

Abstract
Bromodomain and Extra-Terminal domain (BET) family proteins are epigenetic readers that play a critical role in oncogenesis by controlling the expression of oncogenes such as c-Myc. Targeting BET family proteins has recently emerged as a promising anticancer strategy. However, the molecular mechanisms by which cancer cells respond to BET inhibition are not well understood. In this study, we found that inducing the degradation of BET proteins by the Proteolysis Targeting Chimeras (PROTAC) approach potently suppressed the growth of colorectal cancer (CRC) including patient-derived tumors. Mechanistically, BET degradation transcriptionally activates Death Receptor 5 (DR5) to trigger immunogenic cell death (ICD) in CRC cells. Enhanced DR5 induction further sensitizes CRC cells with a mutation in Speckle-type POZ protein (SPOP). Furthermore, DR5 is indispensable for a striking antitumor effect of combining BET degradation and anti-PD-1 antibody, which was well tolerated in mice and almost eradicated syngeneic tumors. Our results demonstrate that BET degradation triggers DR5-mediated ICD to potently suppress CRC and potentiate immune checkpoint blockade. These results provide a rationale, mechanistic insights, and potential biomarkers for developing a precision CRC therapy by inducing BET protein degradation.
Keywords: BET degraders, immunogenic cell death, DR5
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer-related deaths in the United States [1]. CRC patients are typically treated with 5-fluorouracil (5-FU)-based chemotherapy and/or targeted therapy [2]. However, advanced CRCs often progress after initial therapies and become refractory to subsequent treatments [3]. The response to anti-PD-1 immunotherapy is limited to 10-15% of CRCs that are deficient in DNA mismatch repair (dMMR) [4, 5]. Developing new and more effective agents against advanced CRCs is an unmet medical need.
The Bromodomain and Extra-Terminal domain (BET) family proteins function as epigenetic readers by recognizing acetylated lysine residues through their bromodomains to promote transcriptional elongation [6]. They play a critical role in oncogenesis by controlling the expression of oncogenes such as c-Myc [7]. The BET family proteins have recently emerged as attractive cancer targets. Small-molecule BET inhibitors (BETi), such as JQ1, OTX015, IBET-151, and BETi211, have exhibited promising efficacy against a variety of tumors [8]. Several BETi have advanced to clinical testing and shown efficacy against some hematologic malignancies and NUT carcinomas [8]. However, BETi as single agents are generally ineffective against solid tumors [8]. Normal tissue toxicity is a major challenge in developing BET-targeted therapies [9, 10].
A new approach for targeting BET proteins is Proteolysis Targeting Chimeras (PROTAC). Compounds are designed and synthesized by conjugating a BETi to an E3 ubiquitin ligase binder through a flexible linker [11–13]. Such compounds can selectively induce the ubiquitination and proteasomal degradation of BET proteins. Several BET degraders (BETd) have been described [11–15]. Compared with BETi, BETd are much more potent and selective in inhibiting BET proteins and suppressing cancer cell growth [11–13]. It is less likely for tumor cells to generate mutant variants to become resistant to BETd [16]. Furthermore, BETd can be designed on a structural basis for specific targeting of a particular BET protein [17]. The improved potency, selectivity, and specificity may lower a threshold dose for therapeutic efficacy and minimize normal tissue toxicity, thereby improving the therapeutic window necessary for clinical translation.
BET-targeting agents have several anticancer activities such as induction of tumor cell apoptosis and enhancement of antitumor immunity [18–21]. Apoptosis can be initiated by the extrinsic pathway through activation of death receptors (DRs), or by the intrinsic pathway via Bcl-2 family proteins and mitochondrial dysfunction, leading to caspase activation and cell death [22, 23]. DR5 (TRAILR2) is a cell-surface death receptor that is activated upon binding to ligands such as Tumor necrosis factor-Related Apoptosis-Inducing Ligand (TRAIL). DR5 can also be induced by p53 upon DNA damage [24] or by C/EBP homologous protein (CHOP) in response to endoplasmic reticulum (ER) stress [25]. Different modes of cell death have distinct immunological consequences [26, 27]. ER stress-induced apoptosis often shows characteristics of immunogenic cell death (ICD) and can stimulate a robust immune response against dead-cell antigens [28]. BET inhibition can potentiate antitumor immune response by downregulating PD-L1 expression [20, 21]. Furthermore, mutations in SPOP, which encodes a subunit of the E3 ubiquitin ligase of BET proteins, can either enhance or reduce BETi sensitivity depending on tumor types and specific residues affected [29–31].
Despite many previous studies, the molecular mechanisms by which cancer cells respond to BET inhibition remain poorly understood. In this study, we investigated BET targeting in CRC and identified a new mechanism of DR5-mediated ICD, which is essential for the antitumor and immunogenic effects of BETd against CRC.
Results
BET protein degradation potently suppresses CRC cell growth via apoptosis induction.
To determine the functional role of BET family proteins in CRC, we analyzed their mRNA expression in TCGA databases and found that BRD2, BRD3 and BRD4 are significantly overexpressed in CRCs including recurrent and metastatic tumors (Fig. 1A, S1A and S1B). High BRD2, BRD3 and BRD4 expression is significantly associated with shorter survival of CRC patients (Fig. 1A, S1A and S1B). This result is in line with the reported role of BET proteins in CRC [32–34], and prompted us to explore their targeting in CRC.
Figure 1. BET degraders exhibited potent single-agent activity against colorectal cancer cells.

(A) Left, Illumina HiSeq analysis of BRD4 mRNA expression in TCGA 551 colon adenocarcinoma (COAD) samples. FPKM (fragments per kilobase of transcript per million mapped reads) is shown). Right, Kaplan-Meier curves for comparing survival probability of 597 patients with COAD or rectal adenocarcinoma (READ) expressing high and low mRNA levels of BRD4. (B) MTS analysis of HCT116 cells treated with JQ1 and indicated BET degraders at different concentrations for 72 hr. (C) MTS analysis of indicated CRC cell lines and NCM356 non-transformed colonic epithelial cells treated with BETd260 at different concentrations for 72 hr. (D) Western blotting of indicated proteins in HCT116 cells treated with BETd260 and BETd246 at indicated concentrations for 36 hr. (E) Apoptosis in HCT116 cells treated with 5 μM of indicated BETi (JQ1, BET151) or 0.5 μM of indicated BETd (BETd246, BETd260) for 36 hr was analyzed by counting cells containing condensed and fragmented nuclei after nuclear staining with Hoechst 33258. (F) Western blotting of cleaved caspases (C Casp) in HCT116 cells treated with 0.5 μM BETd260 at indicated time points. (G) MTS analysis of HCT116 cells treated with BETd260 and BETd246 at indicated concentrations with or without pretreatment with the pan-caspase inhibitor z-VAD-fmk (z-VAD; 10 μM) for 72 hr. Results in (E) and (G) were expressed as means ± SD of three independent experiments. *, P < 0.05; **, P <0.01; ***, P <0.001.
