

Abstract
The SWI/SNF chromatin remodeling complex, via nucleosome topology modulation, regulates transcription. The SMARCA4 (BRG1) subunit codes for the ATPase energy engine of the SWI/SNF complex. SMARCA4 is a tumor suppressor that is aberrant in ~5–7% of human malignancies. Class I SMARCA4 alterations (truncating mutations, fusions, and homozygous deletion) lead to loss of function while class II alterations (missense mutations) have a dominant negative/gain-of-function effect and/or loss-of function. SMARCA4 alterations typify the ultra-rare small cell carcinomas of the ovary hypercalcemic type (SCCOHT) and SMARCA4-deficient thoracic and uterine sarcomas; they are also found in a subset of more common tumors, e.g., lung, colon, bladder, and breast carcinomas. Germline variants in the SMARCA4 gene lead to various hereditary conditions: rhabdoid tumor predisposition syndrome-2 (RTPS2), characterized by loss-of-function alterations and aggressive rhabdoid tumors presenting in infants and young children; and Coffin-Siris syndrome, characterized by dominant negative/gain-of function alterations and developmental delays, microcephaly, unique facies, and hypoplastic nails of the fifth fingers or toes. A minority of rhabdoid tumors have a germline SMARCA4 variant as do >40% of women with SCCOHT. Importantly, immune checkpoint blockade has shown remarkable, albeit anecdotal, responses in SCCOHT. Additionally, there is ongoing research into BET, EZH2, HDAC, CDK4/6, and FGFR inhibitors, as well as agents that might induce synthetic lethality via DNA damage repair impairment (ATR inhibitors and platinum chemotherapy), or via the exploitation of mitochondrial oxidative phosphorylation inhibitors or AURKA inhibitors, in SMARCA4-aberrant cancers.
Keywords: immune checkpoint inhibitors, precision oncology, SMARCA4, chromatin remodeling, cancer
