

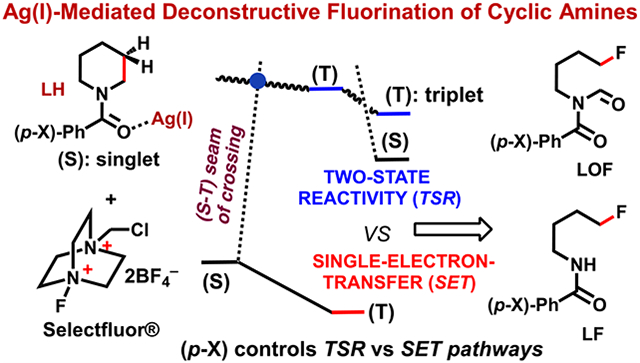
Abstract
Density functional calculations have provided evidence that a Ag(I)-mediated deconstructive fluorination of N-benzoylated cyclic amines (LH) with Selectfluor [(F–TEDA)-(BF4)2] begins with an association of the reactants to form a singlet state adduct {[(LH)–Ag]–[F–TEDA]2+}. The subsequent formation of an iminium ion intermediate, [L+–Ag]–HF–[TEDA]+, is, formally, a Ag(I)-mediated hydride abstraction event that occurs in two steps: (a) a formal oxidative addition (OA) of [F–TEDA]2+ to the Ag(I) center that is attended by an electron transfer (ET) from the substrate (LH) to the Ag center (i.e., OA + ET, this process can also be referred to as a F-atom coupled electron transfer), followed by (b) H-atom abstraction from LH by the Ag-coordinated F atom. The overall process involves lower-lying singlet and triplet electronic states of several intermediates. Therefore, we formally refer to this reaction as a two-state reactivity (TSR) event. The C─C bond cleavage/fluorination of the resulting hemiaminal intermediate via a ring-opening pathway has also been determined to be a TSR event. A competing deformylative fluorination initiated by hemiaminal to aldehyde equilibration involving formyl H-atom abstraction by a TEDA2+ radical dication, decarbonylation, and fluorination of the resulting alkyl radical by another equivalent of Selectfluor may also be operative in the latter step.
Graphical Abstract
INTRODUCTION
The functionalization of “inert” C─H bonds by converting them to C─C or C─X bonds (where X is a heteroatom such as O, N, B, etc.) has revolutionized the synthesis and production of pharmaceuticals, agrochemicals, materials, and fuels.1,2 Similarly, the utilization of C─C and C─X bonds to form new bonds by cleavage and functionalization of their constituent groups may lead to products which cannot be prepared efficiently by other means (i.e., these products possess high synthetic complexity).3-7 In particular, the development of methodologies for the deconstructive functionalization of cyclic amines (i.e., scaffold cleavage/functionalization) may provide new opportunities for diversifying these structural motifs that are abundant in pharmaceuticals and agrochemicals. Among the many deconstructive strategies that have been developed is a method by Sarpong and co-workers that is proposed to proceed through a distinct mechanism.8,9 This method transforms N-acylated saturated aza-cycles (e.g., 1, Scheme 1) into versatile fluorine-containing acyclic amine derivatives (e.g., 3) using commercially available reagents Selectfluor (2) and AgBF4. Presumably, the transformation occurs through selective C(sp3)─C(sp3) bond cleavage in the presence of a C(sp3)─N bond. An in-depth understanding of the mechanism of this unusual transformation should (a) facilitate the development of a more general strategy for the deconstructive functionalization of cyclic amines, and (b) enable the identification of alternative oxidizing salts and fluorinating reagents that are less expensive and may improve the functional group compatibility of the process.
Scheme 1.
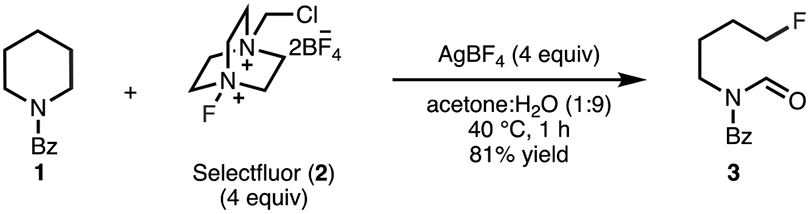
Silver-Mediated Deconstructive Fluorination of N-Benzoylated Cyclic Amine 1
Sarpong and co-workers proposed a two-stage mechanism, each mediated by a silver salt and Selectfluor, for the transformation of 1 → 3 (Figure 1).8,9 In the first stage, cyclic amine 1 is oxidized by the combination of AgBF4 and Selectfluor to the corresponding iminium ion (A), which is trapped by H2O to form hemiaminal B (Figure 1D). Selectfluor and AgBF4 did not react in the absence of a substrate, indicating the importance of Ag(I) binding to the amide moiety of 1 to reactivity. In line with previous studies10-26 as well as their own mechanistic analysis, Sarpong and co-workers proposed that the Ag(I) center binds to 1 to form adduct 4 (Figure 1A). Upon interaction of this adduct with 2, a single electron transfer occurs from the ligated AgBF4 of 4 to 2 to generate a Ag(II) center and radical dication 5. The resulting Ag(II) then oxidizes 1 equiv of 1 through single-electron transfer (SET),27 and subsequent hydrogen-atom abstraction by 5 delivers iminium ion A (Figure 1A,B). An alternative pathway, where radical dication 5 effects α-amino C–H abstraction from 1 to generate an α-amino radical (E) followed by single-electron transfer to Ag(II) to generate A, was also proposed (Figure 1C). In the next stage, iminium ion A is trapped by H2O to give hemiaminal B (Figure 1D).
Figure 1.