Upon analyzing a number of BETi and BETd, we found two recently developed PROTAC BETd, including BETd260 and BETd246 (Fig. S1C) [13, 15], are the most potent BET-targeting agents in CRC cells (Fig. 1B and S1D). They have 10-120 fold lower IC50 (BETd260, 0.28 μM; BETd246, 0.45 μM) compared to BETi such as JQ1 (12.2 μM) and IBET-151 (15.78 μM), and other BETd such as dBET6 (7.2 μM), ARV-825 (32.5 μM) and MZ1 (4.98 μM) [12, 14, 35] in HCT116 CRC cells (Fig. 1B). BETd260 and BETd246 are highly potent across different CRC cell lines (IC50 0.1-0.6 μM), but much less toxic to non-transformed colonic epithelial cells (IC50 36 μM) (Fig. 1C and S1E). Treating HCT116 cells with sub-μM BETd260 or BETd246 depleted BRD2, BRD3, BRD4, and c-Myc (Fig. 1D), and markedly induced apoptosis shown by increased nuclear fragmentation (Fig. 1E), Annexin V staining (Fig. S1F), and activation of caspases 8, 9 and 3 (Fig. 1F). Caspase 8 activation occurred at 24 hr prior to caspase 9 and 3 activation (Fig. 1F), suggesting crosstalk between the extrinsic and intrinsic apoptotic pathways [22, 23]. In contrast, JQ1 and IBET-151 even at 5 μM did not induce substantial apoptosis or affect BET protein expression in CRC cells (Fig. 1E, S1F and S1G). Furthermore, pan caspase inhibitor z-VAD-fmk suppressed, but did not completely block growth inhibition and cell death induced by BETd260 and BETd246 (Fig. 1G and S1H). Inhibiting necroptosis or ferroptosis did not affect cell death induced by BETd260 (Fig. S1I). These results indicate that BETd260 and BETd246 potently suppress CRC cell growth in part via apoptosis induction.
BETd induce DR5 expression via CHOP-mediated transcriptional activation.
To determine the mechanism by which BETd induce apoptosis in CRC cells, we performed mRNA sequencing (RNA-Seq) on HCT116 cells treated with BETd260. Analysis of differentially expressed genes revealed a marked induction of DR5 mRNA in response to BETd260 (P< 0.01; Fig. S2A, left panel), which was also found in cells with BRD4 knockdown, suggesting an on-target effect of BET inhibition. The induction of DR5 in HCT116 cells was confirmed by RT-PCR (Fig. 2A and S2B), and by Western blotting showing upregulation of both DR5 isoforms (Fig. 2B and S2C) in a time- and dose-dependent manner. In addition to DR5, other death receptor family members, including DCR1, DCR2, and TNFR2, were also upregulated by BETd (Fig. 2A and S2A). In contrast, other apoptosis regulators did not significantly or consistently change upon BETd260 treatment (Fig. 2A, S2A and S2D). Treating HCT116 cells with other BET degraders, including dBET6, MZ-1, and ARV-825, also induced DR5 and apoptosis in a dose-dependent manner, albeit at higher concentrations than BETd260 and BETd246 (Fig. S2E). DR5 induction by BETd260 and BETd246 was also observed in other CRC cell lines (Fig. S2, F and G; data not shown).
Figure 2. The potent activity of BET degraders in colon cancer cells is mediated by ER stress-and DR5-dependent apoptosis.

(A) RT-PCR analysis of indicated genes related to the death receptor pathway in HCT116 cells treated with 0.5 μM of BETd260 or BETd246 for 36 hr. (B) Western blotting of indicated proteins in HCT116 cells treated with BETd260 at indicated concentrations for 36 hr. Right: Western blotting of indicated proteins at indicated time points in HCT116 cells treated with 0.5 μM BETd260. p-eIF2α: phospho-eIF2α, Ser51. (C) MTS analysis of WT and DR5-KO HCT116 cells treated with BETd246 (left) or BETd260 (right) at indicated concentrations for 72 hr. (D) WT and DR5-KO HCT116 cells treated with 0.5 μM of BETd246 or BETd260 for 36 hr. Apoptosis was analyzed by counting cells containing condensed and fragmented nuclei after nuclear staining with Hoechst 33258. (E)-(H) WT and DR5-KO HCT116 and RKO cells were treated with 0.5 μM of BETd260 for 36 hr. (E) Colony formation of WT and DR5-KO HCT116 cells was analyzed by crystal violet staining. Left, representative pictures of colonies; right, enumeration of colony numbers. (F) Western blotting of indicated proteins in WT and DR5-KO HCT116 and RKO cells. (G) Mitochondrial membrane integrity of WT and DR5-KO HCT116 cells was analyzed by MitoTracker Red CMXRos staining followed by flow cytometry. (H) Cytochrome c (Cyto c) release in WT and DR5-KO HCT116 cells was analyzed by Western blotting of mitochondrial and cytosolic fractions isolated from treated cells. β-Actin and cytochrome oxidase subunit IV (COX IV) were used as a control for loading and fractionation, respectively. Results in (A), (C), (D) and (E) were expressed as means ± SD of three independent experiments. *, P < 0.05; **, P <0.01; ***, P <0.001.
We analyzed the DR5-activating pathways and found the ER stress pathway is responsible for DR5 induction by BETd. Consistent with the RNA-Seq data (Fig. S2A, right panel), BETd260 and BETd246 treatment induced ER stress markers [36], including CHOP, ATF3 and ATF6, and phosphorylation of eIF2α (p-eIF2α; Ser51) and PERK (p-PERK; Thr980) (Fig. 2B; Fig. S3, A–C). Chromatin immunoprecipitation (ChIP) analysis showed enhanced binding of CHOP to the DR5 promoter upon BETd260 exposure (Fig. S3D), which also activated a DR5 promoter reporter containing a CHOP-binding site [37], without affecting the control with a mutant binding site [38] (Fig. S3E). The induction of DR5 by BETd260 was abrogated by CHOP knockdown (Fig. S3, F and G) and transfection with the S51A dominant negative mutant of eIF2α [39] (Fig. S3H). Furthermore, the induction of DR5 and apoptosis by BETd260 was suppressed by inhibiting ER stress with the p-eIF2α inhibitor salubrinal [40] (Fig. S3I), or blocking protein synthesis with cycloheximide (CHX) (Fig. S3J). These results indicate DR5 induction via ER stress and CHOP-mediated transcription as an on-target effect of BETd.