INTRODUCTION
The switch/sucrose-non-fermenting (SWI/SNF) complex is an evolutionarily conserved ATP-dependent chromatin remodeling complex that plays important roles in DNA repair, transcriptional activation of genes normally repressed by chromatin, differentiation, and organ development.(1) The SWI/SNF complex specifically changes chromatin structure by altering DNA-nucleosome topology, with nucleosomes being a structural unit of chromosomes consisting of a length of DNA coiled around a core of histones (Figure 1)(2).
Figure 1: The SWI/SNF chromatin remodeling complex:
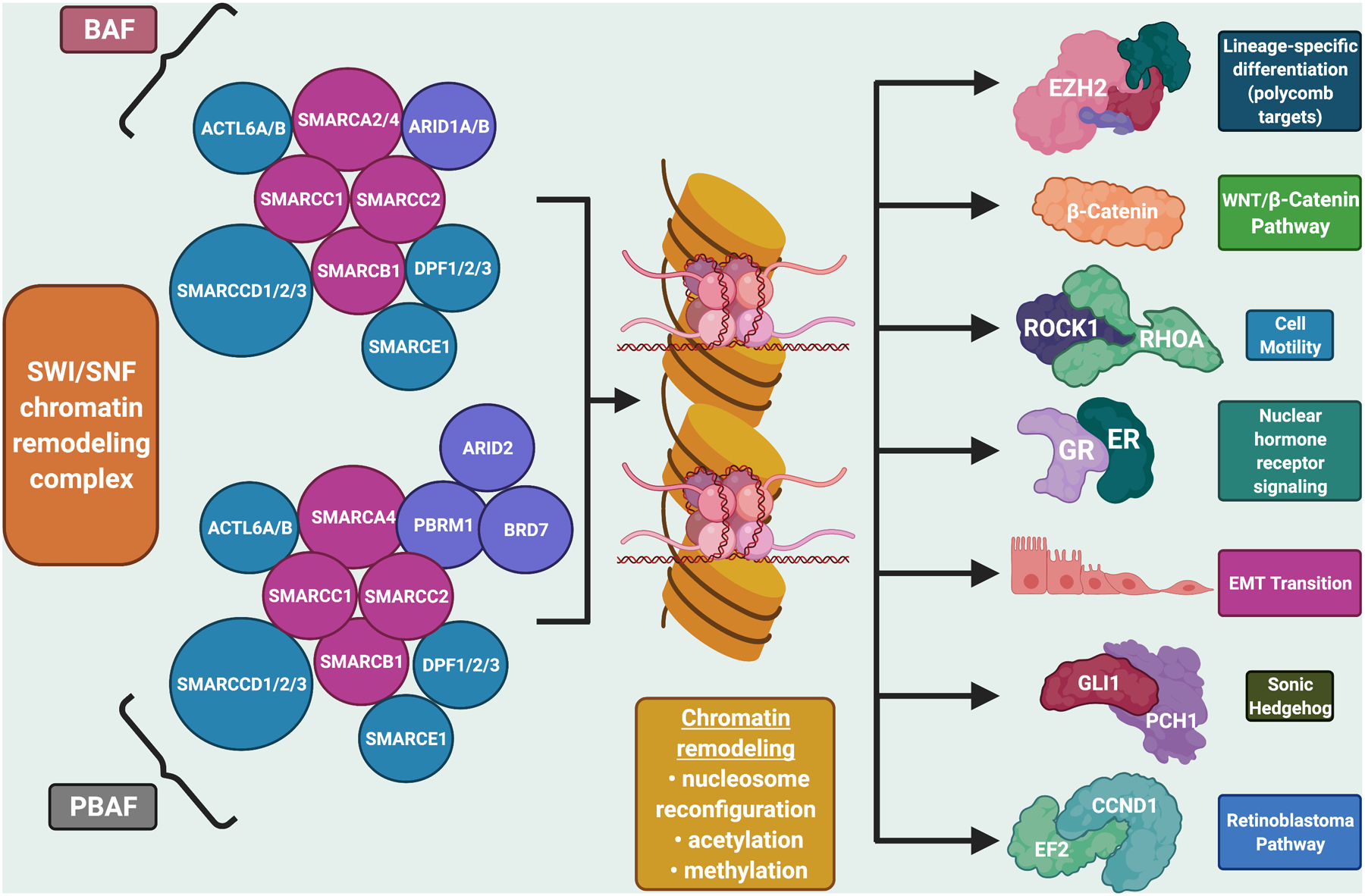
The SWI/SNF complex is a multi-subunit chromatin-remodeling complex that has been implicated in cancer development. The complex interacts with transcription factors to modulate gene expression that contributes to cell differentiation and development. It includes the subunit SMARCA4, which has been shown to function as a tumor suppressor (2). Figure created with biorender.com
Each SWI/SNF complex is comprised of multiple subunits that have an ATP-dependent catalytic unit, either SMARCA4 (BRG1) or SMARCA2 (BRM), as well as a core regulatory subunit such as SMARCB1 (INI1, BAF47), and variable other subunits including ARID1A (Figure 1)(3,4). Recent studies show important tumor suppressor roles for the SWI/SNF complex, with ~20% of human malignancies bearing pathogenic alterations of its subunits.(5–7)
SMARCA4 is also known as transcriptional activator BRG1. It can activate or repress transcription via its ATPase enzymatic role in the chromatin remodeling complex. This protein can also bind BRCA1 (8), as well as regulate the expression of the tumorigenic protein CD44 (a transmembrane glycoprotein implicated in the growth and metastasis of numerous tumors)(9).