Proposed mechanisms for the deconstructive fluorination: (A) overall oxidation sequence, (B) single-electron transfer (SET) occurs first, (C) hydrogen-atom transfer (HAT) occurs first, (D) hemiaminal B formation from iminium ion A, (E) homolytic C–C cleavage, path A, and (F) heterolytic C–N cleavage, path B.
In the second stage of the reaction, the resulting hemiaminal (B) is transformed to the final product (3 or 9; Figure 1E,F). This stage of the overall transformation was proposed to proceed through two possible pathways. In path A (Figure 1E), hemiaminal B reacts with Ag(I) and 2 to form radical C. Presumably, the deprotonation of the hemiaminal and single-electron transfer generate an alkoxy radical intermediate that is homolyzed through selective C(sp3)─C(sp3) bond cleavage to give C.3 A radical fluorination of C by 2 then forms alkyl fluoride product 3. Alternatively, path B (Figure 1F), referred to by Sarpong and co-workers as the “deformylation pathway” in their initial communications, would involve (a) heterolytic C─N bond cleavage of hemiaminal B to linear aldehyde 7 and subsequent oxidation of the formyl group to the corresponding carboxylic acid (8), and finally (b) decarboxylative fluorination to afford 9.8,9
Even though the proposed mechanistic scenarios in Figure 1 are consistent with those previously described for ring-opening functionalization and transition-metal-catalyzed fluorination methods,10-26 the elementary steps, relevant intermediates, and transition states remained to be fully elucidated. We viewed this fundamental knowledge to be vital to identifying simpler, more efficient protocols for the deconstructive fluorination of N-acylated cyclic amines. Therefore, the aims of the computational studies reported here are to provide insight into the mechanism of the Ag(I)-mediated deconstructive fluorination of N-acylated cyclic amine 1 with Selectfuor.
The calculations presented herein, consistent with previous proposals,8,9 show that the formation of iminium ion A from 1, in the presence of AgBF4 and Selectfluor (2), is formally a Ag(I)-mediated hydride abstraction event. We have established, for the first time, that this occurs through (a) a formal oxidative addition (OA) of [F–TEDA]2+ to the Ag(I) center that is attended by an electron transfer (ET) from the substrate (LH) to the Ag center (i.e., OA + ET, this process can also be referred to as F-atom coupled electron transfer, FCET), followed by (b) the abstraction of an H atom from the radical cation of 1 by the Ag-bound F atom. This reaction involves low-lying singlet and triplet electronic states of the reactive intermediates and therefore is characterized as a two-state reactivity (TSR) process28-35 rather than a classical single-electron-transfer (SET) event.
We have shown that the subsequent fluorination of the resulting hemiaminal (B) via the ring-opening mechanism (path A, Figure 1E) begins with H-atom abstraction from the hydroxy group and is also a TSR event. However, the alternative “deformylative” fluorination pathway (i.e., 7 → 9) that may be initiated by the equilibration of the hemiaminal to aldehyde, followed by its oxidation to a carboxylic acid and subsequent decarboxylative fluorination or H-atom abstraction from 7 by 5, decarbonylation, and fluorination by another equivalent of Selectfluor, is not a TSR event. Both net C–C cleavage/fluorination pathways (i.e., paths A and B) are feasible. The operative pathway likely depends on the reaction conditions and the electronic properties of the N-acyl group in 1.
EXPERIMENTAL SECTION: COMPUTATIONAL DETAILS
All reported structures were calculated using the Gaussian 16 suite of programs36 at the B3LYP-D3(BJ)/[6-31G(d,p)+Lanl2dz(Ag)] level of theory with the corresponding Hay–Wadt effective core potential37-39 for Ag. Here we used the B3LYP density functional40-42 with Grimme’s empirical dispersion correction (D3)43 and the Becke–Johnson (BJ) damping correction.44-46 Frequency analyses were used to characterize each minimum with zero imaginary frequency and each transition-state (TS) structure with one imaginary frequency. Intrinsic reaction coordinate (IRC) calculations were performed for all TSs to ensure their true nature. Bulk solvent effects were incorporated for all calculations (including geometry optimizations and frequency calculations) using the self-consistent reaction field polarizable continuum model (IEF-PCM).47,48 We chose water as the solvent. The reported thermodynamic data were computed at a temperature of 298.15 K and 1 atm of pressure. Various lower-lying electronic states, including the open-shell singlet states (where appropriate), were considered for all key species. Unless otherwise stated, energies are given as ΔH/ΔG in kcal/mol.
The open-shell singlet states of 5c and 10c are only slightly higher in free energy (see Supporting Information for details) as compared to the corresponding triplet states, which enabled us to characterize the 5c-s → 5c-t and 10c-s → 10c-t transitions as two-state reactivity events. Since these small energy values are subject to the level of theory employed, adiabatic transitions (i.e., singlet state–singlet state transitions involving high- and low-spin states) cannot be ruled out. A search for transition states associated with adiabatic transitions requires multideterminant approaches which are not practical for such large chemical systems. Triplet states were determined to be more in line with our analyses and allow consistency in our presentation of the major chemical outcomes of this study.
Following an extensive computational survey, we employ dication (F–TEDA)2+, without the two corresponding BF4 counteranions, as a model for Selectfluor (details in Figure S1 in the Supporting Information). Below we use Xc-y labeling to denote calculated structures, where X is a number associated with a structure and c denotes computed. Label y indicates singlet (s), doublet (d), and/or triplet (t) states.
To validate the [B3LYP-D3(BJ)+PCM]/[6-31G(d,p)+Lanl2dz-(Ag)] approach in this study, we have performed a series of calculations at the highest possible levels of theory for critical points along the computed potential energy surfaces. Specifically, the formation of [(LH)–AgBF4] from LH and AgBF4, ΔHcomp/ΔGcomp, and the singlet–triplet energy splitting [i.e., E(S–T)] in complexes 5c and 10c (see below)] were recalculated at the [B3LYP-D3(BJ)+PCM]/[cc-pVTZ+Lanl2dz(f)(Ag)]49 level of theory (to validate the [6-31G(d,p)+Lanl2dz(Ag)] basis sets that we employed) and at the [wB97XD+PCM]/[cc-pVTZ+Lanl2dz(f)(Ag)]50 level of theory (to validate the use of the B3LYP density functional). The results of these calculations are given in the Supporting Information (Table S1). We found that changing the basis sets from [6-31G(d,p)+Lanl2dz(Ag)] to [cc-pVTZ+Lanl2dz(f)(Ag)] reduced the calculated complexation free energy and the E(S–T) of complexes 5c and 10c by ~1 to 2 kcal/mol. In addition, we found that the choice of the density functional strongly impacts several calculated properties. For example, upon going from [B3LYP-D3(BJ)] to wB97XD functionals, the complexation free energy decreased by 3.4 kcal/mol and E(S–T) increased by 1.0 and 6.8 kcal/mol for complexes 5c and 10c, respectively. Importantly, using neither larger basis sets nor the wB97XD functional (instead of [B3LYP-D3(BJ)]) altered our conclusions.
RESULTS AND DISCUSSION
Mechanism of Iminium Ion Formation.
Consistent with previous8,9 experimental findings, our calculations show that AgBF4 (denoted as Ag(I) below) binds the substrate (LH, Figure 2) to form adduct [(LH)–Ag(I)], 4c. For the ground singlet electronic state of this complex (i.e., 4c-s), the calculated interaction between LH and Ag(I) (favorable by 20.5/8.9 kcal/mol) results in a slight elongation of the carbonyl C─O bond (from 1.238 to 1.264 Å) and a shortening of the N─carbonyl bond (from 1.361 to 1.341 Å).52 A charge density analysis indicates that in 4c-s a 0.20 ∣e∣ charge is transferred from LH to Ag(I). (For more details, see Figure S2 in the Supporting Information.)
Figure 2.

Selected structural and electronic parameters (distances are in Å; Mulliken charges, Q, are in ∣e∣). Relative energies (ΔH/ΔG in kcal/ mol) are indicated for LH (or 1), AgBF4, and singlet-state adduct (LH)[AgBF4], 4c-s.
The interaction of 4c-s with (F–TEDA)2+ leads to complex 5c-s. As seen in Figure 3, the geometry and charge distributions in the [(LH)–Ag] and (F–TEDA)2+ fragments did not change noticeably upon interaction of (F–TEDA)2+ and [(LH)–Ag]. In complex 5c-s (Figure 3), a charge of almost +2 is located on (F–TEDA), and only an additional 0.12 ∣e∣ electron is distributed from LH to the AgBF4 unit.
Figure 3.