DR5 is required for apoptosis and chemosensitization by BETd in CRC cells.
We then investigated the functional role of DR5 in response to BETd in CRC cells. Knockout (KO) of DR5 in HCT116 cells [41] abrogated growth suppression by BETd as indicated by 4-6 fold increases in the IC50 of BETd260 (1.23 μM for DR5-KO vs. 0.21 μM for WT) and BETd246 (1.32 μM for DR5-KO vs. 0.32 μM for WT) (Fig. 2C). DR5 KO inhibited BETd260- and BETd246-induced apoptosis analyzed by nuclear fragmentation, annexin V staining, caspase activation, Bid cleavage, cytochrome c release, mitochondrial outer membrane permeabilization, and colony formation (Fig. 2, D–H; Fig. S4, A and B), further suggesting crosstalk of the extrinsic and intrinsic apoptotic pathways. Indeed, KO or knockdown (KD) of key mediators of crosstalk, including FADD, caspase 8, Bid, and BAX [42–44], suppressed BETd-induced apoptosis (Fig. S4, B–G). The requirement of DR5 was confirmed by analyzing BETd260-treated DR5-KO RKO cells generated by CRISPR/Cas9 (Fig. S4A). In contrast, KO of other apoptosis regulators, such as Bim and Noxa, did not affect apoptosis induced by BETd (Fig. S4E). Structural analogs of BETd260 and BETd246, including BETd228 and BETd570 [15], showed similar dependence on DR5, FADD, Bid, and BAX to induce apoptosis in HCT116 cells (Fig. S4E).
We then analyzed the therapeutic efficacy of BETd in combination with the FDA-approved CRC drugs 5-FU and oxaliplatin, or the DR5 ligand TRAIL. BETd260 combined with 5-FU, oxaliplatin, or TRAIL at different concentrations were highly synergistic (combination indexes <0.015) in HCT116 cells (Fig. S5, A–C). The combinations at low, non-apoptotic doses markedly enhanced DR5 induction, caspase activation and apoptosis in WT HCT116 cells, but not in DR5-KO cells (Fig. S5, D and E), indicating the enhanced induction of DR5 and apoptosis mediates the chemosensitization effect of BETd. Together, these results demonstrate that the apoptotic and chemosensitization effects of BETd260 and BETd246 in CRC cells are dependent on DR5 via crosstalk between the two apoptotic pathways.
SPOP activating mutations increase BETd sensitivity due to enhanced DR5 induction.
Mutations in SPOP, a substrate-binding subunit of the BRD4 E3 ubiquitin ligase, determine BETi sensitivity in some cancer cells [29–31]. CRC cells with a SPOP-activating mutation, including NCI-H508 (SPOP-E47K) and SNU-407 (SPOP-G75E) [41], are substantially more sensitive to BETd than SPOP-WT cells, as indicated by 3-20-fold lower IC50 of BETd (Fig. S6A). Compared to WT cells, SPOP-mutant CRC cells have reduced BRD4 but slightly higher basal DR5 levels (Fig. S6B) and are much more prone to DR5 induction by BETd260 (Fig. S6, C and D). BETd260 at 0.1 μM robustly induced DR5 and apoptosis in the SPOP-mutant cells but was not sufficient to do so in SPOP-WT HCT116 cells (Fig. S6, D–H). This increased BETd sensitivity in the mutant cells is dependent on DR5 and could be suppressed by DR5 knockdown (Fig. S6, F–H). Furthermore, knockdown of SPOP in NCI-H508 and SNU-407 cells abolished the enhanced DR5 induction, apoptosis, and BETd sensitivity, but only slightly affected DR5 induction in HCT116 cells (Fig. S7, A–C). Transfecting the SPOP mutants into the parental HCT116, but not DR5-KO cells, recapitulated the observed differences in the mutant cells (Fig. S7, D–G). These results indicate that SPOP activating mutations increase BETd sensitivity by downregulating BET proteins, which lowers threshold for their inhibition and enhances DR5 induction.
DR5 mediates the in vivo antitumor effects of BETd260 in isogenic and PDX models.
To determine the functional role of DR5 in mediating the in vivo antitumor effects of BETd, we analyzed xenograft tumors established from WT and DR5-KO HCT116 cells in athymic nude mice. Treating tumor-bearing mice with BETd260 (i.v.; 5 mg/kg) effectively suppressed growth of WT HCT116 tumors (Fig. 3, A and B), accompanied by induction of DR5 (Fig. 3C) and apoptosis detected by positive staining of TUNEL, active caspase 8, and active caspase 3 (Fig. 3, D–F). In contrast, the antitumor and apoptotic effects of BETd260 were largely suppressed in DR5-KO tumors (Fig. 3, A–F), suggesting a key role of DR5-mediated apoptosis in tumor suppression.
Figure 3. BETd260 suppresses in vivo tumor growth via DR5-mediated apoptosis.