Importantly, SMARCA4 is one of the most frequently aberrant chromatin remodeling ATPases in cancer; it is altered in approximately 5% to 7% of all human malignancies (Table 1) (7,10–25). Further, mutations of this gene are also the hallmark of certain cancers. For instance, inactivating mutations in SMARCA4 can be identified in the vast majority of small cell carcinomas of the ovary, hypercalcemic type (SCCOHT), and studies showed high utility of SMARCA4 immunohistochemical (IHC) loss in the diagnosis of this rare tumor that afflicts young women.(20,21,26–30) SMARCA4 deficiency is also a hallmark of thoracic sarcomatoid tumors(22,31) and of malignant rhabdoid cancers of the uterus (undifferentiated uterine sarcoma)(24). Germline SMARCA4 variants occur and cause Coffin–Siris syndrome (characterized by abnormalities of the craniofacial area, resulting in a coarse facial appearance, microcephaly, and developmental disabilities) and RTPS2 (characterized by young onset of various poorly differentiated tumors) (32) (Table 2)(32–37).
Table 1.
Examples of tumors with somatic SMARCA4 alterations and/or loss of SMARCA4 expression by IHC*
| Tumors with Altered SMARCA4 | |||||
|---|---|---|---|---|---|
| Tumor Type | Total, N | N | % | Comments | References |
| Undifferentiated/rhabdoid gastrointestinal tract carcinomas | 13 | 2 | 15.4% | 12/13 cases (92%) showed loss of at least one SWI/SNF component Loss of SMARCB1 (5/13), SMARCA2 (10/13), SMARCA4 (2/13), and ARID1A (2/13) by IHC was observed either in combination or isolated. | (10) |
| Gastric cancer | 1199 | 27 | 2% | Exhibited complete loss (N=6 of 27 patients), reduced (N=9), and heterogeneous (N=12) SMARCA4 patterns by IHC; SMARCA4-altered gastric cancer had divergent, often undifferentiated histomorphology | (11) |
| Lung Cancer | 146 | 8 | 5.5% | Lung adenocarcinoma (SMARCA4 loss by IHC) | (12) |
| 115 | 6 | 5.2% | Lung SCCs (SMARCA4 loss by IHC) | ||
| 122 | 46 | 37% | Primary lung tumors: SMARCA4 negative by IHC (confirmed to be due to inactivating and biallelic mutations by ultra-deep sequencing) | (13) | |
| 60 | 6 | 10% | SMARCA4 loss of protein expression (60 tumors includes 41 adenocarcinomas and 19 squamous cancers | (14) | |
| 37 | 13 | 35% | SMARCA4 mutations in NSCLC cell lines | (15) | |
| 19 | 1 | 5% | SMARCA4 mutations in SCLC cell lines | ||
| 103 | 16 | 15.5% | SMARCA4 loss by IHC in NSCLC | (16) | |
| 93 | 11 | 12% | SMARCA4 loss by IHC in lung adenocarcinomas | (17) | |
| 4813 | 407 | 8% | Two types of SMARCA4 mutations in NSCLC: class 1 (truncating mutations, fusions, and homozygous deletion) and class 2 (missense mutations). Protein loss in class 1 mutations (81% vs. 0%, P < 0.001). SMARCA4 alterations correlated with shorter OS, with class 1 alterations associated with shortest OS (P<0.001). ICIs correlated with better outcomes in patients with SMARCA4-mutant tumors (P = 0.01); class 1 mutations had the best response (P = 0.027). |
(18) | |
| Ovarian Cancer | 360 | 15 | 4% | Clear cell carcinoma (SMARCA4 loss by IHC) | (19) |
| 46 12 17 |
42 12 14 |
91% 100% 82% |
Primary tumors, SMARCA4 loss by IHC SMARCA4 biallelic inactivating mutations 14/17 (82%), SMARCA4 loss (IHC); 9/12, SMARCA4 inactivating mutation |
||
| 285 | 2 | 0.4% | Primary ovarian cancers other than SCCOHT | (21) | |
| SMARCA4-deficient thoracic sarcoma (SMARCA4-DTS) | 30 | 30 | 100% | SMARCA4 and SMARCA2 loss with overexpression of SOX2 by IHC; poorly differentiated tumors with rhabdoid features | (22) |
| 12 | 40 | 30% | Rhabdoid thoracic sarcomas had SMARCA4 loss by IHC | (23) | |