Calculated intermediates 5c-s, 5c-t, and 6c-s and triplet transition state TS1 (H–F form)-t along with their important geometry (distances are in Å) and electronic parameters (Mulliken charges, Q, and spin densities, S, are in ∣e∣). For simplicity, the BF4 anion and noninteracting H atoms are omitted. For details, see Figure S3 in the Supporting Information.
Calculations show that the generation of iminium 6c-s from complex 5c-s is highly unfavorable on the singlet energy surface (see below). However, reasonable energies were computed for the reaction proceeding via a singlet–triplet seam of crossing. Here, we were not able to locate/optimize the minimum in the seam of crossing (MSX) for such large and conformationally unrestrained systems.52 However, triplet-state intermediate 5c-t is only 13.4/13.5 kcal/mol higher in energy than 5c-s (Figure 4). As seen in Figure 3, the 5c-s → 5c-t transition results in the cleavage of the F–TEDA bond and the formation of the Ag–F (bond distance = 2.040 Å) and Ag–TEDA (Ag–N2 = 2.246 Å) bonds. Furthermore, in 5c-t, the LH-fragment has 0.96 ∣e∣ positive charge and 1.11 ∣e∣ unpaired α-spin (i.e., it is a radical cation similar to 1ox in Figure 1B), another unpaired α-spin is delocalized on AgF (as 0.39 ∣e∣ and 0.26 ∣e∣ spins on Ag and F, respectively), and a monocationic TEDA+ fragment is coordinated to Ag.53 The Ag center has also lost electron density compared to that in 5c-s: it now bears a +0.68 ∣e∣ positive charge and a 0.39 ∣e∣ unpaired α-spin. Thus, the Ag center is further oxidized in 5c-t. The computed charge, spin distributions, and geometry parameters enabled us to characterize 5c-t as a Ag(II) species with a weak Ag–F interaction [(LH+)•−(AgF•)−(TEDA)+] and the 5c-s → 5c-t transition as a fluorine atom coupled electron transfer (FCET) process. Formally, the 5c-s → 5c-t transition can also be viewed as an oxidative addition (OA) of F–TEDA to Ag(I) coupled with an electron transfer (ET) from LH (an OA + ET). However, the exact nature of this dynamic process (synchronous versus asynchronous) remains undetermined.
Figure 4.

Schematic representation of the calculated energy profile for iminium ion formation upon the interaction of N-protected cyclic amine LH, the AgBF4 catalyst, and Selectfluor. Energies are provided relative to the 5c-s intermediate. Energies given in parentheses are relative to the dissociation limit of [(LH)–Ag(I)], 4c-s, +2[F–TEDA]2+.
Historically, Ag(I) has been implicated in mainly one-electron redox chemistry. Therefore, we propose that the formal oxidative addition involves multiple steps (vide infra). However, the exact nature of this dynamic process remains to be determined since we observe only rapid electron transfer from LH. Reports proposing Ag(III)–F species have remained unsubstantiated. However, recently, Ribas and co-workers have reported the synthesis of well-defined Ag(III)–aryl complexes generated from a Ag(I)/Ag(III) redox cycle.54 Furthermore, Musaev and co-workers have recently identified a critical Ag(III) intermediate in the Cu-catalyzed, Ag-salt mediated, Ullmann-type coupling reaction.55
Since (a) we were not able to locate transition states for the Ag(I) oxidative addition to F–TEDA (neither on the singlet nor triplet state PESs) and (b) the 5c-s → 5c-t transition involves lower-lying singlet and triplet states of the initial 5c-s and product 5c-t complexes, here we describe the 4c-s + [F–TEDA]2+ → 5c-s → 5c-t transformation as a two-state reactivity (TSR) event.28-35
In 5c-t, H─F bond formation leads to iminium ion 6c{[L–Ag]–(HF)–[TEDA]}2+, the ground electronic state of which is the singlet state: complex 6c-s lies 59.2/60.9 kcal/mol lower in energy than prereaction complex 5c-s. The triplet state of 6c (i.e., 6c-t) lies 58.6/61.2 kcal/mol higher in energy than 6c-s (Figure S3 in the Supporting Information). Rigid scanning of the singlet potential energy surface for HF formation indicated that the singlet transition state [TS1(H–F form)-s] that may directly connect 5c-s with 6c-s lies very high in energy (dotted black line in Figure 4). Therefore, a search for TS1(H–F form)-s was not pursued. Gratifyingly, we were able to locate the triplet transition state, TS1(H–F form)-t, that directly connects 5c-t with 6c-t (Figure 5). Our analyses show that TS1(H–F form)-t is an H-atom-abstraction transition state from LH by the Ag-coordinated F atom.56 The reactivity of amidyl radical cation 5c-t is consistent with observations from prior studies wherein an amine participates in a hydrogen-atom-transfer event upon single-electron oxidation.57 Notably, the α-C─H bond of an amine is estimated to be significantly weakened (lower BDE) following single-electron oxidation.57 In resulting product complex 6c-s, where an HF molecule is formed, the [L+–Ag] fragment possesses only one positive charge, which is mostly located on the now oxidized piperidine ring of L. As illustrated in Figure 4, TS1(H–F form)-t lies higher (by 0.8/0.2 kcal/mol) relative to triplet-state complex 5c-t.
Figure 5.

Computed triplet transition state TS1(H–F form)-t for H–F formation that connects intermediates 5c-t and 6c-t (distances are in Å).
The formation of the iminium ion is summarized in Scheme 2 from 5c-s and involves (a) F-atom transfer from [F–TEDA]2+ to the Ag center of the adduct [(LH)–Ag] which is coupled with an electron transfer from the substrate (LH) to the AgF fragment [a fluorine atom coupled electron transfer (FCET); formally, a Ag(I) oxidative addition to N–F coupled with an electron transfer, i.e., OA + ET)], triggered by a singlet-to-triplet (S–T) transition to arrive at 6c-t, followed by (b) H-atom abstraction from LH by the Ag-coordinated F atom. Since this reaction involves lower-lying singlet and triplet electronic states of the reactive intermediates, we characterize it as a two-state reactivity (TSR) process.28-35
Scheme 2.

Proposed Two-State Reactivity Mechanism for 6c-s Formation From N-Benzoylated Cyclic Amine LH, Ag(I) Salt, and [F–TEDA]2+
A plausible alternative mechanism consistent with literature precedent (predominantly in oxidative photoredox as well as in first-row transition-metal catalysis) may also be operative.15,18,25 This may begin by single-electron transfer from Ag(I) to Selectfluor to form aminium dication radical TEDA2+, Ag(II), and a fluoride ion. H-atom abstraction of the α-C─H bond of the substrate (LH) by aminium radical dication TEDA2+ forms an α-amino radical, which can undergo further oxidation to generate an iminium ion. Our calculations show that this process is highly unfavorable (by 36.4/35.0 kcal/mol). Since both our calculations and our empirical observations8,9 indicate that LH and Ag(I) salt form an adduct [(LH)–Ag(I)] (i.e., 4c-s), we also studied the thermodynamics of the reaction
and found that this reaction is endergonic (by 27.8/26.4 kcal/mol). Furthermore, coordination of the TEDA2+ radical to [(LH)–Ag(II)–F] to form the triplet-state complex 5c-t, discussed above, is exergonic by 14.3/25.7 kcal/mol. Our computational data has therefore enabled us to rule out this alternative mechanism which resembles the pathway depicted in Figure 1C. These data support a formal oxidative addition of Ag(I) to F–TEDA, which proceeds in a stepwise fashion (vide supra).
Mechanism for the Conversion of Iminium Ion Complex 6c-s to Hemiaminal Complex 8c-s.
Even though the mechanism for hemiaminal formation was anticipated to be straightforward, we have nonetheless computed energies and structures of the relevant intermediates and products for the completeness of the discussion. In this regard, iminium ion complex 6c-s, [(L+–Ag)–(HF)–(TEDA)+], undergoes HF → H2O exchange to form [(L+–Ag)–(H2O)–(TEDA)+] (7c-s, Figure 6). This process requires 13.3/2.9 kcal/mol of energy for the HF dissociation (Figure 4) and is exergonic by 11.6/12.0 kcal/mol. In 7c-s, the (L+–Ag) fragment bears one positive charge, and another positive charge is delocalized on the [TEDA]+ fragment. The deprotonation of the Ag-bound water by TEDA and the subsequent C2─OH bond formation is expected to be a facile process. Here, we were not able to locate the transition state associated with the conversion of 7c-s to 8c-s, [(LOH)–Ag](H–TEDA)2+. Calculations show that the overall process for the conversion of iminium ion complex 6c-s to hemiaminal complex 8c-s is exergonic by 10.0/8.3 kcal/mol.
Figure 6.