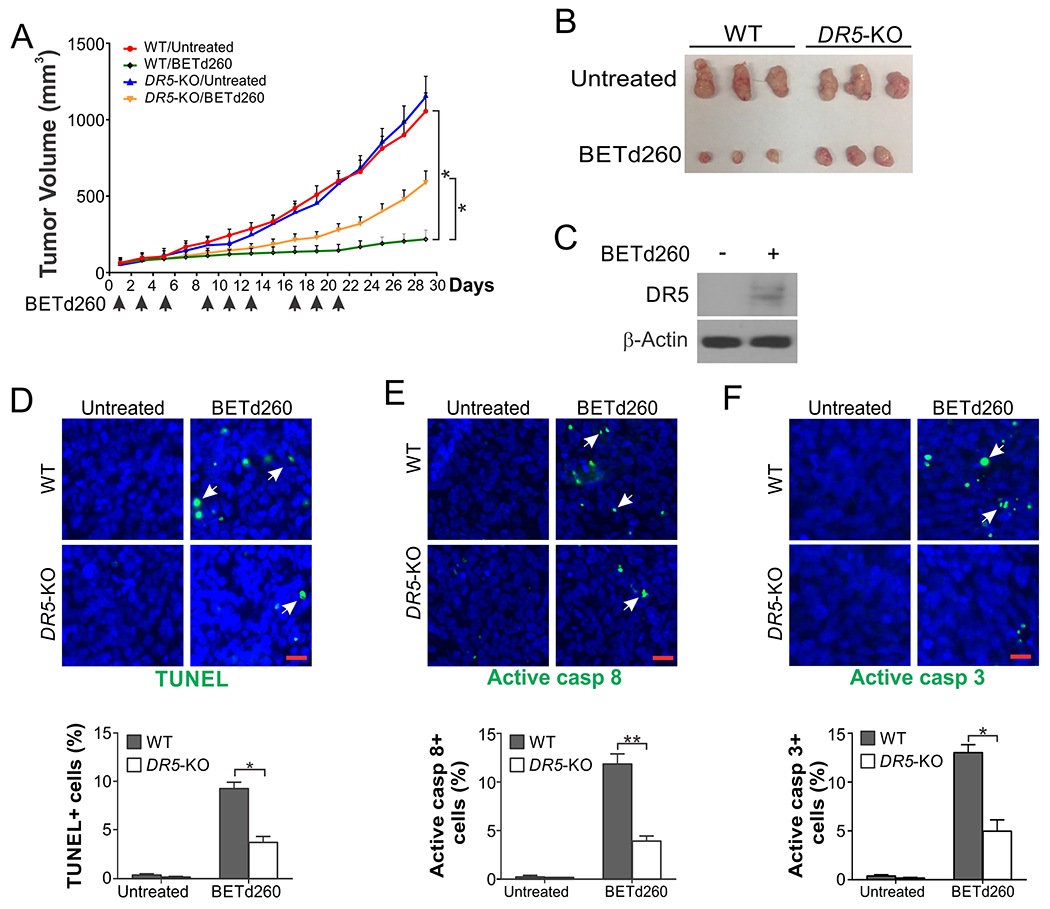
(A) Nude mice were injected subcutaneously with 4×106 WT or DR5-KO HCT116 cells. After tumor growth for 7 days, mice were treated with BETd260 (i.v.; 5 mg/kg; 3×/week). Tumor volume at indicated time points was calculated and plotted with P values for indicated comparisons (N=6 in each group). (B) Representative tumors at the end of the experiment in (A). (C)-(F) Mice bearing WT and DR5-KO HCT116 tumors were treated as in (A) for 5 days. Paraffin-embedded sections of tumor tissues were analyzed by (C) Western blotting of DR5; immunostaining of (D) TUNEL, (E) active caspase 8, and (F) active caspase 3. Upper panels: representative staining pictures with arrows indicating examples of positive signals (scale bars: 25 μm); lower panels: quantification of positive signals showing means ± SEM of 300 cells in each mouse (N=3 in each group). *, P < 0.05; **, P <0.01.
We further analyzed the antitumor effects of BETd260 using PDX models, which better recapitulate histology, heterogeneity, and molecular alterations of original patient tumors than cell line xenografts [45]. Two CRC PDX models established from treatment-naïve primary tumors were analyzed [41], including PDX1 (T4N0M1; KRAS-G13D; NRAS-G12D; MMR-proficient [pMMR]) and PDX2 (T2N0; pMMR). BETd260 treatment markedly suppressed the growth of PDX1 tumors without affecting animal weight (Fig. 4, A–C). The treatment induced DR5 expression, tumor cell loss, and apoptosis analyzed by active caspase 3 staining, as well as proliferation inhibition (Fig. 4, D–H). Furthermore, inhibiting ER stress by salubrinal largely suppressed the antitumor activity of BETd260, as well as DR5 induction and apoptosis (Fig. 4, A–H). BETd260 was also efficacious against PDX2 with ER-stress-dependent DR5 induction (Fig. S8, A–C). These results demonstrate DR5- and ER-stress-mediated antitumor effects of BETd260 against CRC tumors in vivo.
Figure 4. BETd260 suppresses CRC PDX tumors in an ER-stress-dependent manner.

NSG mice subcutaneously implanted with PDX1 model (T4N0M1; KRAS G13D; NRAS G12D; MMR-proficient) were treated with BETd260 (i.v.; 5 mg/kg) +/− salubrinal (i.p.; 1 mg/kg) 3 times per week as indicated for 19 days. (A) Tumor volume at indicated time points with P values indicated (N=4 in each group). (B) Representative tumors at the end of the experiment. (C) Animal weight. (D)-(H) NSG mice bearing PDX1 tumors were treated as in (A) for 10 consecutive days. Tumor tissues were analyzed by (D) H&E staining, (E) Western blotting of DR5, (F) TUNEL, (G) active caspase 3, and (H) Ki67 immunostaining. In (F)-(H), left panels: representative staining pictures with arrows indicating examples of positive signals (scale bars: 25 μm); right panels: quantification of positive signals showing means ± SEM of 300 cells in each mouse (N=3 in each group). **, P <0.01.
BETd induce DR5-dependent immunogenic cell death (ICD) in CRC cells.
Recent studies showed that BET proteins regulate the expression of the PD-1 ligand PD-L1 in some cancer cells, and BET inhibition promotes antitumor immune response [20, 21]. However, BETd260 treatment did not affect the expression of PD-L1 in CRC cells (Fig. 5A). The involvement of ER stress promoted us to test if BETd-induced apoptosis is ICD [28], in which a small fraction of dying tumor cells expose or release Damage Associated Molecular Pattern (DAMP) molecules, such as the ER chaperone calreticulin (CRT) [46], which promote dendritic cell (DC) maturation and subsequent immune attack of remaining tumor cells [26, 27]. We found that treating different CRC cells with BETd260 or BETd246 markedly induced cell-surface CRT (Fig. 5B), which was suppressed in DR5-KO cells (Fig. 5C). We then tested if BETd-induced dying CRC cells can be engulfed by DCs through phagocytosis. Upon co-culture of green-labeled, BETd-treated HCT116 cells with red-labeled DCs prepared using peripheral blood mononuclear cells (PBMCs) from healthy donors, we observed a significant increase in DC phagocytosis of BETd-treated HCT116 cells, which was inhibited by DR5 KO (Fig. 5, D and E), suggesting a key role of ER stress- and DR5-dependent cell killing in mediating the immunogenic effects of BETd.
Figure 5. BET degraders induce DR5-dependent immunogenic cell death.

