concentration was significantly higher in patients with pneumonia (Figure 1B; USAH control group vs. USAH pneumonia group, P < 0.05), which is consistent with the worse clinical outcomes among these patients, including longer ICU and hospital stays. These findings indicate that plasma CA IX does not predict pneumonia, the need for mechanical ventilation, or the ICU/hospital length of stay within 24 hours of medical ICU admission.
Table 1.
Baseline Characteristics of USAH Patients
| Characteristic | Control (n = 20) | Pneumonia (n = 20) | Total (N = 40) |
|---|---|---|---|
| Age, yr, median (range) | 61.5 (21–82) | 63 (34–91) | 62 (21–91) |
| Male sex, n/N (%) | 9/20 (45) | 9/20 (45) | 18/40 (45) |
| Race, n/N (%) | |||
| Black | 9/20 (45) | 12/20 (60) | 21/40 (52.5) |
| White | 11/20 (55) | 8/20 (40) | 19/40 (47.5) |
| Primary diagnosis, n/N (%) | |||
| Sepsis | 0 | 18/20 (90) | 18/40 (45)* |
| Stroke | 10/20 (50) | 0 | 10/40 (25)* |
| Seizure/encephalopathy | 4/20 (20) | 1/20 (5) | 5/40 (12.5) |
| Hypertensive urgency | 3/20 (15) | 0 | 3/40 (7.5) |
| Gastrointestinal bleed | 2/20 (10) | 0 | 2/40 (5) |
| Acute coronary syndrome | 0 | 1/20 (5) | 1/40 (2.5) |
| Electrolyte imbalance | 1/20 (5) | 0 | 1/40 (2.5) |
| Mechanical ventilator, n/N (%) | 3/20 (15) | 17/20 (85) | 20/40 (50)* |
| Vasopressor, n/N (%) | 2/20 (10) | 6/20 (30) | 8/40 (40) |
| ICU stay, d, median (range) | 2 (2–20) | 7 (3–30) | 4 (2–30)* |
| Hospital stay, d, median (range) | 4.5 (2–53) | 13.5 (5–37) | 8.5 (2–53)* |
| Deceased, n/N (%) | 1/20 (5) | 3/20 (15) | 4/40 (10) |
Definition of abbreviations: ICU = intensive care unit; USAH = University of South Alabama University Hospital.
Chi-square tests (or Fisher exact tests for small sample sizes) and Wilcoxon rank sum tests were used for categorical and quantitative data, respectively.
Significant difference (P < 0.05) for control versus pneumonia.
Figure 1.
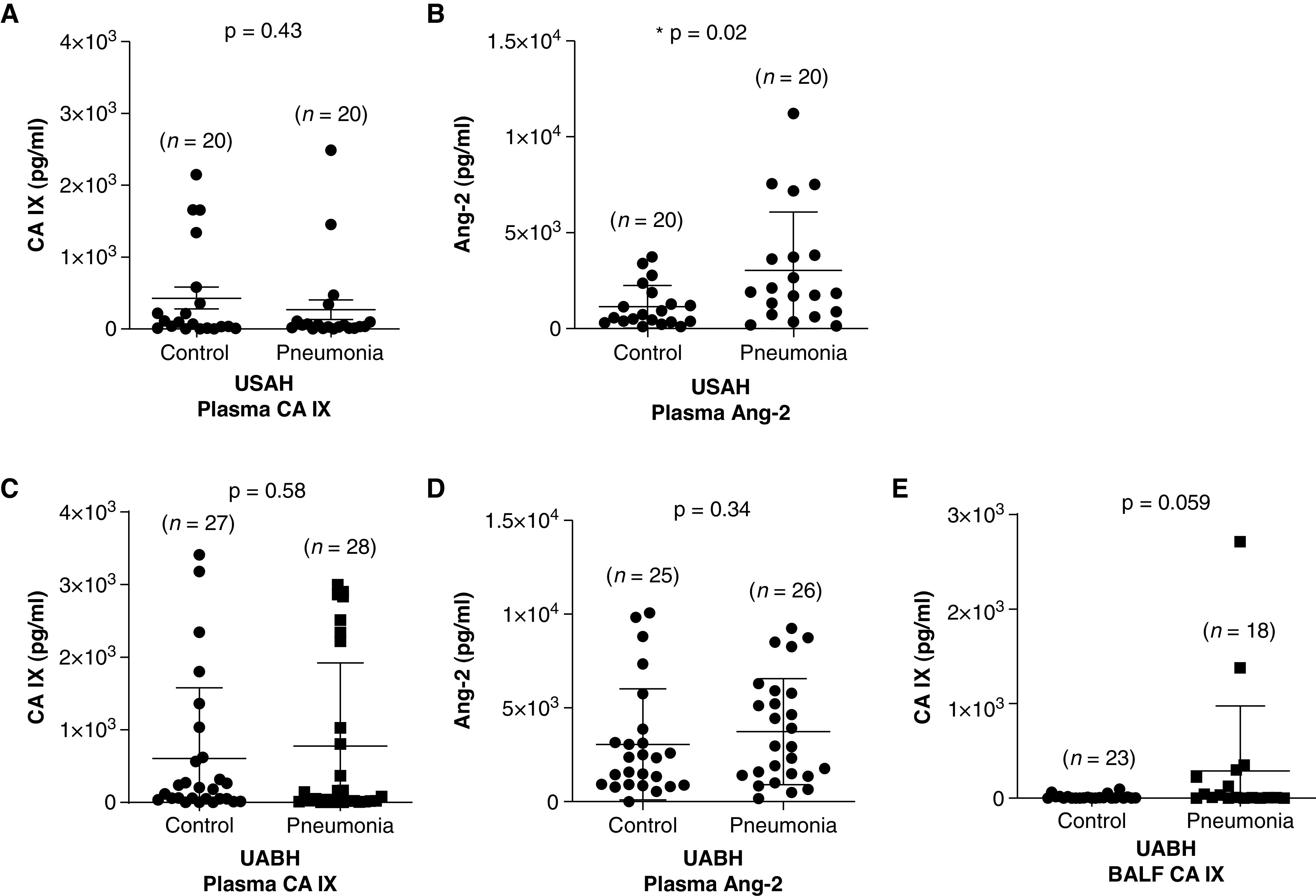
CA (carbonic anhydrase) IX does not change in the plasma but may increase in the BAL fluid (BALF) of mechanically ventilated patients with pneumonia within 24 hours of ICU admission. CA IX was measured by using an ELISA in the plasma of critically ill patients with and without pneumonia from the University of South Alabama University Hospital (USAH) and the University of Alabama at Birmingham Hospital (UABH) at 24 hours after ICU admission. USAH patients were classified into control and pneumonia groups on the basis of primary diagnoses of noninfectious etiology and clinical pneumonia, respectively. UABH patients were classified into control and pneumonia groups on the basis of BALF culture results, and only mechanically ventilated patients were enrolled for this study. (A) There was no difference in the plasma CA IX concentrations between the USAH control and pneumonia groups. (B) Ang-2 (angiopoietin-2) was additionally measured in these plasma samples, as it is a known endothelium-derived biomarker of pneumonia. Plasma Ang-2 was significantly higher in USAH patients with pneumonia than in the control subjects. (C and D) In UABH patients, neither CA IX concentrations nor Ang-2 concentrations were different between the control and pneumonia groups, but (E) there was a trend toward an increase in the BALF CA IX concentration in the pneumonia group compared with the control group. Data represent the mean ± SD. A Wilcoxon rank sum test was used to compare different groups. *Significant difference (P < 0.05) from control group.
Unlike the USAH pneumonia classification, which was based on the clinical diagnosis without considering the culture results, UABH patients were grouped on the basis of BALF culture positivity, and all patients enrolled were mechanically ventilated. Table 2 shows that the primary diagnoses of UABH patients were mostly surgical, and all baseline characteristics were in balance between the control and pneumonia groups. Notably, compared with USAH patients, UABH control patients showed higher vasopressor use and mortality (Table 2; USAH control group vs. UABH control group, P < 0.05), and both UABH groups had longer ICU and hospital stays than their USAH counterparts. Although there was no difference in the plasma CA IX concentration (Figure 1C; UABH control group vs. UABH pneumonia group, P = ns) or plasma Ang-2 concentration (Figure 1D; UABH control group vs. UABH pneumonia group, P = ns) between UABH groups, plasma Ang-2 concentrations for both UABH groups were higher than those of USAH control patients (Table 2; USAH control group vs. UABH control group and USAH control group vs. UABH pneumonia group, P < 0.05). Interestingly, there was a trend toward an increase in the BALF CA IX concentration in the pneumonia group compared with the control group among the UABH patients (Figure 1E; UABH control group vs. UABH pneumonia group, P = 0.059). These findings suggest that plasma CA IX is not a biomarker of pneumonia in critically ill patients within 24 hours of ICU admission but that the BALF CA IX concentration may be associated with pneumonia in mechanically ventilated patients with pneumonia.
Table 2.
Baseline Characteristics of UABH Patients
| Characteristic | Control (n = 27) | Pneumonia (n = 28) | Total (N = 55) |
|---|---|---|---|
| Age, yr, median (range) | 55 (20–87) | 58.5 (20–83) | 58 (20–87) |
| Male sex, n/N (%) | 13/27 (48.1) | 19/28 (67.9) | 32/55 (58.2) |
| Race, n/N (%) | |||
| White | 17/27 (63) | 22/28 (78.6) | 39/55 (70.9) |
| Black | 8/27 (29.6) | 5/28 (17.9) | 13/55 (23.6) |
| Hispanic | 2/27 (7.4) | 1/28 (3.6) | 3/55 (5.5) |
| Primary diagnosis, n/N (%) | |||
| Trauma | 9/27 (33.3) | 17/28 (60.7) | 26/55 (47.3) |
| Stroke | 10/27 (37) | 4/28 (14.3) | 14/55 (25.5) |
| Pancreatitis/gastrointestinal surgery | 2/27 (7.4) | 2/28 (7.1) | 4/55 (7.3) |
| Cardiovascular surgery | 2/27 (7.4) | 1/28 (3.6) | 3/55 (5.5) |
| Burn | 1/27 (3.7) | 1/28 (3.6) | 2/55 (3.6) |
| Tracheal/lung surgery | 0 | 2/28 (7.1) | 2/55 (3.6) |
| Transplantation (liver/kidney) | 1/27 (3.7) | 1/28 (3.6) | 2/55 (3.6) |
| Seizure/encephalopathy | 1/27 (3.7) | 0 | 1/55 (1.8) |
| Genitourinary surgery | 1/27 (3.7) | 0 | 1/55 (1.8) |
| Mechanical ventilator, n/N (%) | 27/27 (100) | 28/28 (100) | 55/55 (100) |
| Vasopressor, n/N (%) | 17/27 (62.3)* | 14/28 (50) | 31/55 (56.4) |
| ICU stay, d, median (range) | 18 (5–106)* | 14.5 (5–105)* | 16 (5–106) |
| Hospital stay, d, median (range) | 26 (6–108)* | 20 (9–105)* | 21 (6–108) |
| Deceased, n/N (%) | 6/27 (22.2)* | 9/28 (32.1) | 15/55 (27.3) |
| BAL culture, n/N (%) | |||
| P. aeruginosa | — | 8/28 (28.6) | — |
| S. aureus | — | 12/28 (42.9) | — |
| K. pneumoniae | — | 2/28 (7.1) | — |
| H. influenzae | — | 1/28 (3.6) | — |
| E. aerogenes | — | 1/28 (3.6) | — |
| S. aureus and P. aeruginosa | — | 1/28 (3.6) | — |
| S. pneumoniae and H. influenza | — | 1/28 (3.6) | — |
| H. influenzae and S. maltophilia | — | 1/28 (3.6) | — |
| E. cloacae and E. sakazakii | — | 1/28 (3.6) | — |
Definition of abbreviations: E. aerogenes = Enterobacter aerogenes; E. cloacae = Enterobacter cloacae; E. sakazakii = Enterobacter sakazakii; H. influenzae = Haemophilus influenzae; K. pneumoniae = Klebsiella pneumoniae; P. aeruginosa = Pseudomonas aeruginosa; S. aureus = Staphylococcus aureus; S. maltophilia = Stenotrophomonas maltophilia; S. pneumoniae = Streptococcus pneumoniae; UABH = University of Alabama at Birmingham Hospital.