Calculated iminium ion and hemiaminal complexes 7c-s and 8c-s, along with their important geometry and electronic parameters (distances are in Å; Mulliken charges, Q, are in ∣e∣)). For more details, see Figure S4 in the Supporting Information.
A close examination of the calculated Mulliken charges supports the characterization of 8c-s as a [(LOH)–Ag(I)](H–TEDA)2+ complex (Figure 6).58
Fluorination of Hemiaminal Complex 8c-s.
As illustrated in Figure 1, hemiaminal B (i.e., LOH in Figures 6 and 7) can be converted to the final fluorinated products via two competing pathways: homolytic β-C─C cleavage (path A, Figure 1E) or loss of the aldehyde group (by oxidation to the carboxylic acid and decarboxylation; path B, Figure 1F).
Figure 7.

Computed structures for 9c-s, 10c-s, and 10c-t, along with their geometry and electronic parameters (distances are in Å; Mulliken charges, Q, and spin densities, S, are in ∣e∣). Energies of each step of the reaction are provided as ΔH/ΔG in kcal/mol. For more details, see Figure S5 in the Supporting Information.
Homolytic C–C Cleavage Pathway (Path A).
Path A is proposed to be initiated by an (H–TEDA)2+ → (F–TEDA)2+ exchange that converts hemiaminal complex 8c-s to intermediate 10c-s [i.e., [(LOH)–Ag](F–TEDA)2+, Figures 7 and 8]. This process is endergonic by 5.2 kcal/mol and may proceed via either the dissociation of (H–TEDA)2+ and the coordination of (F–TEDA)2+ (i.e., stepwise) or a concerted (H–TEDA)2+ → (F–TEDA)2+ exchange pathway. Calculations show that the stepwise pathway requires 10.3 kcal/mol free energy for the dissociation of (H–TEDA)2+. This energy value can also be taken as an upper limit for the concerted (H–TEDA)2+ → (F–TEDA)2+ exchange. Thus, the free energy required for 8c-s → 10c-s (at a maximum of 10.3 kcal/mol) is unlikely to impact the overall outcome of the reaction.
Figure 8.