(A) Western blotting of PD-L1 in indicated human and mouse CRC cell lines treated with 0.5 μM BETd260 at indicated time points. (B) CRT cell-surface translocation in indicated CRC cell lines treated with 0.5 μM BETd260 or BETd246 for 24 hr was examined by immunostaining followed by flow cytometry. (C) CRT cell-surface translocation in WT and DR5-KO HCT116 cells treated as in (B). (D) and (E) WT and DR5-KO HCT116 cells treated with BETd260 or BETd246 as in (B) and dendritic cells differentiated from healthy donors’ monocytes were labelled with green (CFSE) and red (Far Red) fluorescence, respectively, and co-incubated at 1:1 ratio. DC phagocytosis was analyzed by detecting fused cells by (D) fluorescence microscopy and (E) quantified by flow cytometry. In (D), arrows indicate examples of phagocytic DCs (scale bars: 10 μm). In (F), right quadrants in flow cytometry profiles represent fused cells. Results in (B), (C) and (E) were expressed as means ± SD of three independent experiments. **, P <0.01; ***, P <0.001.
BETd260 potentiates anti-PD-1 immunotherapy in a DR5-dependent manner.
We then used the immuno-competent CT26-BALB/cJ syngeneic tumor model to determine the efficacy of BETd260 in combination with an anti-PD-1 antibody. We observed a striking synergism between BETd260 and anti-mouse PD-1 antibody at doses that either agent alone showed little or no efficacy. WT CT26 tumors did not respond to BETd260 at a pre-determined, reduced dose and frequency (from 3×5 mg/kg/week in Fig. 3A to 2×2 mg/kg/week in Fig. 6A), or anti-mouse PD-1 antibody at a previously described dose and schedule [47] (Fig. 6, A–C). Remarkably, the combination of BETd260 and anti-mouse PD-1 markedly enhanced therapeutic efficacy (Fig. 6, A and B), with 8 out of 10 treated mice had virtually no tumor growth at the endpoint of day 28 (Fig. 6C). Upon analyzing DR5-KO CT26 cells generated by CRISPR/Cas9 (Fig. S9, A and B), we found that the therapeutic efficacy of the combination is DR5-dependent. Mice bearing DR5-KO CT26 tumors treated with the combination, similar to those with untreated tumors or treated with either agent alone, had to be sacrificed around day 20 due to a large tumor size (Fig. 6, A–C). The combination treatment was well tolerated and did not significantly affect body weight (Fig. 6D).
Figure 6. BETd260 combined with anti-PD-1 antibody potently suppresses CT26 tumors in a DR5-dependent manner.

BALB/cJ mice were injected subcutaneously with 4×106 WT or DR5-KO CT26 cells. After tumor growth for 7 days, mice were treated with BETd260 (i.v.; 2 mg/kg; 3×/week), anti-mouse PD-1 (Clone 4H2; i.p.; 200 μg/dose), or their combination. (A) Tumor volume at indicated time points after treatment was calculated and plotted with P values for indicated comparisons (N=8-10 in each group as indicated; ***, P <0.001). (B) Cumulative survival of mice over time. ***, P <0.001 (log-rank test). (C) Growth curves of individual WT and DR5-KO tumors treated with BETd260, anti-PD-1, or their combination as in (A). (D) Body weight of the mice treated as in (A). (E) CD3 and CD8 immunostaining of tumor tissues resected from mice treated as in (A) for 10 days (scale bars: 25μm).
Analysis of tumor sections by immunostaining revealed DR5-dependent increases in infiltration of CD3+ and CD8+ cells (Fig. 6E). Further analysis of immune cell markers by flow cytometry showed that the combination treatment significantly increased infiltration of CD3+, CD4+, and CD8+ T cells, and decreased infiltration of CD25+/FoxP3+ regulatory T cells (Tregs) in WT tumors (Fig. 7, A–D). The combination treatment increased interferon γ (IFNγ) and granzyme-B (GzmB) production by tumor-infiltrating CD8+ T cells in WT tumors (Fig. 7, E and F), and also increased the population of infiltrating CD11c+ DCs expressing higher levels of the activation markers CD86 and MHC class II (MHCII) (Fig. 7, G–I). Strikingly, all of the observed changes in immune cell markers in WT tumors were suppressed in DR5-KO tumors (Fig. 7, A–I), indicating a critical role of DR5-dependent cell death in mediating the antitumor immune response to the combination treatment. Furthermore, depleting CD8+ cytotoxic T cells by treating mice with an anti-CD8 antibody abolished the effects of the combination on WT tumors, confirming the role of CD8+ T cells in tumor suppression by combination therapy (Fig. 7J). Collectively, our in vitro and in vivo data demonstrate a key functional role of DR5 induction in mediating the apoptotic and antitumor effects of BETd in CRC cells.
Figure 7. The BETd260/anti-PD-1 combination increases tumor infiltration of lymphocytes in a DR5-dependent manner in CT26 tumors.

(A)-(I) WT or DR5-KO CT26 tumors treated as in Fig. 6A and resected at day 10 were analyzed by flow cytometry for infiltrating immune cells: (A) CD3+/CD45+, (B) CD8+/CD45+, (C) CD4+/CD45+, (D) CD25+/FoxP3+/CD4+, (E) IFNγ+/CD8+, (F) GzmB+/CD8+, (G) CD11c+/CD45+, (H) CD86+/CD11c+, and (I) MHCII+/CD11c+. (J) Mice injected with anti-CD8 (250 μg/dose indicated; 1st dose 1 day before 1st anti-PD-1) or the control IgG were treated with the BETd260/anti-PD-1 combination and analyzed as in Fig. 6A. In (A)-(J), N=6 in each group. *, P <0.05; **, P <0.01.
Discussion
The majority of advanced CRCs are refractory to conventional chemotherapy and recent targeted therapy [2]. BET family proteins are overexpressed in CRCs including recurrent and metastatic tumors and represent attractive therapeutic targets. Our results show that BETd260 and BETd246 are much more potent than other BET-targeting agents in CRC cells. They exhibited striking in vivo efficacy in cell line, PDX, and syngeneic tumor models. Furthermore, combinations of BETd260 with 5-FU, oxaliplatin, TRAIL, and anti-PD-1 are highly synergistic and markedly improve therapeutic efficacy. The remarkable therapeutic efficacy of BETd260 and BETd246 suggests this class of BET degraders can be further developed and optimized for clinical translation.