Chi-square tests (or Fisher exact tests for small sample sizes) and Wilcoxon rank sum tests were used for categorical and quantitative data, respectively.
Significant difference (P < 0.05) for USAH control versus UABH control or USAH pneumonia versus UABH pneumonia.
CA IX Increases in the Lung Tissue but Not in the Plasma of Pneumonia Rats 24 Hours after Infection
Considering the increasing trend of BALF CA IX in critically ill patients with pneumonia, we further investigated lung CA IX dynamics in a rat pneumonia model. Pneumonia was induced in rats by using intratracheal inoculation of P. aeruginosa, a common cause of bacterial pneumonia among critically ill patients (35). Twenty-four hours later, rats developed multifocal lung inflammation and edema, as indicated by consolidation and B-line patterns on lung ultrasound images (Figure 2A), an increased lung tissue wet-to-dry ratio (Figure 2B; control rats vs. pneumonia rats, P < 0.05) and increased nuclear (hematoxylin) and nonspecific protein (eosin) staining intensity on histology images (Figure 2C; control rats vs. pneumonia rats, P < 0.05). Echocardiography showed no change in the ejection fraction (data not shown) or cardiac output estimated from the parasternal long axis view M-mode (Figure 2D; control rats vs. pneumonia rats, P = ns). The aortic arch flow, estimated by using the velocity time integral (VTI), was unchanged (Figure 2E; control rats vs. pneumonia rats, P = ns), but the abdominal aortic VTI was significantly lower in pneumonia (Figure 2F; control rats vs. pneumonia rats, P < 0.05). The disproportionately decreased abdominal aortic VTI compared with the aortic arch VTI may indicate a redistribution of blood flow that prioritizes upper-body perfusion, including perfusion to the brain (36). Although parameters of left ventricular function were unaffected, the tricuspid annular plane systolic excursion (Figure 2G; control rats vs. pneumonia rats, P < 0.05) and the tricuspid tissue Doppler systolic positive wave velocity (Figure 2H; control rats vs. pneumonia rats, P < 0.05), which are sensitive markers of right ventricular systolic function, were both decreased in pneumonia. Together, these physiological findings in a rodent pneumonia model mimic what is seen in critically ill patients with pneumonia.
Figure 2.
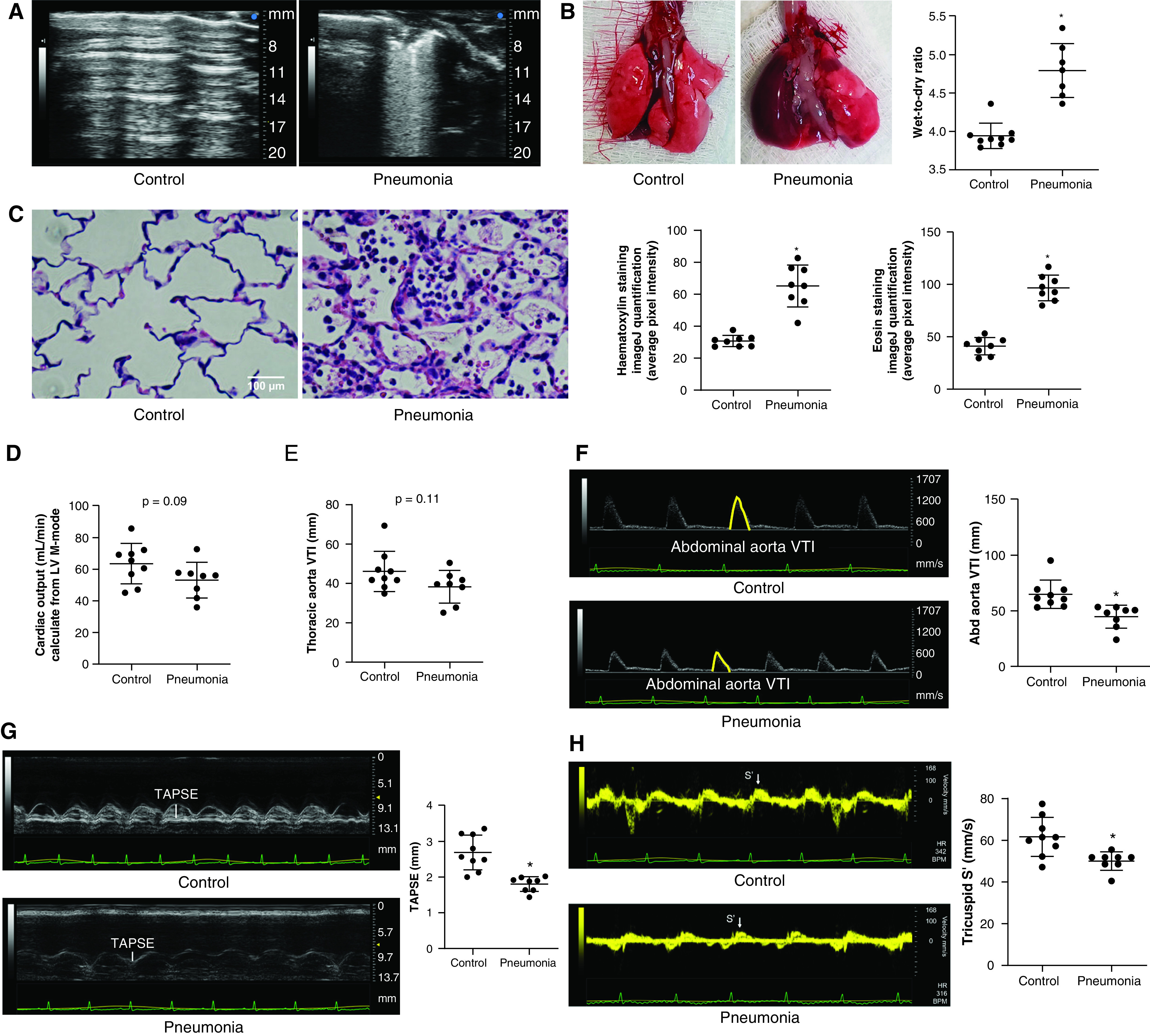
A Pseudomonas aeruginosa pneumonia rat model induces heterogeneous lung injury and cardiovascular changes mimicking human sepsis. To mimic human pneumonia in rats, P. aeruginosa (1 × 107 cfu in 200 μl of PBS) was introduced into the tracheas of Fischer rats. Twenty-four hours later, pneumonia rats (A) developed multiple areas of consolidation and B-line patterns on lung ultrasound images, (B) showed an increased lung tissue wet-to-dry ratio, and (C) showed an increased degree of inflammation as demonstrated by using lung tissue hematoxylin and eosin staining, compared with PBS control rats. For hematoxylin and eosin staining, four evenly spaced axial sections were made throughout the left lungs obtained from two rats per group. Each dot represents an average of two images from each section. Echocardiography revealed no significant change in (D) the cardiac output or(E) the thoracic aortic flow as estimated by using the velocity time integral (VTI), but the (F) Abd aortic VTI, (G) TAPSE, and (H) tricuspid S′ were decreased, indicating potential redistribution of blood flow away from lower body and compromised right ventricular systolic function in pneumonia rats. Scale bar, 100 μm. Data represent the mean ± SD from n = 8–9 animals per group. A Student’s t test was used to compare different groups. *Significant difference (P < 0.05) from control. Abd = abdominal; cfu = colony-forming unit; LV = left ventricular; S′ = tissue Doppler systolic positive wave velocity; TAPSE = tricuspid annular plane systolic excursion.
With this animal model, we measured plasma and major organ tissue CA IX concentrations by using Western blotting. Consistent with our human data, there was no significant difference in the plasma CA IX concentration between control and pneumonia rats (Figure 3A; control rats vs. pneumonia rats, P = ns). However, the CA IX protein concentration was profoundly increased in the lung tissue (Figure 3B; control rats vs. pneumonia rats, P < 0.05) but was not increased in any other major organs, including the kidney, heart, brain, liver, and diaphragm, in pneumonia rats (Figures 3C–3G; control rats vs. pneumonia rats, P = ns). These findings indicate that P. aeruginosa pneumonia increases lung tissue CA IX without significantly affecting circulating CA IX at 24 hours after infection.
Figure 3.
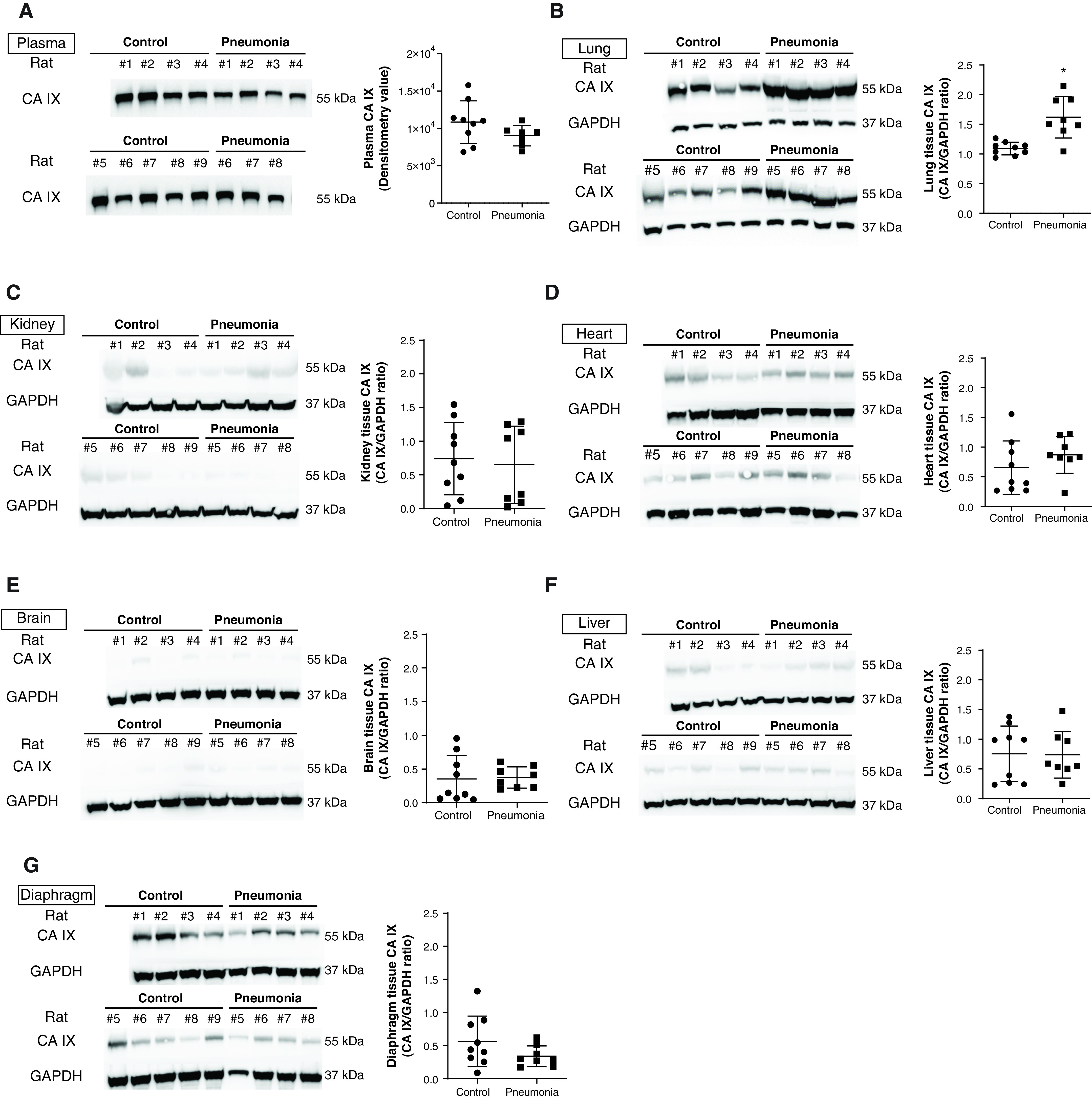
CA IX increases in the lung tissue but not in the plasma or other major organ tissue of rats with pneumonia. P. aeruginosa (1 × 107 cfu in 200 μl of PBS) was introduced into the tracheas of Fischer rats. Twenty-four hours later, after ultrasound was completed, blood samples were drawn from the Abd aorta, and lung, kidney, heart, brain, liver, and diaphragm tissues were harvested. (A) Western blot analysis showed no change in plasma CA IX concentrations with pneumonia, which is consistent with human data. CA IX was significantly increased in (B) lung tissue but not in (C–G) other organs examined in pneumonia rats. Data represent the mean ± SD. A Student’s t test was used to compare different groups. *Significant difference (P < 0.05) from control. Readers may view the uncut gels for B, D, E, and G in the data supplement.