Energy profile of reaction 8c-s + [F–TEDA]2+ → LOF (or 3) + Ag(I) + [H–TEDA]2+. Energies given in parentheses are relative to the dissociation limit of [(LH)–Ag(I)], 4c-s, +2[F–TEDA]2+.
Ring opening from 10c-s could, in principle, proceed through either direct H─F bond formation on the singlet-state energy surface or a two-state reactivity (TSR) mechanism initiated by a singlet-to-triplet seam of crossing (i.e., via the (S–T) transition). Our studies indicate that HF formation in 10c-s via a TSR mechanism is more favorable and requires about 11.0–12.0 kcal/mol of free energy (Figures 7 and 8). As depicted in Figure 7, the transition from 10c-s to 10c-t results in not only a ground electronic state change but also significant geometry alterations: in 10c-t, the Ow─H and N2─F bonds are significantly elongated, and the H─F bond (0.997 Å) and the Ag─Ow bond (2.168 Å) are almost fully formed. Spin, charge density, and geometry analyses of 10c-t show that the 10c-s → 10c-t transition leads to simultaneous F-atom and H-atom coupling to form HF along with dicationic TEDA2+ and [(LO)–Ag] radicals. In the TEDA2+ radical, a 0.75 ∣e∣ unpaired electron is located on the proximal N2 center. Importantly, in the [(LO)–Ag] fragment, the Ag center has acquired more positive charge (compared to that in 10c-s) and has a 0.25 ∣e∣ unpaired α-spin. These findings are indicative of Ag being partially oxidized in complex 10c-t. On the basis of these analyses, we characterize 10c-t as diradical intermediate [(LO)–Ag(II)]•–(HF)–(TEDA)2+•.
In the next stage, intermediate 10c-t is converted to alkoxide complex 11c, which is {[(LO)–Ag]–(FH)–(TEDA)}2+ featuring a hydrogen and TEDA interaction. This transition is expected to be a facile process since it mostly involves the breaking and formation of weak O…HF and FH─TEDA hydrogen bonds, respectively. Therefore, we assume the energy difference between the 10c-s and 10c-t intermediates to be an approximate energy (11 to 12 kcal/mol) required for H─F bond formation between Selectfluor (i.e., F–TEDA2+) and AgBF4-coordinated hemiaminal (LOH). Notably, the open-shell singlet and triplet electronic states of resulting adduct 11c are very similar in energy. As illustrated in Figure 8, overall 8c-s → 11c-s is endergonic by 9.8/11.4 kcal/mol.59
Interestingly, a comparison of the 5c-s → 5c-t and 10c-s → 10c-t transitions shows that the 5c-s → 5c-t transition is a F-atom transfer from (F–TEDA)2+ to the Ag center (or formal N–F oxidative addition) with an attendant electron transfer from the substrate to the AgF unit. This results in the oxidation of both the Ag center and the substrate [from LH to LH+]. In contrast, the 10c-s → 10c-t transition is a simultaneous HF formation with only a slight oxidation of the Ag center.
The conversion of alkoxide intermediate 11c-s to the final alkyl fluoride product (i.e., LOF or 3; Figure 8) is a complex multicomponent process. It may occur through several pathways, including (a) direct reaction with another equivalent of Selectfluor
| (1) |
and/or (b) directly by the HF byproduct
| (2) |
Since the reaction depicted in eq 1 is less exergonic than the reaction in eq 2 (by 0.1/20.1 kcal/mol vs 21.2/51.2 kcal/mol, calculated relative to complex 11c-s), below we discuss eq 2 in detail and include all calculated data for the reaction depicted in eq 1 in the Supporting Information (Figures S7 and S8). Path 3 depicts direct conversion of 11c-s by treatment with an equivalent of selectfloro. This pathway resembles the reaction depicted in Figure 1E. Unfortunately, we were not able to locate the transition state associated with this radical fluorination process.
The reaction depicted in eq 2 can proceed through multiple pathways. One of them is a stepwise or dissociative–associative pathway (path 1), which is initiated by the dissociation of [(FH)–TEDA]+ (i.e., by the following reaction):
| (3) |
Our calculations show that the dissociation of [(FH)–TEDA]+ from 11c-s is endergonic by 30.8/13.1 kcal/mol and leads to the formation of 12c-t and [(FH)–TEDA]+. Complex 12c-t, [(LO)+–Ag], where fragment (LO) bears almost one positive charge, has a triplet ground electronic state. Close analysis shows that most of the 1.70 ∣e∣ unpaired spin of the fragment (LO) is localized on the O atoms (0.72 ∣e∣ and 0.37 ∣e∣ on the Ow and Oamide, respectively). The C2 and C3 centers also have acquired unpaired spins of 0.08 ∣e∣ and 0.17 ∣e∣, respectively. Importantly, the C2─C3 bond is elongated from 1.545 to 1.613 Å upon going from intermediate 11c-s to 12c-t. Thus, the oxidation of the (LO) unit of 11c-s is critical for the facile C2–C3 selective deconstructive fluorination of N-benzoylated cyclic amine 1. In the next step, the [(FH)–TEDA]+ fragment coordinates to the C3 center of 12c-t and initiates the heterolytic cleavage of HF by the C3 center of 12c-t and the TEDA+ monocation. Reaction 12c-t + [(FH)–TEDA]+ → 13c-s + [H–TEDA]2+ is calculated to be highly exergonic (by 61.1/70.1 kcal/mol). However, it is associated with an additional energy barrier at the triplet–singlet seam of the crossing transition state. This transition state was not located because path 1 is energetically more uphill than path 2, which does not require the dissociation of [(FH)–TEDA]+ from 11c-s and has a lower associated energy barrier.
Indeed, path 2 starts by translation of the [FH–TEDA] fragment to the vicinity of C3 followed by fluoride–C3 coupling and C3─C2 bond cleavage via the fluoride-transfer mechanism. All of our efforts to identify relevant intermediates and transition states and their associated energies were unsuccessful. The scanning of the potential energy surface for F─C3 bond formation in 11c-s led to the direct formation of [(LOF)–Ag(I)], (13c-s), and [H–TEDA]2+ species with a low associated energy barrier (Figure S10 in the Supporting Information). The overall reaction
| (4) |
is calculated to be exergonic by 30.3/57.0 kcal/mol. The dissociation of Ag(I) from [(LOF)–Ag(I)] completes the formation of alkyl fluorinated product LOF (or 3), which requires only 5.8 kcal/mol of free energy.
On the basis of our computational findings, we propose a radical fluorination from 11c-s (path 3), or electron transfer followed by fluoride trapping by the nascent cation occurs (i.e., for the transformation of 11c-s to LOF). This mechanistic scenario is consistent with that previously postulated by Sammis and co-workers.60
The data presented thus far shows that both stages of the deconstructive fluorination of LH (or 1) (i.e., the hemiaminal formation (Figure 1A-D) and the subsequent β-C–C cleavage and fluorination (Figure 1E, path A)) proceed via TSR mechanisms (triggered by the (S–T) seam of crossing). Since electron transfer from the substrate to the Ag center is vital to the success of these reactions, the electronic properties of the N-benzoyl group of LH is expected to impact the nature of the reaction. Therefore, we extended our studies to substrates bearing para-NO2 and NH2 substituents on the benzoyl group. The calculated structures of (p-X)-(5c-s), (p-X)-(5c-t), (p-X)-(10c-s), (p-X)-(10c-t), (p-X)-TS1(H–F form)-t, and (p-X)-TS1(H–F form)-t are given in the Supporting Information. Our calculations show that the reaction for (p-NO2)–N-benzoylated cyclic amine will occur through a TSR mechanism, but barriers for both iminium ion formation (i.e., analogous to 5c-s → 5c-t) and the β-C–C cleavage/fluorination (i.e., analogous to 10c-s → 10c-t) increase to 10.7/12.1 and 18.3/16.9 kcal/mol, respectively (Figure 8). Thus, the AgBF4-catalyzed fluorination of cyclic amines bearing an electron-withdrawing para substituent on the N-benzoylated ring requires slightly higher energy barriers but still proceeds via a TSR mechanism.
On the other hand, the presence of electron-donating groups on the N-benzoyl ring (for example, para-NH2 substitution) not only changes the calculated energy barriers but also switches the mechanism of the reaction from TSR to the classical SET. As seen in Figure 8, for a para-NH2-substituted N-protected cyclic amine [(p-NH2)-LH], the triplet electronic state of (p-NH2)-5c is more stable than its singlet electronic state by 14.5/12.9 kcal/mol. Therefore, upon the interaction of (p-NH2)-(4c-s) with Selectfluor simultaneous spin decoupling occurs, and electron transfer from (p-NH2)-LH to (AgF)+ takes place via the classical SET mechanism. The hydrogen-atom-transfer/fluorine-atom-transfer coupling energy barrier at the triplet transition state [(p-NH2)-TS1(H–F form)-t] is only 4.3/3.8 kcal/mol, relative to the triplet state in prereaction complex (p-NH2)-(5c-t).
Similarly, we found that the ground electronic state of (p-NH2)-(10c) is the triplet state, which is 11.2/11.5 kcal/mol more stable than its singlet state. This results in a mechanism switch from TSR to SET in the C–C cleavage/fluorination of the hemiaminal via the ring-opening pathway. However, the calculated hydrogen-atom transfer and fluorine-atom transfer (HAT/FAT coupling) barrier for (p-NH2)-TS2(H–F form)-t is 10.0/10.3 kcal/mol, which is only slightly lower than the 11.0–12.0 kcal/mol barrier assumed for the reaction of LH, where the benzoyl group does not bear any substituents.
On the basis of these computations, we conclude that deconstructive fluorination (via the β C–C cleavage pathway) of electron-poor N-benzoylated cyclic amines has a higher energy barrier and proceeds through a two-state reactivity mechanism. On the contrary, increased electron density on the N-benzoylated cyclic amine may not only slightly enhance its ring-opening fluorination by Selectfluor but may also introduce a mechanism switch into the broadly accepted SET mode.
Deformylative Fluorination Pathway (Path B).
We have also investigated the alternative pathway for C─C bond cleavage/fluorination that begins from the hemiaminal complex {[(LOH)–Ag](H–TEDA)}2+, 8c-s, (i.e., the “deformylative” fluorination pathway). This pathway is initiated by equilibration of the hemiaminal (LOH) to the corresponding aldehyde (Ald; Figure 9), which may occur either directly from complex 8c-s or following the dissociation of (H–TEDA)2+ (i.e., in 9c-s; see Figures 7 and 9). While computations cannot unambiguously support either of these possibilities, they show that the conversion of (LOH) to linear aldehyde (l-Ald) is exergonic by 6.0 kcal/mol in the absence of other coordinating groups and by 2.0 kcal/mol for the Ag-coordinated complex (i.e., complexes 9c-s and 14c-s, in Figure 9).
Figure 9.