We identified ER stress- and CHOP-mediated DR5 induction as a critical activity of BET-targeting agents in CRC cells. The DR5 induction likely results from downregulation of c-Myc, a key oncogenic driver aberrantly activated by the defective APC/β-catenin pathway in CRCs [48]. c-Myc levels were shown to correlate with BETi sensitivity in CRC cells [34]. While c-Myc is often considered to be undruggable, inhibiting its expression using BETd offers an exciting opportunity for targeting this key oncogene. DR5 induction is critical not only for the activity of BETd alone, but also for the synergistic effects of BETd combined with different classes of anticancer agents. This is consistent with the role of DR5 as a nodule point of different apoptotic pathways, including DNA damage-induced and p53-dependent apoptosis, ER stress-mediated apoptosis, and cytokine-induced apoptosis in antitumor immune response. Enhanced DR5 induction may also be related to the inhibitory effect of BETi on protective feedback kinase signaling, which mediates the potentiating effects of BETi on kinase inhibitors [8]. Our data suggest that in addition to apoptosis, non-apoptotic cell death is also involved in the killing of CRC cells by BETd260 and BETd246 (Fig. 1G; Fig. S1, H and I), which will be further examined in our future studies.
Our data indicate that SPOP mutations may be useful biomarkers for predicting BETd sensitivity in CRCs. Despite their low frequency (2-5%) [30], analyzing the effects of SPOP mutations in CRC may have important translational implications as SPOP-mutant tumors may represent “super responders” particularly sensitive to BETd at a clinically achievable and non-toxic dose, similar to lung cancers with rare EGFR mutations or ALK translocation [49]. However, caution should be taken as SPOP has many substrates, and SPOP mutations have complex and context-dependent effects in different types of cancer cells [29–31]. Several other features are associated with differential BETi sensitivity, including epigenetic reprograming, the LncRNA CCAT1, hyper-phosphorylation of BRD4 [50], and CpG island methylator phenotype (CIMP) [32]. Further studies on how SPOP and other drivers modulate the sensitivity to BET-targeting agents may provide a panel of biomarkers for designing a personalized CRC therapy.
Sustained BET inhibition causes intestinal stem cell depletion and both lymphoid and hematopoietic abnormalities [9, 10]. A major challenge in targeting BET proteins is optimizing the therapeutic window to minimize normal tissue toxicity. BETd260 at 5 mg/kg every other day, while robustly inhibiting tumor growth, was well-tolerated and did not cause animal weight loss (Fig. 6 and 7). The potent efficacy of BETd260/246 alone or in combination may provide a therapeutic window necessary for BETd dosing to minimize normal tissue toxicity. In addition to biomarker-based stratification, development of isoform-specific degraders may provide improved specificity for minimizing the toxicity of BET degraders. It has been shown isoform-specific BETd can be designed based on the structural information of BET family proteins [17].
Our results indicate that both cell-intrinsic and immunogenic effects contribute to the therapeutic efficacy of BETd. In contrast to studies on PD-L1 downregulation [9, 10], BETd treatment did not affect PD-L1 expression in CRC cells (Fig. 5A). In CRC patients, PD-L1 is expressed at low levels in a small fraction of tumors, and PD-L1 levels do not predict clinical response to anti-PD-1 immunotherapy [4, 5]. Our results showed that BETd treatment induced DR5-dependent cell killing with hallmarks of ICD, suggesting a critical role of DR5 in ICD. Consistent with our findings, other anticancer agents, such as a combination of EGFR antibody and chemotherapy, also induce ICD via ER stress and apoptosis [28]. The DR5-dependent immunogenic effects of BETd can be further verified by other approaches such as in vivo vaccination [26, 27], in which mice vaccinated with BETd260-treated WT, but not DR5-KO, CT26 cells are expected to be protected against subsequent tumor development.
Immune checkpoint inhibitors (ICIs) have revolutionized cancer treatment [51]. However, the clinical benefit of ICIs is limited in only 10-15% of CRCs with dMMR [5]. A burning issue is to covert immunologically “cold” tumors into “hot” tumors. We demonstrate remarkable synergism of BETd260 and anti-PD-1 antibody, which nearly eradicated mouse CT26 syngeneic tumors in a DR5-and CD8-dependent manner. As CT26 is pMMR [52], our results suggest that BETd are potentially useful for sensitizing the majority (>85%) of CRCs to anti-PD-1 immunotherapy [4, 5]. Our study supports a critical role of DR5-mediated cell death in the tumor microenvironment in priming the T cell response. Identification of specific drugs or drug combinations that activate DR5-dependent ICD may be broadly applicable in CRCs. The translational potential of the BETd/ICI combinations remains to be evaluated in future animal and clinical studies.
Collectively, our results demonstrate a striking efficacy of BETd against CRCs through DR5-mediated cell death and antitumor immunity. These results provide a rationale, proof-of-principle evidence, new mechanistic insights, and potential biomarkers for further developing BETd for improving CRC therapies.
Materials and Methods
Cell culture and drug treatment
Cell lines are described in Supplemental Methods. Cell lines were authenticated by genotyping and analysis of protein expression by Western blotting, and routinely checked for Mycoplasma contamination by PCR. All CRC cell lines were maintained in a 37°C incubator at 5% CO2. Cell culture media were supplemented with 10% defined FBS (HyClone, Logan, UT), 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). For drug treatment, cells were plated in 12-well plates at 20-30% density 24 hr before treatment.
BETd246, BETd260, BETd228, and BETd570 are previously described [13, 15]. All other chemicals and drugs are described in Table S1. DMSO stocks were prepared and diluted in cell culture media.
Analysis of cell viability and apoptosis
MTS assay was performed as described in Supplemental Methods. Apoptosis was measured by counting cells with condensed and fragmented nuclei after nuclear staining with Hoechst 33258 [53]. A minimum of 300 cells were counted for each sample. Apoptosis was also analyzed by flow cytometry of cells stained with Annexin V/propidium iodide (PI) [54]. Crystal violet staining of viable cells was performed on cells treated for 36 hr in 12-well plates. Long-term cell survival was analyzed by colony formation assay as described [54]. Cytochrome c release was analyzed by cytochrome c Western blotting of mitochondrial and cytosolic fractions isolated by differential centrifugation. Each assay was conducted in triplicate and repeated three times.
Western blotting
Western blotting was performed using antibodies listed in Table S1 as previously described [43].
Analysis of mRNA expression
Total RNA was isolated from cells using the Mini RNA Isolation II Kit (Zymo Research) according to the manufacturer’s protocol. RNA-Seq was performed as described in Supplemental Methods. For Real-time reverse Transcriptase (RT) PCR, one μg of total RNA was used to generate cDNA by the SuperScript II reverse transcriptase (Invitrogen). PCR was performed using primers listed in Table S2 and previously described cycle conditions [55].