Rat PMVECs Expressing Domain-Specific CA IX Functional Mutants Confirm a Unique Role of the IC Domain in Mediating CA IX Membrane Localization
Our previous finding of abundant CA IX expression in PMVECs (22) suggests that pulmonary capillaries may contribute to the increased concentrations of CA IX in the lung tissue of pneumonia rats. To investigate secretory mechanisms and physiological roles of CA IX during infection, we first generated PMVECs conditionally expressing either WT CA IX or functional mutants, including ΔPG, ΔCA, and ΔIC mutants. Schematic representations of these functional mutants are depicted in Figure 4A. Western blot analysis confirmed the expression of CA IX functional mutants in PMVECs, indicated by molecular weight shifts correlating with the size of each domain deleted. Because the CA domain is the largest (residues 137–391) domain, and because the IC domain is the smallest domain, the band shift is the largest with the ΔCA mutant and it is the smallest with the ΔIC mutant (Figure 4B). Interestingly, although the crystal structure of CA IX suggests that its multimer formation is mainly mediated by disulfide bonds between the CA domain of each monomer (37), our results showed preserved multimer formation in all mutant PMVECs, including the ΔCA mutant PMVECs. Upon introduction of doxycycline, the expression of CA IX variants in rescued cells was suppressed (Figure 4B), suggesting successful establishment of the Tet-Off conditional expression system.
Figure 4.
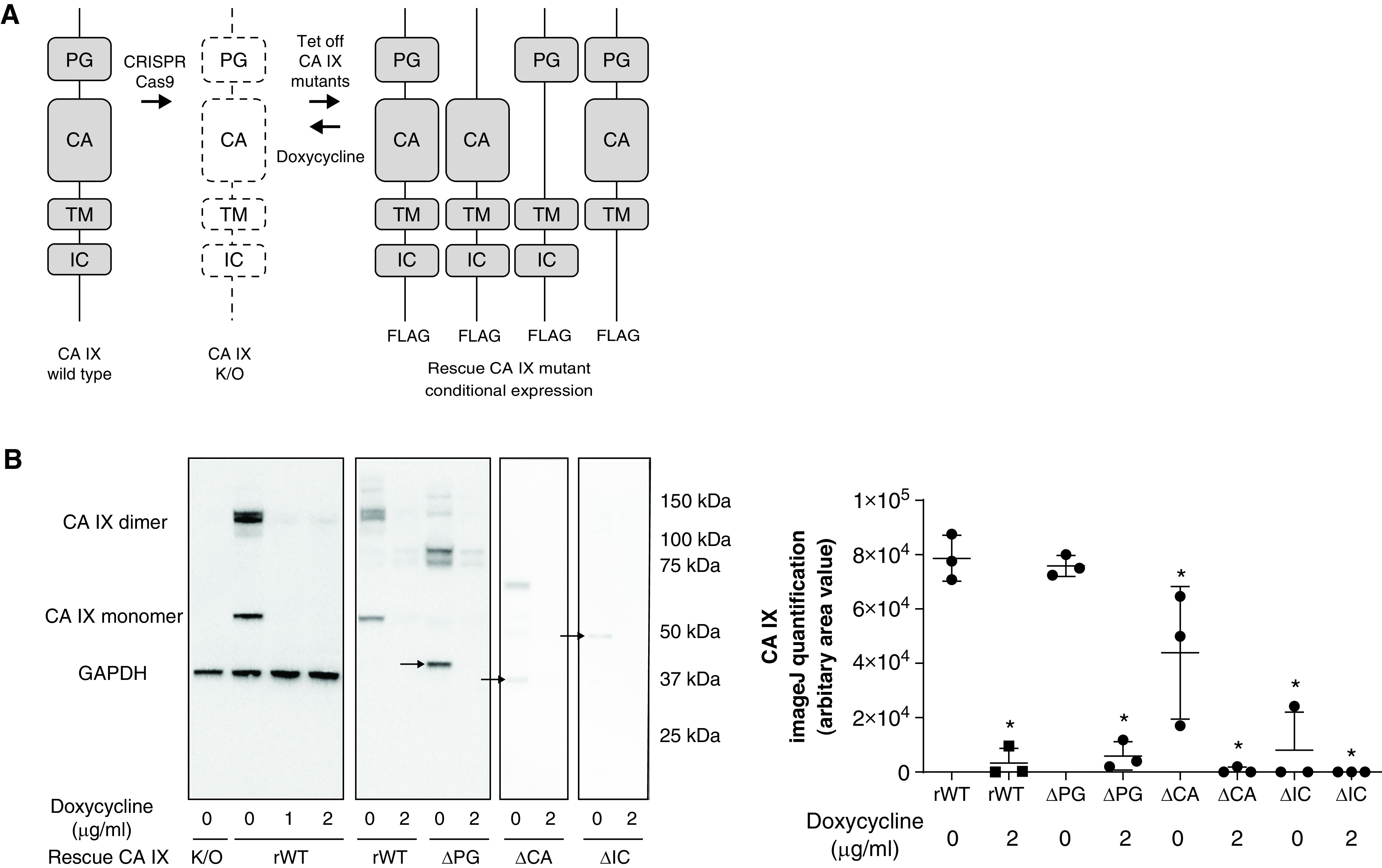
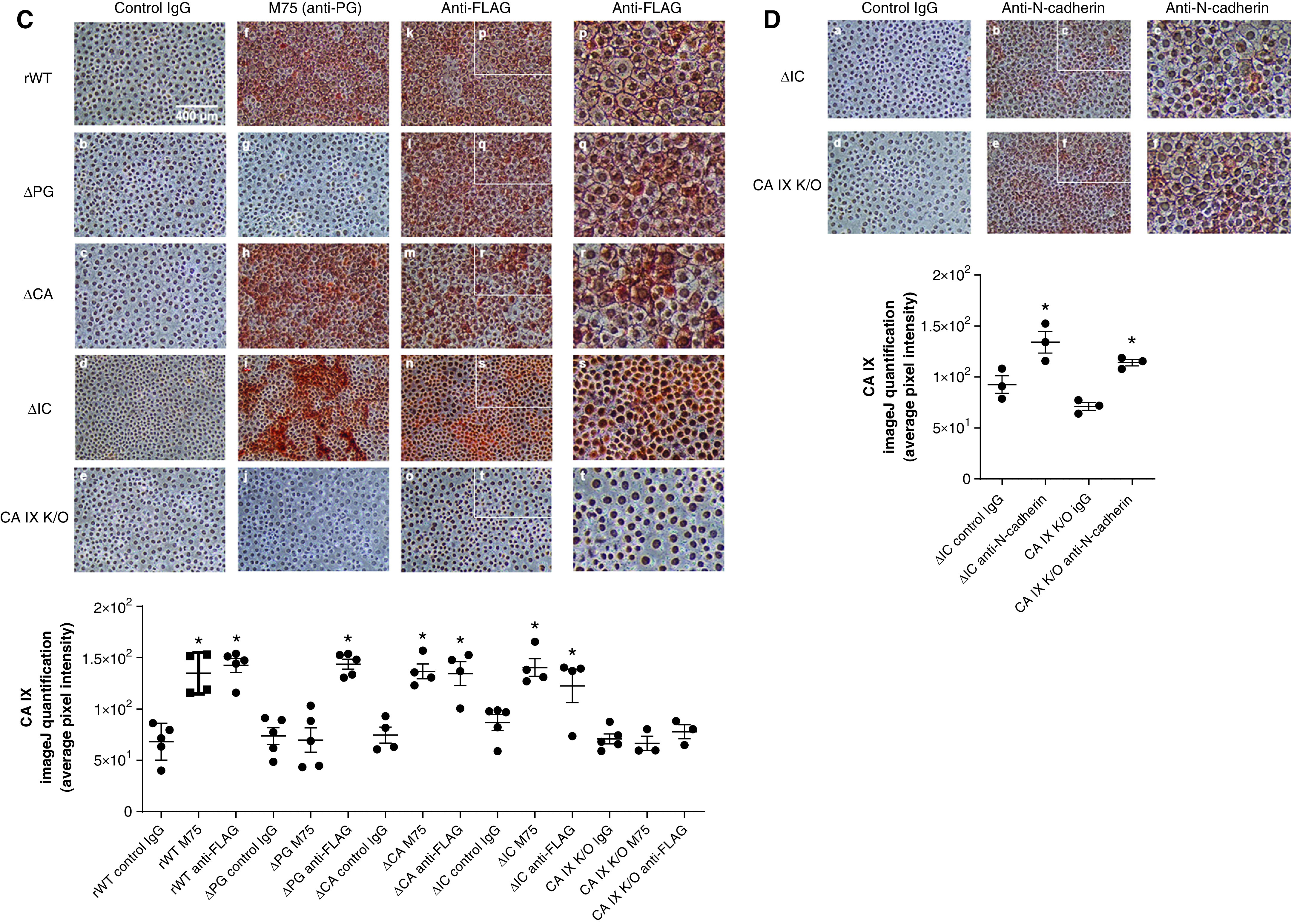
Rat pulmonary microvascular endothelial cells (PMVECs) expressing domain-specific CA IX functional mutants confirm a unique role of the intracellular (IC) domain in mediating CA IX membrane localization. (A) The CA IX functional mutant-generation strategy is schematically illustrated. CA IX consists of four functional domains: a proteoglycan-like (PG) domain, a catalytic CA domain, a transmembrane (TM) domain, and an IC domain. (B) Double transfection with a doxycycline-induced Tet-off conditional expression system and domain-specific CA IX mutant constructs was performed on previously generated CA IX–K/O PMVECs. CA IX protein abundance was assessed and quantified in cell lysates by using Western blotting with the FLAG-tag antibody. CA IX functional mutant PMVECs were successfully generated indicated by CA IX monomer molecular weight shifts (arrows) and protein abundance decreases upon doxycycline introduction. (C) To further visualize CA IX mutant protein expression, we performed immunocytochemistry on these CA IX mutant cells by using an HRP (horseradish peroxidase) and 3,3'-diaminobenzidine detection method. Both M75 and FLAG tag antibody staining were consistent with the presence and absence of the epitope of each mutant cell line. Notably, ΔIC (IC domain-deleted) PMVECs showed a distinctive staining pattern in which the proteins are localized to perinuclear areas, saving intercellular borders. (D) N-cadherin membrane localization was unaffected by the absence of the IC domain, indicating that the mediating role of the IC domain in protein trafficking and membrane stabilization is specific to the CA IX protein. At least three separate experiments were performed. Scale bar, 400 μm (for C and D). Data represent the mean ± SD. One-way ANOVA and Bonferroni post hoc tests were used to compare different groups. *Significant difference (P < 0.05) from rescued wild-type (rWT) dose of 0 μg/ml doxycycline (B), rWT control IgG (C), and ΔIC control IgG (D). ΔCA = CA domain deleted; ΔPG = PG domain deleted; K/O = knockout; Tet-off = tetracycline off.