Energy surfaces (energies are given as ΔH/ΔG in kcal/mol) of the iminium ion formation and fluorination of hemiaminal via a ring-opening pathway for the unsubstituted (X = H) and para-X substituted N-protected cyclic amines (where X = NO2 and NH2).
Because the interaction of [(LOH)–Ag] and [(Ald)–Ag] with Selectfluor [i.e., (F–TEDA)2+] has a minimal impact on the calculated geometries and energies, we began our analyses from the [(LOH)–Ag] and [(Ald)–Ag] complexes, which possess several isomers that are similar in energy. A few of the energetically most favorable isomeric forms of these species are shown in Figure 9 (Figure S11 in the Supporting Information).
As seen in Figure 9, in [(l-Ald)–Ag], 14c-s, the aldehyde group is coordinated to the Ag center via the amide oxygen. In the lowest-energy isomer of the nonlinear aldehyde (n-Ald) and Ag-salt complex (i.e., 15c-s, iso-1), Ag is coordinated to the oxygen atom (Ow) of the formyl group and the Ph ring of the benzoyl group. This isomer is 3.8–3.9 kcal/mol more stable than iso-2 (i.e., complex 16c-s), where Ag is coordinated to the aldehyde and amide carbonyl groups.
In principle, the formyl group could be oxidized to the corresponding carboxylic acid under the reaction conditions. The mechanism of the Ag(I)-catalyzed decarboxylative fluorination of aliphatic carboxylic acids by Selectfluor has been previously investigated,15,19 and it was established that these processes start with carboxylate coordination to the Ag(I) center followed by oxidation of the resulting Ag-carboxylate by Selectfluor:
| (5) |
| (6) |
To the best of our knowledge, no detailed mechanistic studies on Ag-catalyzed deformylative fluorinations of hemiaminals by Selectfluor have been reported in the literature. In our previous studies, attempts to monitor these processes only led to line broadening in the 1H NMR and the appearance of carboxylic acid and aldehyde.8,9 Therefore, the direct deformylative pathway cannot be ruled out. To investigate this possibility, we studied the Ag-catalyzed deformylative fluorination of aldehydes by Selectfluor initiated from [(n-Ald)-Ag], 15c-s.
Our analyses indicate that the deformylative fluorination of 15c-s may proceed through several pathways. (See Figure 11 and the Supporting Information for more details.) We found that a pathway initiated by H-atom abstraction from 15c-s by previously generated radical dication TEDA2+ (Figure 10) has the lowest associated energy barrier. The initial step of this pathway (eq 7)
| (7) |
occurs with almost no associated energy barrier and is exergonic by 20.3/20.0 kcal/mol. This result is in line with the findings of MacMillan and co-workers,61 who have demonstrated a facile aldehyde H-atom abstraction by a quinuclidinium radical cation.
Figure 11.

Calculated representative structures, along with their key geometry parameters (distances are in Å), for the proposed deformylative fluorination pathway. Energies (in kcal/mol) are provided relative to the prereaction complex as ΔH/ΔG.
Figure 10.

Calculated representative structures of hemiaminal–AgBF4, 9c-s, linear aldehyde–AgBF4, 14c-s, and two nonlinear aldehyde–AgBF4 complexes, 15c-s and 16c-s, along with their key geometry parameters (distances are in Å) and relative energies given as ΔH/ΔG in kcal/mol.
In the resulting complex (17c-d), the unpaired electron is localized on the CO fragment (by 0.65 ∣e∣), whereas the C3 center bears only 0.12 ∣e∣ unpaired spin. From this radical intermediate, the loss of a CO molecule (that requires only 11.4 kcal/mol of free energy, see Figure 10) leads to [L–Ag], (18c-d). In intermediate 18c-d, one unpaired electron is distributed between the C3 and Ag centers (0.71 ∣e∣ and 0.23 ∣e∣, respectively), indicating that the C3 center is slightly oxidized and the Ag(I) center is slightly reduced. (See Figure 10c in the Supporting Information for more details.)
On the basis of the preceding discussion, it is anticipated that the C3 radical center of 18c-d will undergo facile fluorination by another equivalent of Selectfluor. A full scan of the reaction path (using the C3–[F–TEDA]2+ distance as a reaction coordinate) demonstrates that this process has a very small associated energy barrier and proceeds with the participation of the Ag center. However, we were not able to locate any intermediates that possess a Ag─F bond (Figures S12 and S13 in the Supporting Information).21 The overall reaction
| (8) |
is exergonic by 67.3/55.2 kcal/mol. On the basis of spin density analyses, we characterize 19c-d as an (LF)–[Ag(I)]–[(TEDA)2+.], which possesses a dicationic TEDA radical. The dissociation of LF (i.e., 9, in Figure 1) from 19c-d is endergonic by 35.7/23.0 kcal/mol (Figure 10).
In Figure 11, we compare the initial steps of the ring-opening (i.e., β-C–C cleavage) and deformylative fluorination of hemiaminal 9c-s by Selectfluor. Overall, the free-energy barrier required for the ring-opening pathway, leading to the alkyl fluorinated product (LOF, or 3), is 11.0 kcal/mol (using the energy span approach62). The initial steps of the deformylative fluorination of the aldehyde intermediate, leading to LF (i.e., 9), have an even smaller free-energy barrier. Since we were not able to identify an energy barrier required for the hemiaminal → aldehyde equilibration (which is expected to be small), here we conclude that both pathways are feasible and the preference of one over the other depends on the reaction conditions and the substrates that are employed.
CONCLUSIONS
Computational studies on the mechanism of the Ag(I)-mediated deconstructive fluorination of N-benzoylated piperidines (LH) described here provide the following evidence:
The first stage of the reaction (i.e., iminium ion formation) is formally a hydride abstraction event and proceeds via subsequent formation of an iminium ion intermediate, [L+–Ag]–HF–[TEDA]+, which is formally a Ag(I)-mediated hydride-abstraction event that occurs in two steps: (a) a formal oxidative addition (OA) of [F–TEDA]2+ to the Ag(I) center that is attended by an electron transfer (ET) from the substrate (LH) to the Ag center (i.e., OA + ET, this process can also be referred to as a fluorine atom coupled electron transfer, FCET) and (b) H-atom abstraction from LH by the Ag-coordinated F atom. The overall process involves lower-lying singlet and triplet electronic states of several intermediates and is therefore best characterized as a two-state reactivity (TSR) event.28-35
The second stage of the reaction is fluorination of the hemiaminal intermediate. This process may occur through either ring opening or deformylative fluorination pathways. We found that a ring-opening fluorination (i.e., via β-C–C cleavage/fluorination) is also a two-state reactivity (TSR) event. However, a competing deformylative fluorination is not a TSR event. Rather, it is initiated by a hemiaminal to aldehyde equilibration, followed by a formyl H-atom abstraction by a TEDA2+ radical dication, decarbonylation, and fluorination of the C3-radical center by another equivalent of Selectfluor. Both fluorination pathways are feasible, and preference for one over the other is subject to the reaction conditions and the substrates that are employed.
Facile oxidation of the substrate is critical for both stages (i.e., the iminium ion formation and hemiaminal fluorination) of the N-benzoylated cyclic amine deconstructive fluorination. We have shown that ring-opening fluorination of the substrates bearing para electron-withdrawing substituents on the benzoyl group has a higher free-energy barrier. On the contrary, substrates bearing electron-donating substituents on the N-benzoyl group enhance ring-opening fluorination by Selectfluor.
The insights presented here are expected to aid in (a) identifying simpler, more efficient protocols for the deconstructive fluorination of N-acylated cyclic amines, (b) elucidating conditions that will effect deconstructive functionalization in aqueous solvent mixtures, and (c) the widespread adoption of this method for late-stage skeletal diversification.
Supplementary Material
Figure 12.

Comparison of the relative free energies of the initial steps of the ring-opening and deformylative fluorination of hemiaminal by Selectfluor.
Scheme 3.