Analysis of immunogenic cell death (ICD)
CRC cells treated with 0.5 μM BETd260 or BETd246 for 24 hr were analyzed for ICD markers, including cell-surface calreticulin (CRT) and phagocytosis of CRC cells by dendritic cells (DCs), as described in Supplemental Methods.
Animal experiments
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Sample size estimation was based on previous experience [41, 43]. All treated animals were analyzed without randomization and blinding to the group allocation. Mice were housed in a sterile environment with micro isolator cages and allowed access to water and chow ad libitum. BETd260 was dissolved in 10% PEG400: 3% Cremophor: 87% PBS. Tumor growth was monitored by calipers, and tumor volumes were calculated according to the formula ½×length×width2.
Cell line xenografts were established by subcutaneously injecting 4×106 WT or DR5-KO HCT116 cells into both flanks of 5-6-week old female Nu/Nu mice (Charles River). Xenograft tumors were allowed to grow for 7 days to reach ~60 mm3 in size before treatment. Tumor-bearing mice were treated with BETd260 at 5 mg/kg by intravenous injection (i.v.) for 3 times/week. Six mice were used for each group.
Patient derived xenograft (PDX) tumors were established from treatment-naïve primary tumors in 5-6-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory) as described [41]. PDX1 was from a KRAS-mutant (G13D), NRAS-mutant (G12D), and MMR-proficient sigmoid colon tumor (T4N0M1). PDX2 was established from an MMR-proficient right colon tumor (T2N0). Tumors passaged and expanded for two generations (P2) in NSG mice were used. Tumor-bearing mice were randomized into 4 groups and subjected to following treatments for 3 weeks: 1) untreated; 2) BETd260; 3) Salubrinal; 4) BETd260 + Salubrinal. Four mice were used for each group. Salubrinal (10 mg/mL) was freshly diluted in 50/50 Poly (ethylene glycol) (PEG)/saline solution before administration and i.p. injected at 1 mg/kg.
Syngeneic tumors were established by subcutaneously injecting 5×105 WT or DR5-KO CT26 cells into both flanks of 5-6-week-old BALB/cJ mice (Jackson Laboratory). After tumor growth for 7 days, tumor-bearing mice were randomized and treated with 2 mg/kg BETd260, anti-PD-1 antibody (200 μg/dose), or their combination. The ethical endpoint was reached when a tumor reached 2 cm or more in any dimension.
Immunostaining of tumor tissues
Tumor tissues were dissected and fixed in 10% formalin and embedded in paraffin. Immunostaining was performed on 5-μm paraffin-embedded tumor sections using kits and antibodies listed in Table S1 as described [53], with AlexaFluor 488-conjugated secondary antibody for signal detection and 4’ 6-Diamidino-2-phenylindole (DAPI) for nuclear counter staining.
Flow cytometry analysis of tumor-infiltrating lymphocytes
WT and DR5-KO CT26 tumors treated with BETd260 with or without combination with anti-PD-1 antibody were dissected 24 hr after the third treatment and dispersed in 5 mL of base RPMI-1640 media with 10× dissociation cocktail containing collagenase I, collagenase IV, and DNase I for 1 hr at 37 °C with shaking. Single-cell suspensions were prepared by passing cells through a 70-μm cell strainer. Cells were stained with combinations of fluorochrome-conjugated antibodies listed in Table S1 and analyzed by flow cytometry as described [56].
Statistical analysis
Statistical analysis was carried out using the GraphPad Prism 9 software. For cell culture experiments, P values were calculated using the Student’s t-tests. Means ± standard deviation (SD) are reported in the figures. For animal experiments, P values were calculated by repeated measures (RM) ANOVA with Fisher’s LSD post-hoc tests. Means ± standard error of the mean (SEM) are reported in the figures. Differences were considered significant if P < 0.05.
Supplementary Material
Acknowledgments
The authors thank our lab members for discussion and critical reading. This work was supported by the U.S. National Institutes of Health grants (R01CA203028, R01CA217141, R01CA236271, R01CA247231, and R01CA248112 to L. Zhang; R01CA215758 to S. Wang; U19AI068021 and R01CA215481 to J. Yu; T32GM133332 to K. Ermine). This project used the Hillman Cancer Center Animal Facility, Cytometry Facility, and Tissue and Research Pathology Services, which are supported in part by P30CA047904.
Abbreviations:
- 5-FU
5-fluorouracil
- BET
Bromodomain and Extra-Terminal domain
- BETd
BET degraders
- BETi
BET inhibitors
- CFSE
carboxyfluorescein succinimidyl ester
- CHOP
C/EBP homologous protein
- ChIP
chromatin immunoprecipitation
- CHX
cycloheximide
- CIMP
CpG island methylator phenotype
- CRC
colorectal cancer
- CRT
calreticulin
- DAMP
damage associated molecular pattern
- DAPI
4’ 6-Diamidino-2-phenylindole
- DC
dendritic cell
- dMMR
deficient in DNA mismatch repair
- DR5
Death Receptor 5
- ER
endoplasmic reticulum
- Fer-1
Ferrostatin-1
- FPKM
fragments per kilobase of transcript per million mapped reads
- GzmB
granzyme-B
- ICD
immunogenic cell death
- ICIs
immune checkpoint inhibitors
- IFNγ
interferon γ
- KD
knockdown
- KO
knockout
- MHCII
MHC class II
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NSA
necrosulfonamide
- NSG
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ
- PBMCs
peripheral blood mononuclear cells
- PDX
patient-derived xenograft
- PI
propidium iodide
- pMMR
MMR-proficient
- PROTAC
Proteolysis Targeting Chimeras
- RM
repeated measures
- RNA-Seq
mRNA sequencing
- RT
reverse transcriptase
- SD
standard deviation
- SPOP
speckle-type POZ protein
- TRAIL
Tumor necrosis factor-Related Apoptosis-Inducing Ligand
- TUNEL
terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling
- WT
wild-type
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020; 70: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Chu E An update on the current and emerging targeted agents in metastatic colorectal cancer. Clin Colorectal Cancer 2012; 11: 1–13. [DOI] [PubMed] [Google Scholar]
- 3.Fakih MG. Metastatic colorectal cancer: current state and future directions. J Clin Oncol 2015; 33: 1809–1824. [DOI] [PubMed] [Google Scholar]
- 4.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015; 372: 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017; 357: 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 2014; 54: 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011; 146: 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stathis A, Bertoni F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov 2018; 8: 24–36. [DOI] [PubMed] [Google Scholar]
- 9.Bolden JE, Tasdemir N, Dow LE, van Es JH, Wilkinson JE, Zhao Z et al. Inducible In Vivo Silencing of Brd4 Identifies Potential Toxicities of Sustained BET Protein Inhibition. Cell Rep 2014; 8: 1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee DU, Katavolos P, Palanisamy G, Katewa A, Sioson C, Corpuz J et al. Nonselective inhibition of the epigenetic transcriptional regulator BET induces marked lymphoid and hematopoietic toxicity in mice. Toxicol Appl Pharmacol 2016; 300: 47–54. [DOI] [PubMed] [Google Scholar]