Although each CA IX mutant expression was confirmed, immunoblot band intensities for these mutants suggested that there were potentially fewer CA IX mutant proteins in ΔCA and ΔIC cells than in rWT PMVECs (Figure 4B; rWT vs. ΔCA and ΔIC in the absence of doxycycline, P < 0.05). However, this protein abundance difference between mutants was ns when quantified through immunocytochemistry by using the M75 antibody raised against the PG domain (Figure 4C; rWT M75 vs. ΔCA M75 vs. ΔIC M75, P = ns) and the anti-FLAG antibody (Figure 4C; rWT anti-FLAG vs. ΔPG anti-FLAG vs. ΔCA anti-FLAG vs. ΔIC anti-FLAG, P = ns). Importantly, immunocytochemistry revealed a strikingly different CA IX mutant protein subcellular distribution pattern in ΔIC PMVECs. Unlike all other CA IX mutants that spread throughout the cytoplasm and intercellular junctions (Figure 4C panel p, q, and r), ΔIC CA IX was exclusively localized to the perinuclear areas (Figure 4C panel s). These findings are consistent with Hulikova and colleagues’ report (38) and suggest an important role of the IC domain in mediating CA IX membrane localization. This IC domain role in protein trafficking and/or membrane stabilization is most likely specific to CA IX because the deletion of the IC domain or the whole CA IX protein did not affect N-cadherin intercellular junction staining (Figure 4D; positive intercellular junction staining).
CA IX Is Released from Rat PMVECs Unprocessed, and It Is Cleaved Extracellularly by MMPs
Having established PMVECs that express CA IX variants, we investigated the secretory mechanisms of CA IX, which may help identify whether pulmonary capillaries contribute to the increase in lung tissue CA IX concentrations in pneumonia rats. We first confirmed the presence of the CA IX mutant proteins in both cell lysates and supernatants at baseline. The anti-PG M75 antibody primarily detected the cleaved ectodomain (PG–CA domain) CA IX monomers and dimers, indicated by the band shift (Figures 5A and 5B), compared with the unprocessed molecules detected by the FLAG tag antibody (Figures 5A and 5B). This ectodomain cleavage event was further supported by the appearance of a new low-molecular-weight band that represents the TM–IC domain fragment after ectodomain cleavage, as detected by the FLAG tag antibody (Figures 5C and 5D and Figure 6B). These findings indicate that CA IX is constitutively released and cleaved at the ectodomain–TM domain junction.
Figure 5.
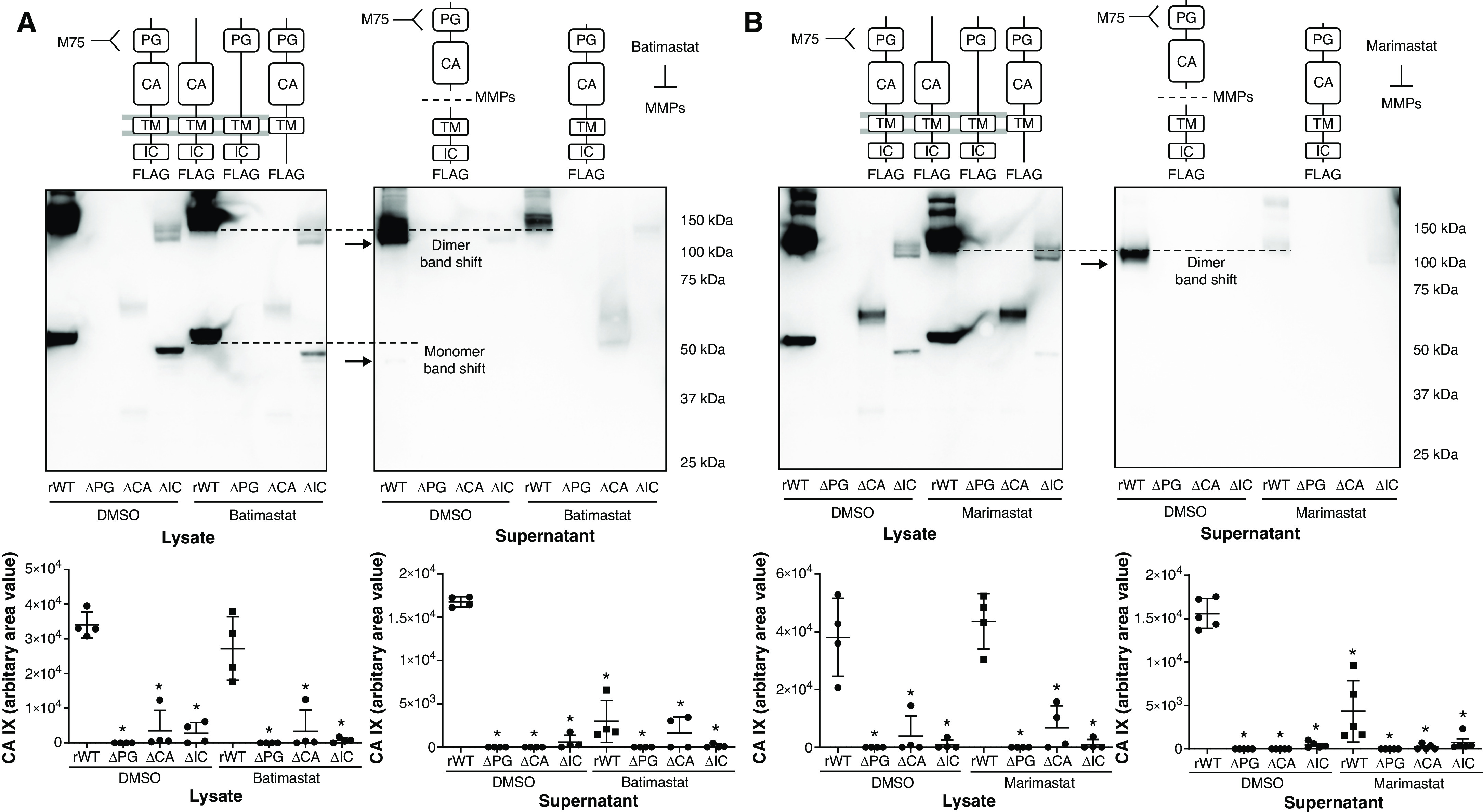
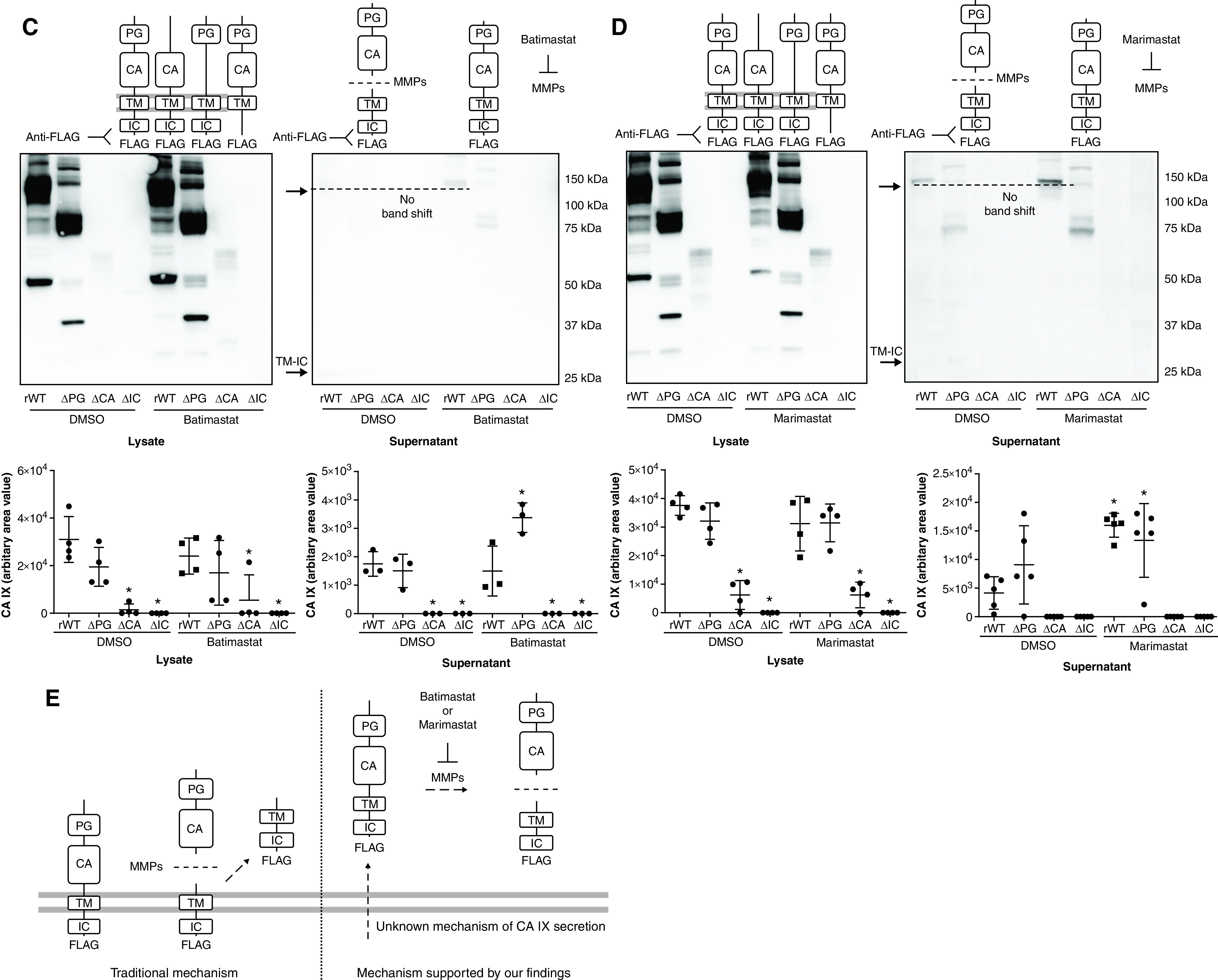
CA IX is released at full length and is cleaved extracellularly by MMPs (metalloproteinases) in PMVECs. (A–D) PMVEC CA IX mutant cell lines were seeded at 5.0 × 105 cells/well on 6-well plates on bicarbonate-buffered media. Two days after cell seeding, media were changed to Hanks’ balanced salt solution (HBSS) and treated with DMSO (0.1%), batimastat (10μM), or marimastat (10μM). Twenty-four hours later, CA IX protein abundance was measured in cell lysates and supernatants by using Western blotting. (A and B) An M75 antibody was used to detect monomer and dimer band shifts in supernatants, whereas (C and D) a FLAG tag antibody was used to detect full-length CA IX and a new low-molecular-weight band. The CA IX band shift detected by using the M75 antibody and the appearance of a new low-molecular-weight band detected by using the FLAG tag antibody were inhibited by (A and C) batimastat or (B and D) marimastat treatment, indicating that the band shift and the appearance of a new low-molecular-weight band represent a cleaved PG–CA ectodomain and remnants of the TM–IC domain, respectively. (E) A potential mechanism of CA IX release and MMP-mediated ectodomain cleavage in PMVECs is schematically illustrated. Considering that MMP inhibitors only inhibit extracellular cleavage, as evidenced by no molecular weight shift in lysate CA IX and the presence of a TM–IC fragment only in supernatants but not in cell lysates, unprocessed release followed by extracellular ectodomain cleavage is most likely the mechanism of CA IX release in PMVECs. At least three separate experiments were performed. One-way ANOVA and Bonferroni post hoc tests used to compare different groups. *Significant difference (P < 0.05) from rWT DMSO. Readers may view the uncut gels for B and C in the data supplement.