| Endometrial stromal sarcomas (uterine) | 52 | 4 | 8% | Endometrial stromal sarcomas (SMARCA4 loss by IHC) | (19) |
| SMARCA4-deficient undifferentiated uterine sarcoma (malignant uterine rhabdoid tumor) | 4 | 4 | 100% | SMARCA4 loss by IHC. Uterine tumors with morphologic, IHC, and genetic similarities to SCCOHT | (24) |
A complete list of tumor types with altered SMARCA4 can be found in work by Fernando and colleagues(25)
Abbreviations: ICI = immune checkpoint inhibitor; IHC = immunohistochemistry; NSCLC = non-small cell lung cancer; OS = overall survival; SCC = squamous cell carcinoma; SCCOHT=small cell carcinoma of the ovary hypercalcemic type; SCLC=small cell lung cancer; SMARCA4-DTS = SMARCA4-deficient thoracic sarcoma
Table 2:
Germline SMARCA4 alterations
| Name of disorder | Features | % with SMARCA4 versus other alterations | Associated cancers | Comments | References |
|---|---|---|---|---|---|
| Rhabdoid tumor predisposition syndrome (RTPS) | Significantly increased risk of rhabdoid tumors before the age of 3. | SMARCA4 variants: ~5% to 15% (RTPS2) SMARCB1 variants: ~85%–95% (RTPS1) | Rhabdoid tumors can occur in almost any location, most commonly in the central nervous system; >50% in the cerebellum. Other common locations: extracranial extrarenal malignant rhabdoid tumors (eMRT)--heart, bladder, liver, retroperitoneum, head and neck, paravertebral muscles, mediastinum, pelvis; rhabdoid tumor of the kidney (RTK); and SCCOHT (seen only in RTPS2, which is due to germline SMARCA4 alterations (and not in RTPS1 (with germline SMARCB1 alterations) |
These highly aggressive cancers are designated rhabdoid because their cells look like rhabdomyoblasts, which are cells that are normally seen in embryos and develop into muscles. ~35% of pediatric malignant rhabdoid tumors are linked to germline SWI/SNF alterations SMARCA4 mutation type Mostly loss-of-function SMARCA4 alterations |
(32) |
| Coffin-Siris syndrome | Rare and clinically heterogeneous congenital disorder Hallmarks include developmental disability, microcephaly, abnormalities of the fifth fingers or toes, and characteristic facial features. | ~7–11% | Case report with SCCOHT (patient had mild Coffin-Siris syndrome with inactivating SMARCA4 alteration (non-traditional for Coffin-Siris syndrome since most SMARCA4 alterations in this syndrome are dominant negative/gain-of function).) | Associated with mutations in ARID1A, ARID1B, SMARCA4, SMARCA2, SMARCB1 or SMARCE1 or de novo SOX11 mutations SMARCA4 mutation type Mostly non-truncating (either missense or small in-frame deletions) within the highly conserved ATPase/helicase domain, thus suggesting dominant-negative or gain-of-function effects (though some of these mutations may also show loss of function). |
(33–36) |
| SCCOHT | Rare and very aggressive type of undifferentiated ovarian cancer | Most patients have SMARCA4 alterations; ~43% germline |
Mostly affects young women (mean age at diagnosis = 24 years). Morphologically similar to rhabdoid tumors. Ultra-rare, aggressive cancer. Also known as malignant rhabdoid tumor of the ovary SMARCA4 mutation type: Generally, loss-of-function SMARCA4 mutations. |
(37) |
Abbreviations: SCOOHT = small cell carcinoma of the ovary, hypercalcemic type; RTPS = rhabdoid tumor predisposition syndrome type 2
Herein, we review the landscape of SMARCA4 abnormalities, their association with cancer, and the emerging treatment implications of these gene alterations suggesting sensitization to certain therapies, such as immune checkpoint inhibitors (ICIs).