- 11.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015; 348: 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 2015; 22: 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res 2017; 77: 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell 2017; 67: 5–18 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J Med Chem 2018; 61: 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.An S, Fu L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018; 36: 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gadd MS, Testa A, Lucas X, Chan K-H, Chen W, Lamont DJ et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chemical Biology (Article) 2017; 13: 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conery AR, Centore RC, Spillane KL, Follmer NE, Bommi-Reddy A, Hatton C et al. Preclinical Anticancer Efficacy of BET Bromodomain Inhibitors Is Determined by the Apoptotic Response. Cancer Res 2016; 76: 1313–1319. [DOI] [PubMed] [Google Scholar]
- 19.Yao W, Yue P, Khuri FR, Sun SY. The BET bromodomain inhibitor, JQ1, facilitates c-FLIP degradation and enhances TRAIL-induced apoptosis independent of BRD4 and c-Myc inhibition. Oncotarget 2015; 6: 34669–34679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep 2016; 16: 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep 2017; 18: 2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashkenazi A Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 2008; 7: 1001–1012. [DOI] [PubMed] [Google Scholar]
- 23.Bhola PD, Letai A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol Cell 2016; 61: 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu GS, Burns TF, McDonald ER 3rd, Jiang W, Meng R, Krantz ID et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 1997; 17: 141–143. [DOI] [PubMed] [Google Scholar]
- 25.He K, Zheng X, Li M, Zhang L, Yu J. mTOR inhibitors induce apoptosis in colon cancer cells via CHOP-dependent DR5 induction on 4E-BP1 dephosphorylation. Oncogene 2016; 35: 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017; 17: 97–111. [DOI] [PubMed] [Google Scholar]
- 27.Wang YJ, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis 2018; 5: 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pozzi C, Cuomo A, Spadoni I, Magni E, Silvola A, Conte A et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat Med 2016; 22: 624–631. [DOI] [PubMed] [Google Scholar]
- 29.Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med 2017; 23: 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med 2017; 23: 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai X, Gan W, Li X, Wang S, Zhang W, Huang L et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 2017; 23: 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCleland ML, Mesh K, Lorenzana E, Chopra VS, Segal E, Watanabe C et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J Clin Invest 2016; 126: 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Y, Zhou J, Ye F, Xiong H, Peng L, Zheng Z et al. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int J Mol Sci 2015; 16: 1928–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Togel L, Nightingale R, Chueh AC, Jayachandran A, Tran H, Phesse T et al. Dual Targeting of Bromodomain and Extraterminal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol Cancer Ther 2016; 15: 1217–1226. [DOI] [PubMed] [Google Scholar]
- 35.Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 2015; 10: 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS et al. Cell death. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014; 345: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 2004; 279: 45495–45502. [DOI] [PubMed] [Google Scholar]
- 38.Oh YT, Yue P, Zhou W, Balko JM, Black EP, Owonikoko TK et al. Oncogenic Ras and B-Raf proteins positively regulate death receptor 5 expression through co-activation of ERK and JNK signaling. J Biol Chem 2012; 287: 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Li M, Ruan H, Qiu W, Xu X, Zhang L et al. Co-targeting translation and proteasome rapidly kills colon cancer cells with mutant RAS/RAF via ER stress. Oncotarget 2017; 8: 9280–9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005; 307: 935–939. [DOI] [PubMed] [Google Scholar]
- 41.Tan X, Tong J, Wang YJ, Fletcher R, Schoen RE, Yu J et al. BET inhibitors potentiate chemotherapy and killing of SPOP-mutant colon cancer cells via induction of DR5. Cancer Res 2019; 79: 1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han B, Yao W, Oh YT, Tong JS, Li S, Deng J et al. The novel proteasome inhibitor carfilzomib activates and enhances extrinsic apoptosis involving stabilization of death receptor 5. Oncotarget 2015; 6: 17532–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tong J, Zheng X, Tan X, Fletcher R, Nikolovska-Coleska Z, Yu J et al. Mcl-1 Phosphorylation without Degradation Mediates Sensitivity to HDAC Inhibitors by Liberating BH3-Only Proteins. Cancer Res 2018; 78: 4704–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leibowitz B, Qiu W, Buchanan ME, Zou F, Vernon P, Moyer MP et al. BID mediates selective killing of APC-deficient cells in intestinal tumor suppression by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A 2014; 111: 16520–16525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012; 9: 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 2009; 28: 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selby MJ, Engelhardt JJ, Johnston RJ, Lu LS, Han M, Thudium K et al. Preclinical Development of Ipilimumab and Nivolumab Combination Immunotherapy: Mouse Tumor Models, In Vitro Functional Studies, and Cynomolgus Macaque Toxicology. PLoS One 2016; 11: e0161779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr., Kinzler KW. Cancer genome landscapes. Science 2013; 339: 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin JJ, Shaw AT. Resisting Resistance: Targeted Therapies in Lung Cancer. Trends Cancer 2016; 2: 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016; 529: 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 2019; 16: 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017; 552: 116–120. [DOI] [PubMed] [Google Scholar]
- 53.Chen D, Wei L, Yu J, Zhang L. Regorafenib inhibits colorectal tumor growth through PUMA-mediated apoptosis. Clin Cancer Res 2014; 20: 3472–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tong J, Wang P, Tan S, Chen D, Nikolovska-Coleska Z, Zou F et al. Mcl-1 Degradation Is Required for Targeted Therapeutics to Eradicate Colon Cancer Cells. Cancer Res 2017; 77: 2512–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang P, Yu J, Zhang L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci U S A 2007; 104: 4054–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, Jie HB, Lei Y, Gildener-Leapman N, Trivedi S, Green T et al. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res 2015; 75: 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.