Figure 6.
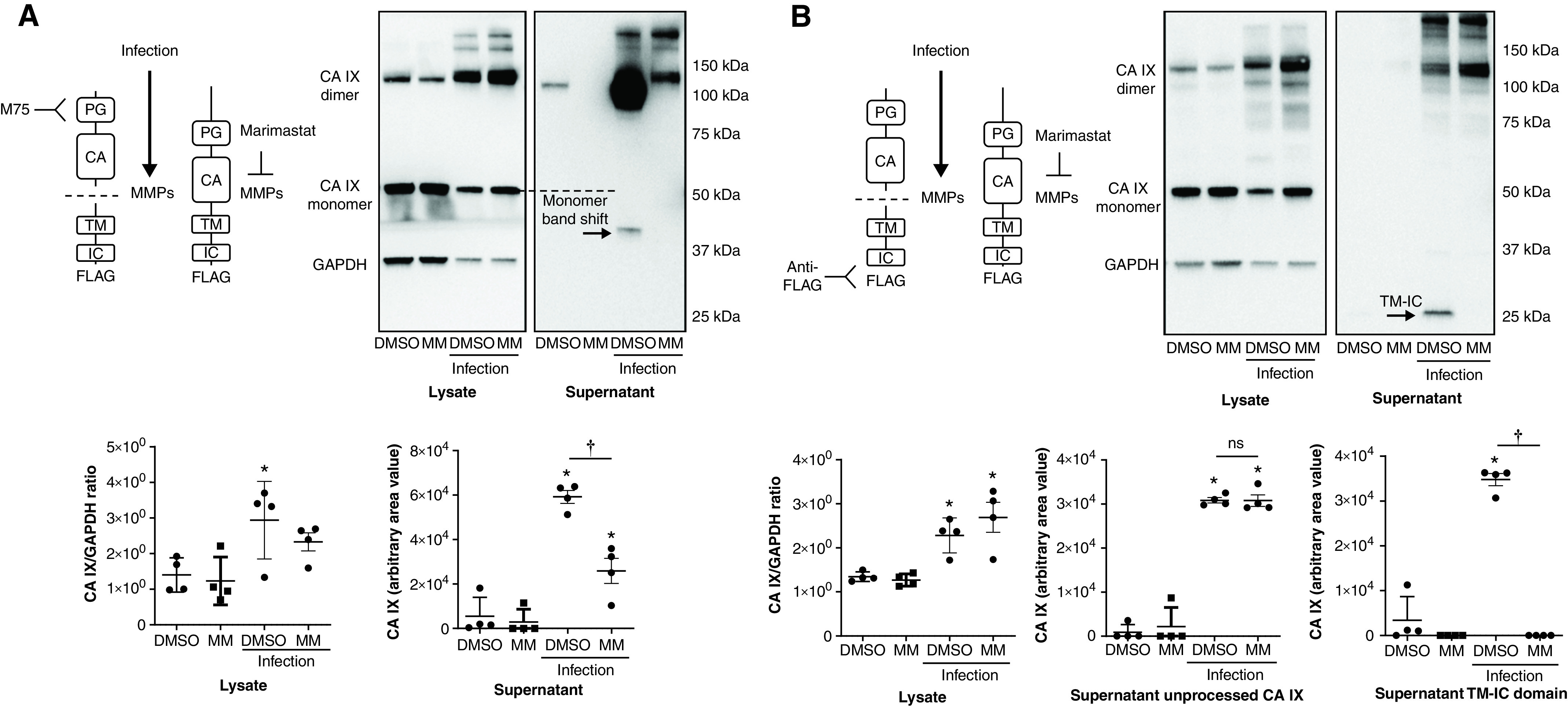
P. aeruginosa infection increases CA IX expression and release in PMVECs. (A and B) CA IX mutant PMVEC cell lines were seeded at 5.0 × 105 cells/well on 6-well plates on bicarbonate-buffered media. Two days after cell seeding, media were changed to HBSS, and cells were treated with P. aeruginosa at a multiplicity of infection of 0.055 and with DMSO (0.1%) or marimastat (10μM). Six hours later, the CA IX protein abundance was measured in cell lysates and supernatants by using Western blotting. P. aeruginosa infection significantly increased CA IX protein expression and release, and marimastat (MM) reduced extracellular ectodomain cleavage. At least five separate experiments were performed. One-way ANOVA and Bonferroni post hoc tests were used to compare different groups. *Significant difference (P < 0.05) from DMSO control. †Significant difference (P < 0.05) from DMSO infection. Readers may view the uncut gels for B in the data supplement. ns = not significant.
To identify whether CA IX ectodomain cleavage is mediated by MMPs, as described by other groups (39), we used two different MMP inhibitors, including batimastat and MM. These drugs nonspecifically inhibit multiple MMP subtypes; their inhibitory profiles partially overlap. In PMVECs, neither batimastat nor MM affected the abundance of CA IX mutant proteins in cell lysates (Figures 5A–5D; DMSO rWT vs. batimastat or MM rWT, P = ns), but both inhibitors profoundly reduced the cleaved ectodomain (Figures 5A and 5B; DMSO rWT vs. batimastat or MM rWT, P < 0.05) and increased uncleaved CA IX in the supernatants (Figures 5C and 5D; DMSO rWT vs. batimastat or MM rWT, P < 0.05). Although the M75 antibody can detect full-length CA IX, in the absence of an MMP inhibitor, it predominantly detected cleaved CA IX. We believe this may be related to the relative abundance of the cleaved form and/or increased affinity of the cleaved form to the antibody. Notably, the cleaved TM–IC domain was only detected in the supernatants and not in the lysates, indicating that MMP-mediated CA IX proteolytic cleavage occurs in the extracellular space after unprocessed CA IX is released from the cell, rather than occurring at the cell surface while the enzyme is still membrane bound (Figure 5E).
P. aeruginosa Infection Increases CA IX Protein Abundance and Release in Rat PMVECs
To further investigate whether infection promotes CA IX expression and release from PMVECs that subsequently contribute to the lung tissue CA IX increase in pneumonia, PMVECs were infected with P. aeruginosa, and cell lysate and supernatant CA IX was measured at 6 hours. P. aeruginosa infection increased cell lysate CA IX and the supernatant ectodomain (detected by using M75), TM–IC domain (detected by using FLAG tag antibody), and uncleaved CA IX in rWT PMVECs (Figures 6A and 6B; DMSO vs. infection DMSO, P < 0.05). MMP inhibition by MM treatment decreased the ectodomain and TM–IC domain (Figures 6A and 6B; infection DMSO vs. infection MM, P < 0.05), without significantly affecting unprocessed CA IX in the supernatant (Figure 6B; infection DMSO vs. infection MM, P = ns). This finding supports the idea that CA IX ectodomain cleavage occurs extracellularly after unprocessed CA IX is released, and that elevated lung tissue CA IX in pneumonia rats can be attributed to increased CA IX expression and release from PMVECs.
The CA IX IC Domain and Hypoxia Decreases Necrotic Cell Death, whereas Hypercapnia Attenuates the Protective Effect of Hypoxia, during P. aeruginosa Infection in Rat PMVECs
Prior studies have reported that CA IX can be cleaved from the cell surface (40). However, our data suggest that infection leads to CA IX release from PMVECs with subsequent extracellular proteolysis. Although both processes would increase the amount of extracellular soluble ectodomains, the fate of the TM–IC domain and its consequences can be dramatically different. Because the IC domain determines CA IX membrane localization and promotes extracellular acidification during hypoxia (38) and because its phosphorylation mediates the migration of tumor cells (41), it may also influence PMVEC survival during infection. To address this question, CA IX rWT and ΔIC PMVECs were treated with P. aeruginosa, and their survival was assessed by using an LDH cytotoxicity assay at 3 hours and 6 hours. Indeed, the lack of the IC domain accelerated PMVEC necrotic death in response to both serum deprivation and P. aeruginosa infection at 3 hours (Figures 7A and 7B; rWT control vs. ΔIC control and rWT infection vs. ΔIC infection, P < 0.05) and 6 hours (Figures 7C and 7D; rWT control vs. ΔIC control and rWT infection vs. ΔIC infection, P < 0.05). Considering that the morphological cellular damage is more severe in ΔIC PMVECs and is out of proportion to the LDH increases compared with those in rWT PMVECs, we measured ATP concentrations at 6 hours. ATP was profoundly and similarly decreased in both infected rWT PMVECs and infected ΔIC PMVECs at 6 hours (Figure 7E; rWT control vs. rWT infection and rWT control vs. ΔIC infection, P < 0.05; rWT infection vs. ΔIC infection, P = ns). To address these conflicting results between the LDH and ATP assays, we performed an alternative necrosis assay, propidium iodide staining, at an earlier time point of 5.5 hours after infection, while the monolayers were mostly still intact. Consistent with the LDH results, propidium iodide staining intensity was higher in infected ΔIC PMVECs than in infected rWT PMVECs (Figure 7F; rWT infection vs. ΔIC infection, P < 0.05). These findings indicate the CA IX IC domain fulfills a critical role in PMVEC survival during environmental stress, including during serum starvation and infection.
Figure 7.