Schematic Presentation of Elementary Reactions Involved in the Proposed Electron Transfer [from Alkoxide to (FH–TEDA)2+], Followed by the Fluoride Trapping (by the C3 Center of the Alkoxy Group) Mechanism of the 11c-s Transformation to [(LOF)–[Ag(I)]
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1700982). R.S. is grateful to the NIGMS (R35 GM130345A) for support of the experimental work that was the basis for this computational study. The authors gratefully acknowledge the use of the resources of the Cherry Emerson Center for Scientific Computation at Emory University. J.B.R. thanks Bristol-Myers Squibb for a graduate fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c13061.
Cartesian coordinates of all reported structures and Figures S1-S13 (PDF)
The authors declare no competing financial interest.
Contributor Information
Jose B. Roque, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Richmond Sarpong, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Djamaladdin G. Musaev, Cherry L. Emerson Center for Scientific Computation, and Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States.
REFERENCES
- (1).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late-Stage Functionalization of Drug-Like Molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (2).Hartwig JF Evolution of C─H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc 2016, 138, 2–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang B; Perea MA; Sarpong R Transition Metal-Mediated C─C Single Bond Cleavage: Making the Cut in Total Synthesis. Angew. Chem., Int. Ed 2020, 59, 18898–18919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Murakami M; Ishida N Potential of Metal-Catalyzed C─C Single Bond Cleavage for Organic Synthesis. J. Am. Chem. Soc 2016, 138, 13759–13769. [DOI] [PubMed] [Google Scholar]
- (5).O’Reilly ME; Dutta S; Veige AS β-Alkyl Elimination: Fundamental Principles and Some Applications. Chem. Rev 2016, 116, 8105–8145. [DOI] [PubMed] [Google Scholar]
- (6).Souillart L; Cramer N Catalytic C─C Bond Activations via Oxidative Addition to Transition Metals. Chem. Rev 2015, 115, 9410–9464. [DOI] [PubMed] [Google Scholar]
- (7).Roque JB; Kuroda Y; Jurczyk J; Xu L-P; Ham JS; Göttemann LT; Roberts C; Adpressa D; Saurí J; Joyce LA; Musaev DG; Yeung CS; Sarpong R ACS Catal 2020, 10, 2929–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Roque JB; Kuroda Y; Göttemann LT; Sarpong R Deconstructive Fluorination of Cyclic Amines by Carbon-Carbon Cleavage. Science 2018, 361, 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Roque JB; Kuroda Y; Göttemann LT; Sarpong R Deconstructive Diversification of Cyclic Amines. Nature 2018, 564, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yu C; Shoaib MA; Iqbal N; Kim JS; Ha H-J; Cho EJ Selective Ring-Opening of N-Alkyl Pyrrolidines with Chloroformates to 4-Chlorobutyl Carbamates. J. Org. Chem 2017, 82, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (11).Feraldi-Xypolia A; Gomez Pardo D; Cossy J Ring Contraction of 3-Hydroxy-3 (trifluoromethyl)piperidines: Synthesis of 2-Substituted 2-(Trifluoromethyl)pyrrolidines. Chem. - Eur. J 2015, 21, 12876–12880. [DOI] [PubMed] [Google Scholar]
- (12).Wang F; He Y; Tian M; Zhang X; Fan X Synthesis of α-Formylated N-Heterocycles and Their 1,1-Diacetates from Inactivated Cyclic Amines Involving an Oxidative Ring Contraction. Org. Lett 2018, 20, 864–867. [DOI] [PubMed] [Google Scholar]
- (13).Huang F-Q; Xie J; Sun J-G; Wang Y-W; Dong X; Qi LW; Zhang B Regioselective Synthesis of Carbonyl-Containing Alkyl Chlorides via Silver-Catalyzed Ring-Opening Chlorination of Cyclo-alkanols. Org. Lett 2016, 18, 684–6877. [DOI] [PubMed] [Google Scholar]
- (14).Zhao H; Fan X; Zhu C Silver-Catalyzed Ring-Opening Strategy for the Synthesis of β- and γ-Florinated Ketones. J. Am. Chem. Soc 2015, 137, 3490–3493. [DOI] [PubMed] [Google Scholar]
- (15).Patel NR; Flowers RA II Mechanistic Study of Silver-Catalyzed Decarboxylative Fluorination. J. Org. Chem 2015, 80, 5834–5841. [DOI] [PubMed] [Google Scholar]
- (16).Michaudel Q; Thevenet D; Baran PS Intermolecular Ritter-Type C–H Amination of Unactivated sp3-Carbons. J. Am. Chem. Soc 2012, 134, 2547–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yayla HG; Wang H; Tarantino KT; Orbe HS; Knowles RR Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O─H Bonds. J. Am. Chem. Soc 2016, 138, 10794–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pitts CR; Bloom S; Woltornist R; Auvenshine DJ; Ryzhkov LR; Siegler MA; Lectka T Direct, Catalytic Monofluorination of sp3 Bonds: A Radical-Based Mechanism with Ionic Selectivity. J. Am. Chem. Soc 2014, 136, 9780–9791. [DOI] [PubMed] [Google Scholar]
- (19).Yin F; Wang Z; Li Z; Li C Silver-Catalyzed Decarboxylative Fluorination of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc 2012, 134, 10401–10404. [DOI] [PubMed] [Google Scholar]
- (20).Huang X; Hooker JM; Groves JT Targeted Fluorination with the Fluoride Ion by Manganese-Catalyzed Decarboxylation. Angew. Chem., Int. Ed 2015, 54, 5241–5245. [DOI] [PubMed] [Google Scholar]
- (21).Brandt JR; Lee E; Boursalian GB; Ritter T Mechanism of Electrophilic Fluorination with Pd(IV): Fluoride Capture and Subsequent Oxidative Fluoride Transfer. Chem. Sci 2014, 5, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhu Q; Yang P; Chen M; Hu J; Yang L A Rapid Method to Aromatic Aminoalkyl Esters via the Catalyst-Free Difunctionalization of C─N Bonds. Synthesis 2018, 50, 2587–2594. [Google Scholar]
- (23).Romero-Ibañez J; Cruz-Gregorio S; Sandoval-Lira J; Hernández-Pérez JM; Quintero L; Sartillo-Piscil F Transition-Metal-Free Deconstructive Lactamization of Piperidines. Angew. Chem., Int. Ed 2019, 58, 8867–8871. [DOI] [PubMed] [Google Scholar]
- (24).Zhang Y; Sun S; Su Y; Zhao J; Li Y-H; Han B; Shi F Deconstructive di-functionalization of unstrained, benzo cyclic amines by C─N bond cleavage using a recyclable tungsten catalyst. Org. Biomol. Chem 2019, 17, 4970–4974. [DOI] [PubMed] [Google Scholar]
- (25).Hua AM; Mai DN; Martinez R; Baxter RD Radical C–H fluorination using unprotected amino acids as radical precursors. Org. Lett 2017, 19, 2949–2952. [DOI] [PubMed] [Google Scholar]
- (26).Ito R; Umezawa N; Higuchi T Unique Oxidation Reaction of Amides with Pyridine-N-oxide Catalyzed by Ruthenium Porphyrin: Direct Oxidative Conversion of N-Acyl-L-proline to N-Acyl-L-glutamate. J. Am. Chem. Soc 2005, 127, 834–835. [DOI] [PubMed] [Google Scholar]
- (27).Strictly speaking, this an electron-transfer process. However, in the organic chemistry literature, such processes are referred to as SET events, analogous to the use of these terms in electrocatalysis and photocatalysis. For consistency, here we also use the term single-electron transfer (SET).