The SMARC family
The SWI/SNF-related, matrix-associated, actin-dependent regulators of chromatin (SMARC), also called BRG1-associated factors, are components of human SWI/SNF-like chromatin-remodeling protein complexes. SMARC family members include SMARCA4 (BRG1), SMARCA2 (BRM), SMARCB1, SMARCC1, SMARCC2, SMARCD1, SMARCD2, SMARCD3, SMARCE1. SMARCA4 maps to chromosome 19p13.2 (https://www.omim.org/entry/603254)
SMARCA4 structure and function
The SMARCA4 gene encodes a transcriptional activator protein, which has also been called BRG1 (Figures 2). The SMARCA4 protein features a bromodomain and helicase/ATPase activity (Figure 3). A bromodomain is an approximately 110 amino acid protein domain that recognizes acetylated lysine residues, such as those on the N-terminal tails of histones; these domains play a key role in regulating gene transcription. Helicases are enzymes that bind and can remodel nucleic acid or nucleic acid protein complexes. ATPases are enzymes that catalyze the hydrolysis of a phosphate bond in adenosine triphosphate (ATP) to form adenosine diphosphate (ADP); they harness the energy released from the breakdown of the phosphate bond and use it to execute other cellular reactions.
Figure 2: SMARCA4 is a multifunctional tumor suppressor.
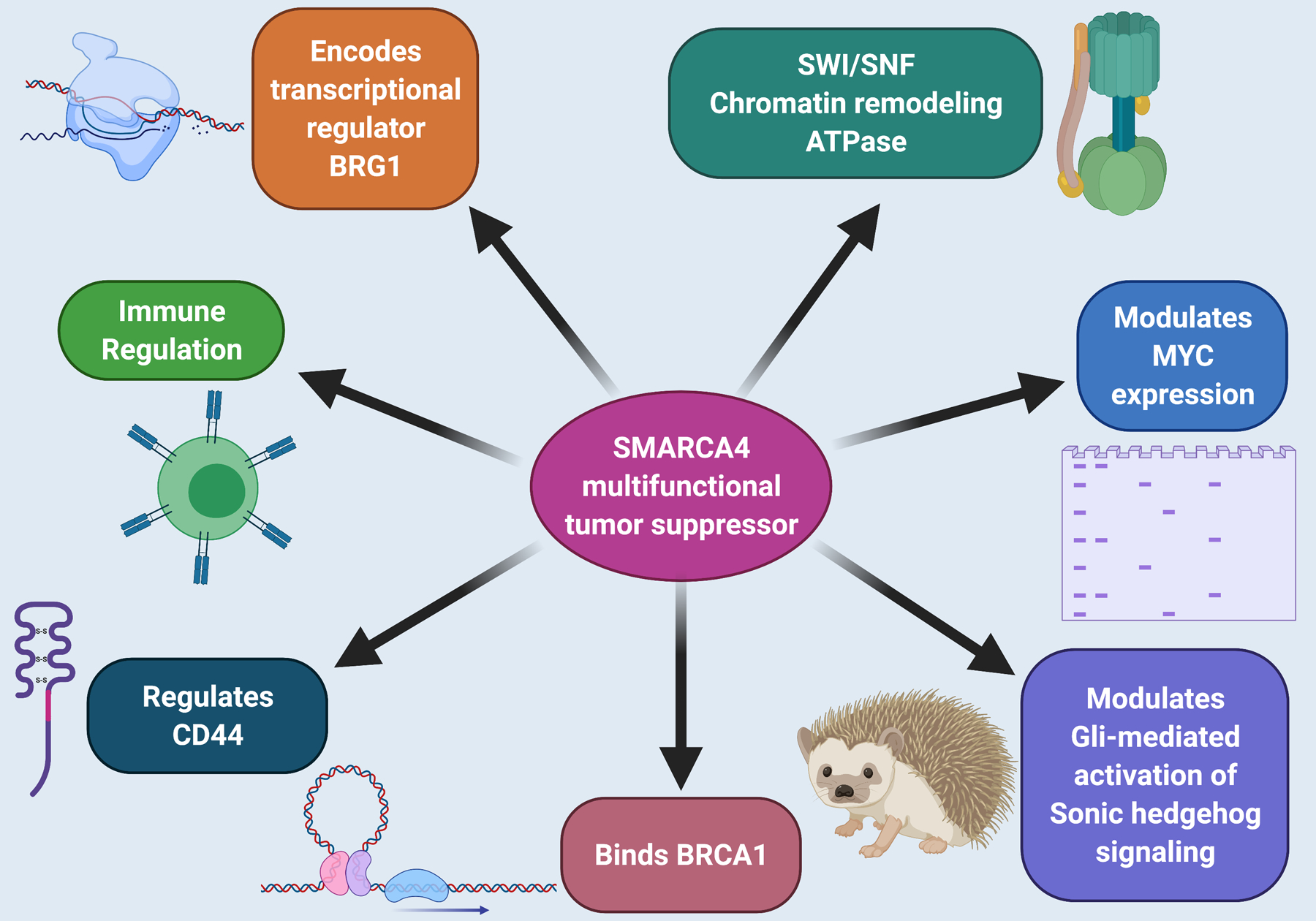