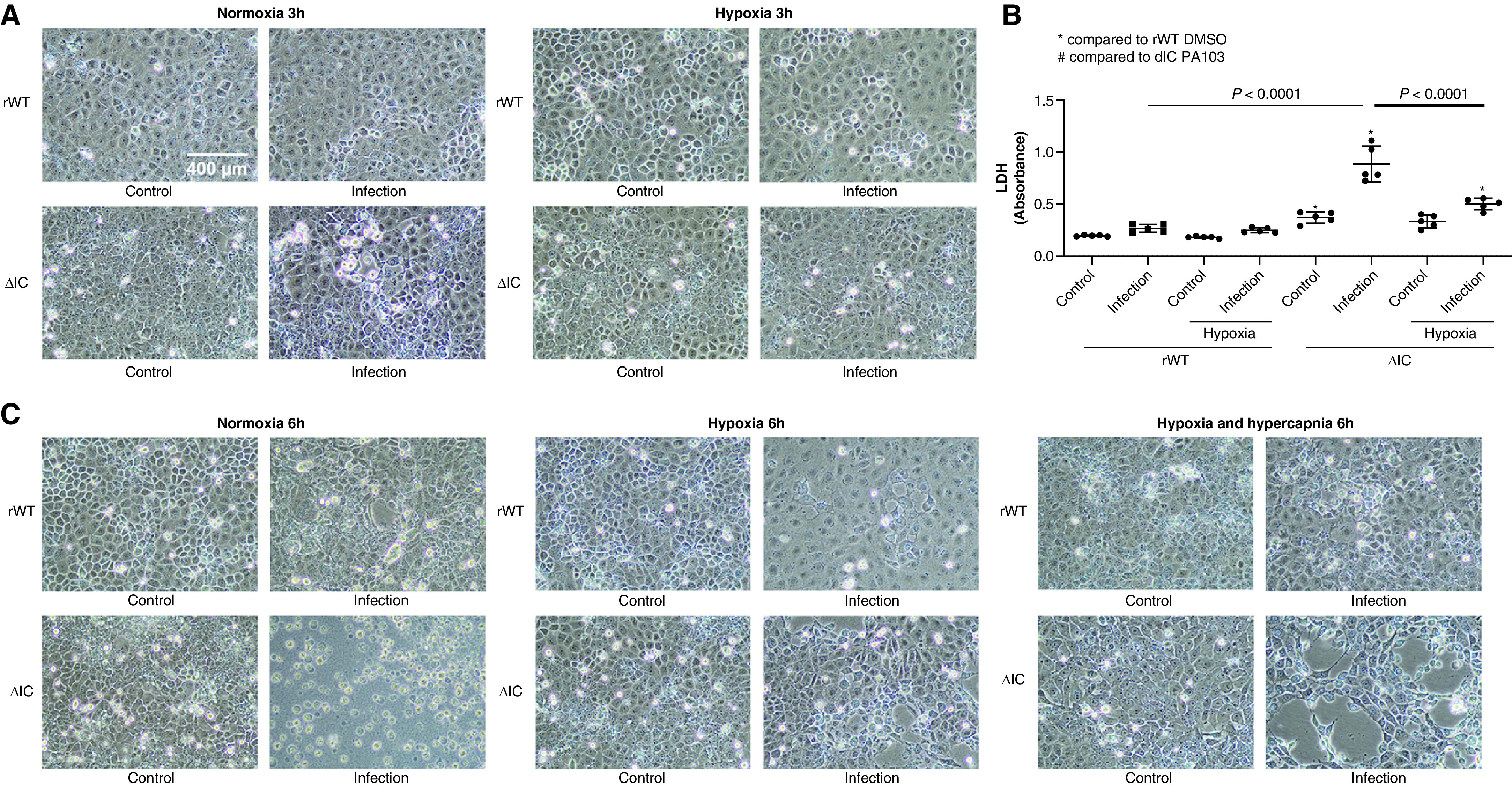
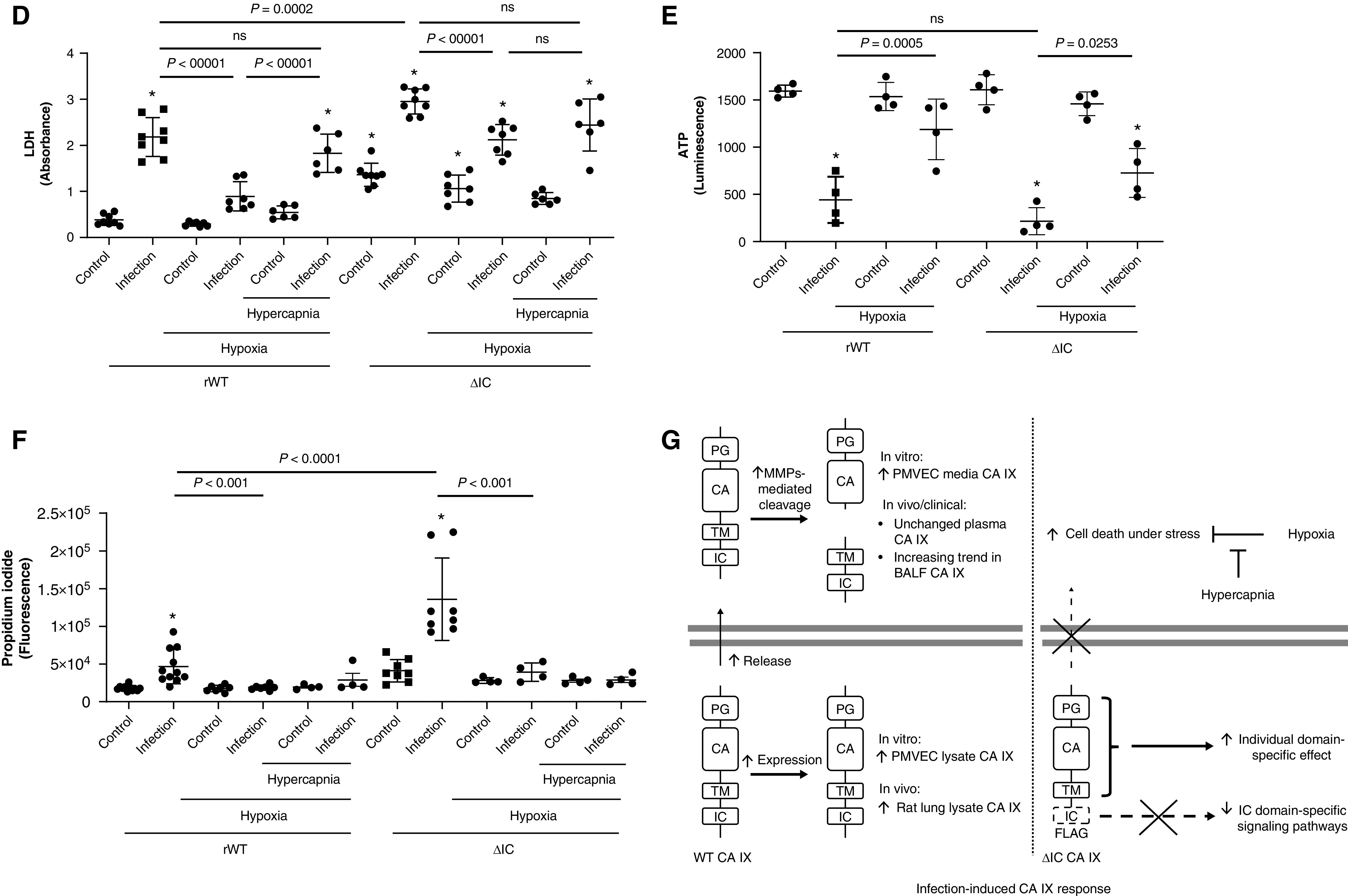
The CA IX IC domain and hypoxia decrease necrotic cell death, whereas hypercapnia attenuates the protective effect of hypoxia, during P. aeruginosa infection in PMVECs. CA IX rWT and ΔIC PMVECs were seeded at 5.0 × 105 cells/well on 6-well plates on bicarbonate-buffered media. Two days after cell seeding, media were changed to HBSS and treated with P. aeruginosa (multiplicity of infection of 0.055) under normoxia (21% O2) and hypoxia (1% O2) with and without additional hypercapnia (10% CO2). Three and 6 hours later, (A and C) images were obtained and (B and D) LDH (lactate dehydrogenase) was measured in cell supernatants. Compared with rWT PMVECs, necrotic cell death was significantly higher in ΔIC PMVECs during serum starvation and during P. aeruginosa infection at (A and B) 3 hours and (C and D) 6 hours, as indicated by the increased number of rounded and floating cells, intercellular gap formation, monolayer damage/lifting, and LDH release. Hypoxia independently decreased P. aeruginosa–induced necrotic cell death, whereas concomitant hypercapnia attenuated the protective effect of hypoxia. This attenuating effect of hypercapnia was less significant in ΔIC PMVECs, indicating a potential mediating role of the IC domain in hypoxic hypercapnic PMVEC injury during infection. Scale bar, 400 μm (for A and C). (E) Considering the significant cytotoxicity at 6 hours and the microscopic differences in the degree of monolayer damage between infected rWT and ΔIC PMVECs, we measured ATP concentrations in the normoxia and hypoxia groups to assess cell viability. The results indicated that both infected rWT cells and infected ΔIC cells are profoundly damaged by 6 hours, and the distinctions discovered by using an LDH assay between infected rWT cells and infected ΔIC cells were not caused by ATP concentrations. Therefore, we performed propidium iodide staining at an earlier time point of 5.5 hours as a supplemental necrosis assay while most cell layers were mostly attached to the cell culture dish. (F) The staining intensity indicated that most cells are intact by 5.5 hours but that infected rWT and ΔIC cells have started undergoing necrosis, with ΔIC PMVECs undergoing more necrosis than rWT PMVECs. At least five separate experiments were performed. Data represent the mean ± SD. One-way ANOVA and Bonferroni post hoc tests were used to compare different groups. *Significant difference (P < 0.05) from rWT control. (G) Schematic representation of the key findings of this paper. Infection induces increased expression and release of CA IX in vitro, induces increased lung tissue CA IX in vivo, and causes and an increasing trend in CA IX concentrations in human BALF. There is no change in the plasma CA IX concentration at 24 hours after bacterial inoculation for the rat pneumonia model or within 24 hours of ICU admission for patients. Deletion of the IC domain traps the CA IX protein in the perinuclear area, which increases susceptibility to stressful conditions, including serum starvation and P. aeruginosa infection. This phenotypic changes of ΔIC PMVECs may be a direct effect of alterations in IC domain–specific signaling pathways or may be an indirect effect of PG–CA–TM IC entrapment and their interaction with IC signaling factors. Independent of the IC domain, hypoxia improves PMVEC survival during infection, whereas hypercapnia attenuates this protective effect of hypoxia in vitro. Overall, the proper functions of the IC domain and hypoxia are protective for PMVEC survival during P. aeruginosa infection in vitro.
How O2 and CO2 independently affect cell physiology in the lung is poorly understood, despite their important clinical implications. Because CA IX is a critical CO2 regulator, we investigated how hypoxic and hypercapnic environments affect PMVEC survival during infection. Most notably, hypoxia (1% O2 and 5% CO2) strikingly decreased P. aeruginosa–induced necrotic cell death, independent of the IC domain at 3 hours (Figure 7B; normoxia ΔIC infection vs. hypoxia ΔIC infection, P < 0.05), 5.5 hours (Figure 7F; normoxia rWT infection vs. hypoxia rWT infection and normoxia ΔIC infection vs. hypoxia ΔIC infection, P < 0.05), and 6 hours (Figures 7D and 7E; normoxia rWT infection vs. hypoxia rWT infection and normoxia ΔIC infection vs. hypoxia ΔIC infection, P < 0.05). However, this protective effect of hypoxia was attenuated by concomitant hypercapnia (1% O2 and 10% CO2) (Figure 7D; hypoxia rWT infection vs. hypercapnia rWT infection and hypoxia ΔIC infection vs. hypercapnia ΔIC infection, P = ns). Although hypercapnia attenuates the protective effect of hypoxia in both rWT- and ΔIC-expressing cells, this hypercapnic effect was less profound in cells expressing the ΔIC mutant (Figure 7D; hypoxia rWT infection vs. hypercapnia rWT infection, P < 0.05; hypoxia ΔIC infection vs. hypercapnia ΔIC infection, P = ns), indicating a potential role of the IC domain in mediating the cytotoxic effects of hypercapnia under hypoxia during infection. Together, these findings suggest that the CA IX IC domain is critical to PMVEC survival during serum starvation and P. aeruginosa infection and that, furthermore, hypoxia is protective, whereas hypercapnia is harmful for PMVEC survival during P. aeruginosa infection.