- (28).Schroder D; Shaik S; Schwarz H Two-state reactivity as a new concept in organometallic chemistry. Acc. Chem. Res 2000, 33, 139–145. [DOI] [PubMed] [Google Scholar]
- (29).de Visser SP; Ogliaro F; Harris N; Shaik S Epoxidation of Ethene by Cytochrome P450: A Quantum Chemical Study. J. Am. Chem. Soc 2001, 123, 3037–3047. [DOI] [PubMed] [Google Scholar]
- (30).Klinker EJ; Shaik S; Hirao H; Que L Jr. Two-state reactivity model explains unusual kinetic isotope effect patterns in CH bond cleavage by non-heme oxoiron(IV) complexes. Angew. Chem., Int. Ed 2009, 48, 1291–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Musaev DG; Morokuma K Ab Initio molecular orbital study of the electronic and geometrical structure of MCH2+ and the reaction mechanism: MCH2+ + H2 ↓ M+ + CH4, (M = Co, Rh and Ir). Isr. J. Chem 1993, 33, 307–316. [Google Scholar]
- (32).Musaev DG; Morokuma K Molecular orbital study of the mechanism of Sc+ with methane. Comparison of the reactivity of early and late first-row transition metal cations and their carbene complexes. J. Phys. Chem 1996, 100, 11600–11609. [Google Scholar]
- (33).Musaev DG; Morokuma K Potential energy surface of transition metal catalyzed chemical reactions. Adv. Chem. Phys 2007, 61–128. [Google Scholar]
- (34).Musaev DG; Morokuma K; Koga N; Nguyen KA; Gordon MS; Cundari TR An Ab Initio study of the molecular and electronic structure of CoCH2+ and the reaction mechanism: CoCH2+ + H2. J. Phys. Chem 1993, 97, 11435–11444. [Google Scholar]
- (35).Musaev DG; Koga N; Morokuma K Ab Initio molecular orbital study of the electronic and geometrical structure of RhCH2+ and the reaction mechanism: RhCH2+ + H2 → Rh+ + CH4. J. Phys. Chem 1993, 97, 4064–4075. [Google Scholar]
- (36).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2019. [Google Scholar]
- (37).Hay PJ; Wadt WR Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys 1985, 82, 270–283. [Google Scholar]
- (38).Hay PJ; Wadt WR Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys 1985, 82, 299–310. [Google Scholar]
- (39).Wadt WR; Hay PJ Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys 1985, 82, 284–298. [Google Scholar]
- (40).Becke AD Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A: At., Mol., Opt. Phys 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- (41).Lee C; Yang W; Parr RG Development of The Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (42).Becke AD A New Mixing of Hartree–Fockand Local Density-Functional Theories. J. Chem. Phys 1993, 98, 1372–1377. [Google Scholar]
- (43).Grimme S; Antony J; Ehrlich S; Krieg H A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys 2010, 132, 154104–154122. [DOI] [PubMed] [Google Scholar]
- (44).Becke AD; Johnson ER A Density-Functional Model of the Dispersion Interaction. J. Chem. Phys 2005, 123, 154101–154106. [DOI] [PubMed] [Google Scholar]
- (45).Becke AD; Johnson ER Exchange-Hole Dipole Moment and the Dispersion Interaction. J. Chem. Phys 2005, 122, 154104–154109. [DOI] [PubMed] [Google Scholar]
- (46).Johnson ER; Becke AD A Post-Hartree-Fock Model of Intermolecular Interactions: Inclusion of Higher-Order Corrections. J. Chem. Phys 2006, 124, 174104–174112. [DOI] [PubMed] [Google Scholar]
- (47).Barone V; Cossi M Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- (48).Cossi M; Rega N; Scalmani G; Barone V Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
- (49).Pritchard BP; Altarawy D; Didier B; Gibson TD; Windus TL A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model 2019, 59 (11), 4814–4820. [DOI] [PubMed] [Google Scholar]
- (50).Chai J-D; Head-Gordon M Long-range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (51).The triplet electronic state of this adduct (i.e., 4c-t, see the Supporting Information) is 77.3/74.3 kcal/mol higher in energy, where unpaired α-spins are mostly located in LH.
- (52).Yarkony D Diabolical Conical Intersections. Rev. Mod. Phys 1996, 68, 985–1013. [Google Scholar]
- (53).Since LH has a lower ionization potential as compared to AgBF4, LH is oxidized more readily than the Ag(I) salt upon interaction of with Selectfluor. Consistently, in 5c-s, an (S–T) transition, mostly, engages an intrasubstrate electron-transfer process.
- (54).Font M; Acuna-Pares F; Parella T; Serra J; Luis JM; Lloret-Fillol J; Costas M; Ribas X Nat. Commun 2014, 5, 4373. [DOI] [PubMed] [Google Scholar]
- (55).Ajitha MJ; Pary F; Nelson TL; Musaev DG Unveiling the role of base and additive in the Ullmann-type of arene-aryl C–C coupling reaction. ACS Catal 2018, 8, 4829–4837. [Google Scholar]
- (56).In this TS, as compared to prereaction complex 5c-t, the nascent H1─F bond is shortened to 1.502 Å from 1.943 Å, whereas the C2─H1 and Ag─F bonds are elongated to 1.177 and 2.100 Å from 1.089 and 2.040 Å, respectively. Of note is the shortening of the N1─C2 and N1─C17 bond distances, which is consistent with the changes in spin densities on the N1 and C2 centers. Indeed, the unpaired spin density at N1 is reduced from 0.67∣e∣ to 0.50∣e∣, whereas the spin density at C2 has increased from 0.12∣e∣ to 0.24∣e∣ upon going from 5c-t to TS1(H-F form)-t.
- (57).Beatty JW; Stephenson CRJ Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res 2015, 48, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).As illustrated in Figure 6, for complex 8c-s, H─TEDA is hydrogen bonded to the hemiaminal (LOH) with an Ow---H(TEDA) bond length of 1.659 Å. Also, the Ag(I) center is coordinated to the amide oxygen of LOH with a Ag─O bond length of 2.219Å. Importantly, in going from 7c-s to 8c-s, the N1─C17 bond is shortened from 1.478 to 1.360 Å, whereas the N1─C2 bond is elongated from 1.297 to 1.462 Å. These geometry changes are consistent with hemiaminal formation.
- (59).The +2 total charge of 11c is mostly localized on the TEDA unit (which has + 1.77∣e∣ and 1.75∣e∣ charges in its singlet and triplet states, respectively). Furthermore, the calculated H─TEDA bond distances are 1.073 and 1.101 Å in 11c-s and 11c-t, respectively.
- (60).Rueda-Becerril M; Sazepin CC; Leung JCT; Okbinoglu T; Kennepohl P; Paquin J-F; Sammis GM Fluorine Transfer to Alkyl Radicals. J. Am. Chem. Soc 2012, 134, 4026–4029. [DOI] [PubMed] [Google Scholar]
- (61).Zhang X; MacMillan DWC Direct Aldehyde C–H Arylation and Alkylation via the Combination of Nickel, Hydrogen Atom Transfer, and Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 11353–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kozuch S; Shaik S How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res 2011, 44, 101–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.