Its cancer-relevant functions include, but are not limited to, binding BRCA1, modulating MYC and sonic hedgehog expression, and regulating transcription via its role in the SWI/SNF chromatin remodeling complex. However, SMARCA4 has pleiotropic functional effects and regulates multiple additional transcriptional pathways and biologies. Figure created with biorender.com
Figure 3: SMARCA4 is the core catalytic subunit (ATPase) of the SWI/SNF chromatin remodeling complex.
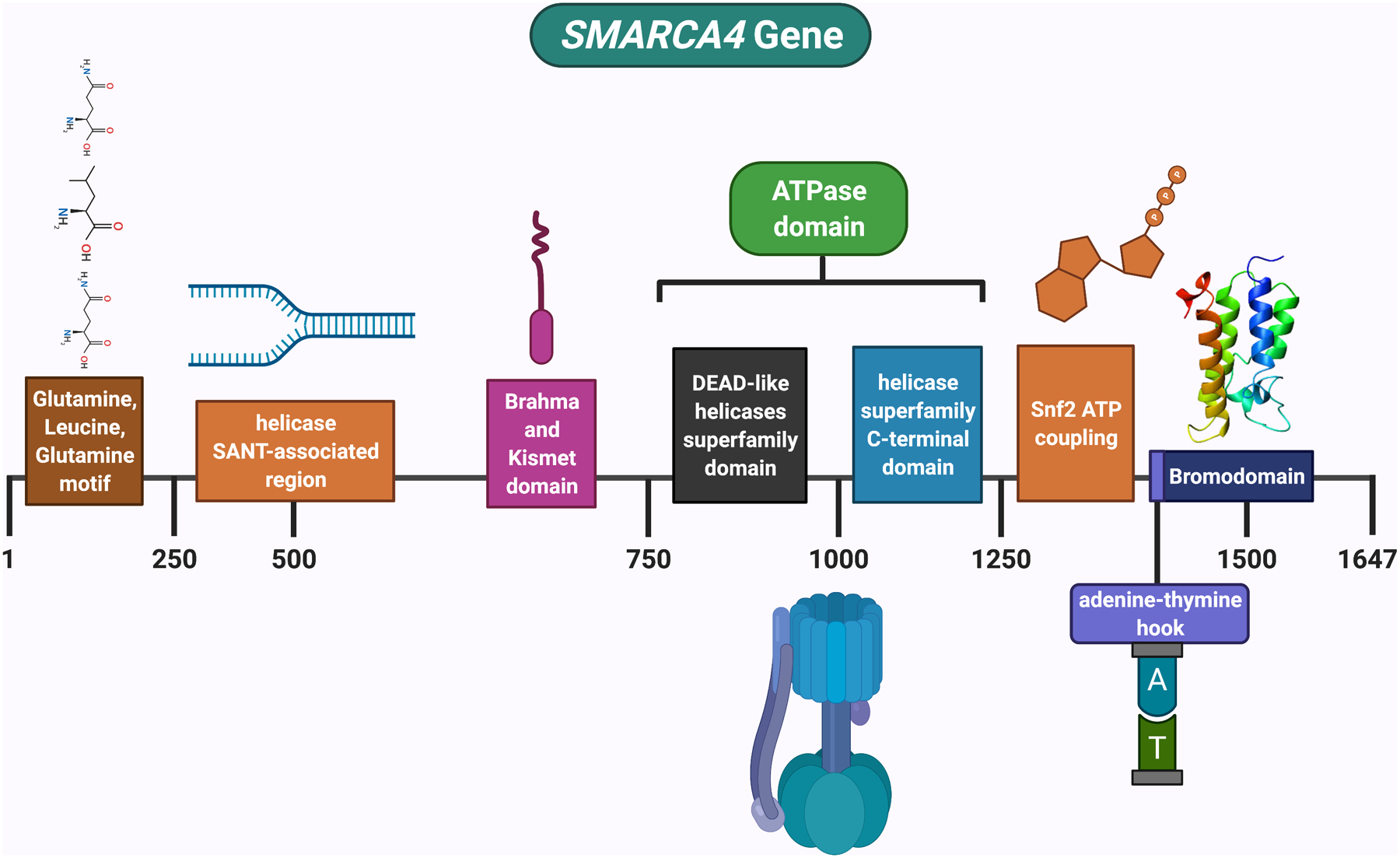
The ATPase utilizes ATP to generate energy that is critical for the nucleosome remodeling function of the complex. SMARCA4 has a bromodomain, which is a domain that recognizes acetylated lysine residues, such as those on the N-terminal tails of histones; these domains regulate gene transcription. It also has a helicase, which is an enzyme that binds and can remodel nucleic acid or nucleic acid protein complexes. Figure created with biorender.com
SMARCA4 (BRG1) forms one subunit of several different protein groupings designated SWI/SNF protein complexes. SWI/SNF complexes regulate gene activity/expression by the chromatin remodeling process. Chromatin is the arrangement of protein and DNA that packages DNA into chromosomes. The architecture of chromatin can be remodeled to adjust how tightly DNA is parceled. Chromatin remodeling is one of the critical manners in which gene expression is controlled during development. When DNA is tightly packed, gene expression is dampened as compared to when DNA is loosely packed. SMARCA4 uses ATP to provide energy for chromatin remodeling.
Through their ability to regulate gene activity, SWI/SNF complexes are involved in many cellular functions (Figures 1 and 2): replicating DNA; modulating the growth, division, and differentiation of cells; and repairing damaged DNA. Via these processes, the SMARCA4 protein acts as a tumor suppressor (Figure 2). Having functional SMARCA4 is also important for development past the pre-implantation stage.(38) SMARCA4 plays a role in the growth of smooth muscle of the heart and gastrointestinal tract.(39) Additionally, SMARCA4 is required for Gli-mediated transcription activation in Sonic hedgehog (Shh) signaling.(40) Mutations of SMARCA4 correlate with context-dependent expression changes at MYC genes, indicating that the SMARCA4 and MYC proteins are functionally related.(41,42) Cell lines lacking SMARCA4 do not respond to retinoic acid or glucocorticoids, while restoration of SMARCA4 restores sensitivity.(42) Finally, as mentioned earlier, SMARCA4 binds BRCA1(8), and regulates the expression of the tumorigenic transmembrane glycoprotein CD44(9).