Discussion
In this study, we investigated the potential role of CA IX in PMVEC survival during infection. We report eight key findings: 1) plasma CA IX does not change in critically ill patients and rats with pneumonia within 24 hours of ICU admission and at 24 hours after bacterial inoculation, respectively; 2) BALF CA IX has an increasing trend in critically ill mechanically ventilated patients with pneumonia; 3) CA IX increases in the lung tissue of rats with pneumonia; 4) the CA IX IC domain mediates CA IX membrane localization; 5) CA IX is constitutively released, and its ectodomain is cleaved extracellularly by MMPs; 6) P. aeruginosa infection increases both the CA IX release and the extracellular ectodomain cleavage; 7) the CA IX IC domain is critical to PMVEC survival during serum deprivation and P. aeruginosa infection; and 8) hypoxia protects PMVECs from necrotic cell death during P. aeruginosa infection, and this protective effect of hypoxia is attenuated by concomitant hypercapnia, which is partly dependent on the CA IX IC domain. These findings suggest that CA IX is a potential mediator of PMVEC survival during P. aeruginosa infection. The key findings are summarized in Figure 7G.
“Shedding” refers to the proteolytic release of ectodomain regions of mature membrane proteins into the extracellular space (42). After ectodomain shedding, the remaining TM and IC domains can be additionally cleaved, released into the extracellular space, or internalized to activate IC signaling pathways (43). In cancer cells, CA IX undergoes ectodomain shedding, which is enhanced by hypoxia and inhibited by nonspecific MMP inhibitors (40). Although our study with primary PMVECs showed a similar finding of MMP-mediated CA IX ectodomain cleavage, a few distinctive points were noted. First, there was a significant amount of uncleaved CA IX released both at baseline and upon P. aeruginosa infection. Second, neither form of CA IX release was affected by hypoxia (data not shown). Third, both forms of CA IX release were increased during infection. Fourth, the ectodomain-cleaved product of the TM–IC domain was detected in cell culture media but not in cell lysates, indicating that CA IX release is not the traditional shedding that occurs at the cell surface but is reflective of unprocessed protein release followed by extracellular MMP-mediated ectodomain cleavage. Therefore, MMP inhibition prevented extracellular CA IX cleavage but had little effect on total CA IX release from PMVECs.
The strong infection-induced increase in CA IX expression and release in vitro and upregulation of lung tissue CA IX in vivo suggest a potential role of CA IX in pneumonia. Although we expected an accompanying increase of plasma CA IX in human and animal pneumonia, there was no significant change in the plasma CA IX concentration in either setting. We consider two major possibilities. First, the sample collection time may have been too early to capture an increase in the plasma CA IX concentration; if delayed, CA IX release from the lung tissue occurred. Second, released CA IX from the pulmonary circulation may undergo rapid degradation/binding before it reaches the systemic circulation, and therefore, it may not alter the arterial plasma CA IX concentration. Nonetheless, to our knowledge, this is first study to report pneumonia-specific CA IX changes in the lung, and further investigation is required to elucidate its clinical implications.
Regardless of whether ectodomain cleavage occurs at the membrane surface or in the extracellular space, a common consequence of this cleavage is the relative loss of the IC domain. Unlike some forms of traditional shedding, which leave the IC domain at the cell surface where it can be used by the cells for IC signaling, a total loss of the IC domain into the extracellular space, as shown in our setting, may adversely affect cellular signaling. In cancer cells, the IC domain contributes to CA IX membrane localization, ectodomain shedding, extracellular acidification, and migration (38, 41). Little is known about whether the IC domain has any role in nontransformed PMVEC function during infection. In our study, the IC domain depletion profoundly increased PMVEC necrotic death during serum starvation and P. aeruginosa infection. However, whether this result is due to an effect of alterations in the signaling pathways directly linked to the IC domain or is an indirect effect related to perinuclear ΔIC CA IX localization that contains PG–CA domains is not clear. Although it is beyond the scope of this paper, our ongoing studies indicate that the IC domain modulates metabolism and other characteristics of PMVECs, which may be a key to understanding the prosurvival role of the IC domain. These findings indicate that modifying IC domain–mediated signaling pathways represents a potential therapeutic target during infection.
Our study has limitations. First, challenges in determining the optimal PaO2 and PaCO2 goal in ARDS are multifactorial, including limited and conflicting clinical data and difficulties in resolving the relationships between arterial versus tissue level gas contents in the setting of a complex O2–CO2–pH interplay (44). Our data showed a protective effect of hypoxia on PMVEC survival during P. aeruginosa infection. Such protection in the context of infection has been reported by other groups, with possible mechanisms that include a reduction of the Pseudomonas virulence factor (45) or bacterial cellular uptake (46), activation of the HIF pathway (47), and tightening of the cellular monolayer (48) by hypoxia. Interestingly, additional hypercapnia negated the protective effect of hypoxia without significantly affecting noninfected cells in our study. Although the interaction between hypoxia and hypercapnia is highly relevant to ARDS, we have not yet tested the impact of lung-protective ventilatory strategies in experimental ARDS in vivo to assess the impact of hypoxia and hypercapnia on lung injury and repair. We have not yet validated this concept in prospective clinical studies. This future work will be relevant to the treatment of patients with ARDS. In the in vivo setting, the context of hypoxia becomes much more diverse and complicated, such as in ARDS and sepsis, in which target organ tissue hypoxia can be secondary to altered gas contents and/or compromised perfusion (49). Considering that hypoxic hypercapnia is a common presentation of ARDS, especially during lung-protective ventilation and permissive hypoxemia/hypercapnia, further elucidating molecular and cellular implications of the differential and combined effects of hypoxia and hypercapnia may help better identify the optimal target PaO2 and PaCO2 ranges in ARDS. Second, our studies found that CA IX was cleaved outside of the endothelium by MMPs. Future studies will be required to determine whether this mechanism occurs in vivo and how MMPs are activated to cleave CA IX during inflammation. Third, CA IX mutant proteins were not all expressed at equal amounts when quantified by using Western blotting. In the future, we can generate clones with equal protein expression by using single-cell cloning, as we have previously reported (28).
In summary, we report that plasma CA IX concentrations did not change within 24 hours of ICU admission in critically ill patients with pneumonia. However, there was a trend toward higher BALF CA IX concentrations in mechanically ventilated ICU patients with pneumonia. In our rat pneumonia model, CA IX did not change in the plasma, but it significantly increased in the lung tissue at 24 hours after infection. In vitro, P. aeruginosa infection increased CA IX expression, release, and MMP-mediated ectodomain cleavage in PMVECs. We also demonstrated important roles of the IC domain in CA IX membrane localization and PMVEC survival during serum starvation and infection. Lastly, hypoxia protected PMVECs from necrotic cell death, and concomitant hypercapnia attenuated the protective effect of hypoxia. The findings suggest CA IX is a potential therapeutic target for critically ill patients with Pseudomonas-induced pneumonia.
Footnotes
Supported by American Heart Association grants 18CDA34080151 (J.Y.L. and T.S.) and 19PRE34380166 (M.S.G.); National Institutes of Health grants HL153958 (J.Y.L. and T.S.), OD010944 (M.F.A.), HL140182 (R.B. and T.S.), HL076124 (S.V.), HL147512 (S.V.), GM127584 (B.M.W.), HL118334 (B.W.F., J.P.A., and D.F.A.), HL148069 (J.Y.L., R.B., J.-F.P., and T.S.), HL66299 (R.B. and T.S.), and HL60024 (T.S.).
Author Contributions: J.Y.L., M.F.A., R.B., and T.S. contributed to the conception and design of the study. J.Y.L., C.Z., P.R., A.K., N.K., V.P., M.S.G., S.V., A.d., B.M.W., J.-F.P., Y.E., W.S., T.N., C.C., B.W.F., J.P.A., D.F.A., and T.S. contributed to the acquisition of data. J.Y.L., R.P.S., M.K., M.F.A., R.B., B.M.W., J.-F.P., B.W.F., J.P.A., D.F.A., and T.S. contributed to the analysis and interpretation of the data. J.Y.L., R.P.S., M.F.A., R.B., B.M.W., J.-F.P., B.W.F., J.P.A., D.F.A., and T.S. contributed to the drafting of the manuscript and review for important intellectual content.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0537OC on July 12, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Fan E, Del Sorbo L, Goligher EC, Hodgson CL, Munshi L, Walkey AJ, et al. American Thoracic Society; European Society of Intensive Care Medicine; Society of Critical Care Medicine. An official American Thoracic Society/European Society of Intensive Care Medicine/Society of Critical Care Medicine clinical practice guideline: mechanical ventilation in adult patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2017;195:1253–1263. doi: 10.1164/rccm.201703-0548ST. [DOI] [PubMed] [Google Scholar]
- 2. Papazian L, Aubron C, Brochard L, Chiche JD, Combes A, Dreyfuss D, et al. Formal guidelines: management of acute respiratory distress syndrome. Ann Intensive Care. 2019;9:69. doi: 10.1186/s13613-019-0540-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A. Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 4. Slutsky AS, Ranieri VM. Mechanical ventilation: lessons from the ARDSNet trial. Respir Res. 2000;1:73–77. doi: 10.1186/rr15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carlo W. Permissive hypercapnia and permissive hypoxemia in neonates. J Perinatol. 2007;27:S64–S70. [Google Scholar]
- 6. Kregenow DA, Swenson ER. The lung and carbon dioxide: implications for permissive and therapeutic hypercapnia. Eur Respir J. 2002;20:6–11. doi: 10.1183/09031936.02.00400802. [DOI] [PubMed] [Google Scholar]
- 7. Abdelsalam M. Permissive hypoxemia: is it time to change our approach? Chest. 2006;129:210–211. doi: 10.1378/chest.129.1.210. [DOI] [PubMed] [Google Scholar]
- 8. Myti D, Gunjak M, Casado F, Khaghani Raziabad S, Nardiello C, Vadász I, et al. Elevated FiO2 increases SARS-CoV-2 co-receptor expression in respiratory tract epithelium. Am J Physiol Lung Cell Mol Physiol. 2020;319:L670–L674. doi: 10.1152/ajplung.00345.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Panwar R, Hardie M, Bellomo R, Barrot L, Eastwood GM, Young PJ, et al. CLOSE Study Investigators; ANZICS Clinical Trials Group. Conservative versus liberal oxygenation targets for mechanically ventilated patients: a pilot multicenter randomized controlled trial. Am J Respir Crit Care Med. 2016;193:43–51. doi: 10.1164/rccm.201505-1019OC. [DOI] [PubMed] [Google Scholar]
- 10. Barrot L, Asfar P, Mauny F, Winiszewski H, Montini F, Badie J, et al. LOCO2 Investigators and REVA Research Network. Liberal or conservative oxygen therapy for acute respiratory distress syndrome. N Engl J Med. 2020;382:999–1008. doi: 10.1056/NEJMoa1916431. [DOI] [PubMed] [Google Scholar]
- 11. Girardis M, Busani S, Damiani E, Donati A, Rinaldi L, Marudi A, et al. Effect of conservative vs conventional oxygen therapy on mortality among patients in an intensive care unit: the oxygen-ICU randomized clinical trial. JAMA. 2016;316:1583–1589. doi: 10.1001/jama.2016.11993. [DOI] [PubMed] [Google Scholar]
- 12. Barnes T, Zochios V, Parhar K. Re-examining permissive hypercapnia in ARDS: a narrative review. Chest. 2018;154:185–195. doi: 10.1016/j.chest.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 13. Morales-Quinteros L, Camprubí-Rimblas M, Bringué J, Bos LD, Schultz MJ, Artigas A. The role of hypercapnia in acute respiratory failure. Intensive Care Med Exp. 2019;7:39. doi: 10.1186/s40635-019-0239-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fuchs H, Rossmann N, Schmid MB, Hoenig M, Thome U, Mayer B, et al. Permissive hypercapnia for severe acute respiratory distress syndrome in immunocompromised children: a single center experience. PLoS One. 2017;12:e0179974. doi: 10.1371/journal.pone.0179974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Swenson ER. Sepsis and therapeutic hypercapnia: sailing too close to the wind? Anesthesiology. 2010;112:269–271. doi: 10.1097/ALN.0b013e3181ca33be. [DOI] [PubMed] [Google Scholar]
- 16. Lemasters JJ, Bond JM, Chacon E, Harper IS, Kaplan SH, Ohata H, et al. The pH paradox in ischemia-reperfusion injury to cardiac myocytes. EXS. 1996;76:99–114. doi: 10.1007/978-3-0348-8988-9_7. [DOI] [PubMed] [Google Scholar]
- 17. Kregenow DA, Rubenfeld GD, Hudson LD, Swenson ER. Hypercapnic acidosis and mortality in acute lung injury. Crit Care Med. 2006;34:1–7. doi: 10.1097/01.ccm.0000194533.75481.03. [DOI] [PubMed] [Google Scholar]
- 18. Tiruvoipati R, Pilcher D, Buscher H, Botha J, Bailey M. Effects of hypercapnia and hypercapnic acidosis on hospital mortality in mechanically ventilated patients. Crit Care Med. 2017;45:e649–e656. doi: 10.1097/CCM.0000000000002332. [DOI] [PubMed] [Google Scholar]
- 19. Nin N, Muriel A, Peñuelas O, Brochard L, Lorente JA, Ferguson ND, et al. VENTILA Group. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med. 2017;43:200–208. doi: 10.1007/s00134-016-4611-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Charney AN, Alexander-Chacko J, Gummaconda R, Egnor RW. Non-catalytic role of carbonic anhydrase in rat intestinal absorption. Biochim Biophys Acta. 2002;1573:141–148. doi: 10.1016/s0304-4165(02)00370-7. [DOI] [PubMed] [Google Scholar]
- 21. Mboge MY, Chen Z, Khokhar D, Wolff A, Ai L, Heldermon CD, et al. A non-catalytic function of carbonic anhydrase IX contributes to the glycolytic phenotype and pH regulation in human breast cancer cells. Biochem J. 2019;476:1497–1513. doi: 10.1042/BCJ20190177. [DOI] [PubMed] [Google Scholar]
- 22. Lee JY, Alexeyev M, Kozhukhar N, Pastukh V, White R, Stevens T. Carbonic anhydrase IX is a critical determinant of pulmonary microvascular endothelial cell pH regulation and angiogenesis during acidosis. Am J Physiol Lung Cell Mol Physiol. 2018;315:L41–L51. doi: 10.1152/ajplung.00446.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ochoa CD, Stevens T. Studies on the cell biology of interendothelial cell gaps. Am J Physiol Lung Cell Mol Physiol. 2012;302:L275–L286. doi: 10.1152/ajplung.00215.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stevens T, Phan S, Frid MG, Alvarez D, Herzog E, Stenmark KR. Lung vascular cell heterogeneity: endothelium, smooth muscle, and fibroblasts. Proc Am Thorac Soc. 2008;5:783–791. doi: 10.1513/pats.200803-027HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee JY, Onanyan M, Garrison I, White R, Crook M, Alexeyev MF, et al. Extrinsic acidosis suppresses glycolysis and migration while increasing network formation in pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2019;317:L188–L201. doi: 10.1152/ajplung.00544.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wagener BM, Anjum N, Christiaans SC, Banks ME, Parker JC, Threet AT, et al. Exoenzyme Y contributes to end-organ dysfunction caused by Pseudomonas aeruginosa pneumonia in critically ill patients: an exploratory study. Toxins (Basel) 2020;12:369. doi: 10.3390/toxins12060369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee JY, Fagan KA, Zhou C, Batten L, Cohen MV, Stevens T. Biventricular diastolic dysfunction, thrombocytopenia, and red blood cell macrocytosis in experimental pulmonary arterial hypertension. Pulm Circ. 2020;10:2045894020908787. doi: 10.1177/2045894020908787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee JY, McMurtry SA, Stevens T. Single cell cloning generates lung endothelial colonies with conserved growth, angiogenic, and bioenergetic characteristics. Pulm Circ. 2017;7:777–792. doi: 10.1177/2045893217731295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Opavský R, Pastoreková S, Zelník V, Gibadulinová A, Stanbridge EJ, Závada J, et al. Human MN/CA9 gene, a novel member of the carbonic anhydrase family: structure and exon to protein domain relationships. Genomics. 1996;33:480–487. doi: 10.1006/geno.1996.0223. [DOI] [PubMed] [Google Scholar]
- 30. Morrow KA, Ochoa CD, Balczon R, Zhou C, Cauthen L, Alexeyev M, et al. Pseudomonas aeruginosa exoenzymes U and Y induce a transmissible endothelial proteinopathy. Am J Physiol Lung Cell Mol Physiol. 2016;310:L337–L353. doi: 10.1152/ajplung.00103.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexeyev MF, Fayzulin R, Shokolenko IN, Pastukh V. A retro-lentiviral system for doxycycline-inducible gene expression and gene knockdown in cells with limited proliferative capacity. Mol Biol Rep. 2010;37:1987–1991. doi: 10.1007/s11033-009-9647-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abul Y, Ozsu S, Mentese A, Durmus I, Bektas H, Pehlivanlar M, et al. Carbonic anhydrase IX in the prediction of right ventricular dysfunction in patients with hemodynamically stable acute pulmonary embolism. Clin Appl Thromb Hemost. 2014;20:838–843. doi: 10.1177/1076029613486540. [DOI] [PubMed] [Google Scholar]
- 33. Hong T-H, Hu F-C, Kuo S-W, Ko W-J, Chow L-P, Hsu L-M, et al. Predicting outcome in patients under extracorporeal membrane oxygenation due to cardiogenic shock through dynamic change of lymphocytes and interleukins. IJC Metab Endocr. 2015;7:36–44. [Google Scholar]
- 34. Gutbier B, Neuhauß AK, Reppe K, Ehrler C, Santel A, Kaufmann J, et al. CAPNETZ and PROGRESS Study Groups. Prognostic and pathogenic role of angiopoietin-1 and -2 in pneumonia. Am J Respir Crit Care Med. 2018;198:220–231. doi: 10.1164/rccm.201708-1733OC. [DOI] [PubMed] [Google Scholar]
- 35. American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171:388–416. doi: 10.1164/rccm.200405-644ST. [DOI] [PubMed] [Google Scholar]
- 36. Ranjan R, Valez EM, Haldipur A, Schiller NB. Relation of velocity-time integral of the left ventricular outflow tract to that of the descending thoracic aorta and usefulness of a fixed ratio for internal validation. Am J Cardiol. 2018;122:166–169. doi: 10.1016/j.amjcard.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 37. Alterio V, Hilvo M, Di Fiore A, Supuran CT, Pan P, Parkkila S, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA. 2009;106:16233–16238. doi: 10.1073/pnas.0908301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hulikova A, Zatovicova M, Svastova E, Ditte P, Brasseur R, Kettmann R, et al. Intact intracellular tail is critical for proper functioning of the tumor-associated, hypoxia-regulated carbonic anhydrase IX. FEBS Lett. 2009;583:3563–3568. doi: 10.1016/j.febslet.2009.10.060. [DOI] [PubMed] [Google Scholar]
- 39. Zatovicova M, Sedlakova O, Svastova E, Ohradanova A, Ciampor F, Arribas J, et al. Ectodomain shedding of the hypoxia-induced carbonic anhydrase IX is a metalloprotease-dependent process regulated by TACE/ADAM17. Br J Cancer. 2005;93:1267–1276. doi: 10.1038/sj.bjc.6602861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vidlickova I, Dequiedt F, Jelenska L, Sedlakova O, Pastorek M, Stuchlik S, et al. Apoptosis-induced ectodomain shedding of hypoxia-regulated carbonic anhydrase IX from tumor cells: a double-edged response to chemotherapy. BMC Cancer. 2016;16:239. doi: 10.1186/s12885-016-2267-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ditte P, Dequiedt F, Svastova E, Hulikova A, Ohradanova-Repic A, Zatovicova M, et al. Phosphorylation of carbonic anhydrase IX controls its ability to mediate extracellular acidification in hypoxic tumors. Cancer Res. 2011;71:7558–7567. doi: 10.1158/0008-5472.CAN-11-2520. [DOI] [PubMed] [Google Scholar]
- 42. Lichtenthaler SF, Lemberg MK, Fluhrer R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018;37:e99456. doi: 10.15252/embj.201899456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 44. West JB. Three classical papers in respiratory physiology by Christian Bohr (1855-1911) whose work is frequently cited but seldom read. Am J Physiol Lung Cell Mol Physiol. 2019;316:L585–L588. doi: 10.1152/ajplung.00527.2018. [DOI] [PubMed] [Google Scholar]
- 45. Schaible B, Rodriguez J, Garcia A, von Kriegsheim A, McClean S, Hickey C, et al. hypoxia reduces the pathogenicity of Pseudomonas aeruginosa by decreasing the expression of multiple virulence factors. J Infect Dis. 2017;215:1459–1467. doi: 10.1093/infdis/jix139. [DOI] [PubMed] [Google Scholar]
- 46. Schaible B, McClean S, Selfridge A, Broquet A, Asehnoune K, Taylor CT, et al. Hypoxia modulates infection of epithelial cells by Pseudomonas aeruginosa. PLoS One. 2013;8:e56491. doi: 10.1371/journal.pone.0056491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ali I, Nanchal R, Husnain F, Audi S, Konduri GG, Densmore JC, et al. Hypoxia preconditioning increases survival and decreases expression of Toll-like receptor 4 in pulmonary artery endothelial cells exposed to lipopolysaccharide. Pulm Circ. 2013;3:578–588. doi: 10.1086/674337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Solodushko V, Parker JC, Fouty B. Pulmonary microvascular endothelial cells form a tighter monolayer when grown in chronic hypoxia. Am J Respir Cell Mol Biol. 2008;38:491–497. doi: 10.1165/rcmb.2007-0127OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manninen PH, Unger ZM. In: Prabhakar H, editor. San Diego, CA: Academic Press; 2016. Hypoxia; pp. 169–180.. [Google Scholar]