SMARCA4 alterations and cancer
The SWI/SNF ATPase SMARCA4 is one of the most frequently mutated chromatin remodeling ATPases in cancer.(43) Mutations are enriched at highly conserved ATPase sequences, which reside on important functional surfaces such as the DNA-binding surface or the ATP pocket.(41) Overall, SMARCA4 is altered in about 5–7% of all malignancies. Table 1 lists some of the tumors best explored in the context of SMARCA4 genomic alterations or loss of expression. Additional tumors in which SMARCA4 alterations have been identified include, but are not limited to, lung cancer, colon adenocarcinoma, bladder urothelial carcinoma, and invasive breast ductal carcinoma.(44)
There are two main categories of SMARCA4 alterations (18)
-
→
Class 1 mutations – truncating mutations, fusions, and homozygous deletion (loss of function usually associated with protein loss)
-
→
Class 2 mutations – missense mutations (postulated to have dominant negative or gain of function effects, but some reports suggest loss of function, especially in lung cancer)(25) Dominant-negative activity may be implicated in the context of a wild-type SMARCA4 allele (when present as heterozygous mutations) or dominant-negative activity with SMARCA2. Missense SMARCA4 mutations can also result in loss of accessibility and loss of chromatin remodeling activity.(41,45)
Rhaboid tumors:
Many of the SMARCA4-altered tumors have rhabdoid features defined by characteristic large cells with eccentrically located nuclei and abundant eosinophilic cytoplasm. Rhabdoid tumors are rare, highly aggressive cancers that emerge most frequently in the brain or in the kidneys; they affect primarily infants and young children between the ages of 1 and 3 years old, but have also been identified in adults. About one-third of rhabdoid tumors are linked to germline SWI/SNF variants, usually in SMARCB1 and less commonly in SMARCA4.(46)
Small cell carcinoma of the ovary, hypercalcemic type, (SCCOHT):
SCCOHT is the most common undifferentiated ovarian cancer that afflicts women aged under 40 years of age. However, it is still very rare. The young women afflicted with SCCOHT usually present with symptoms related to a pelvic mass. The age range at diagnosis is quite wide (7 months to 56 years, with an average age of ~24 years). About 60% of patients with SCCOHT have hypercalcemia. SCCOHT is an aggressive cancer, with long-term survival rates of only~30% in early-stage cases.
SCCOHT is a monogenic illness, harboring somatic and germline SMARCA4 variants. It has morphological and molecular resemblance to malignant rhabdoid tumors, which are often triggered by variants in a related SWI/SNF gene--SMARCB1. Overall, inactivating mutations in the SMARCA4 gene are seen in 75 to 100% of SCCOHT cases and accompanied by SMARCA4 protein loss. In contrast, only about 0.4% (2/485) of other primary ovarian tumors have similar changes in SMARCA4.(21,47)
The incidence of SMARCA4 germline variants is high, causing ~43% of SCCOHTs. Women with germline deleterious variants in SMARCA4 likely have a clinically important risk of SCCOHT up to the age of ~60 years old, particularly in the context of a positive family history. Germline SMARCA4 genetic testing is recommended for all affected women with SCCOHT. Additional cascade testing of at-risk family members upon identification of germline deleterious SMARCA4 variants should be performed.(48)
In light of the fact that surveillance is of unclear benefit, risk-reducing bilateral salpingo-oophorectomy is indicated in unaffected adult women with germline pathogenic SMARCA4 variants and a positive family history.(48) Responses to ICIs have been reported.(48,49)
SMARCA4-deficient thoracic sarcoma (SMARCA4-DTS) (22,23):
SMARCA4-DTS is a recently described aggressive entity with specific genomic alterations in SMARCA4. It represents a distinct subset of thoracic sarcomas with undifferentiated rhabdoid morphology. These sarcomas usually occur in the 30- to 50-year-old age group, with male predominance and a smoking history. Tumors are generally large compressive masses located in the mediastinum, pleura, and/or lung. Median overall survival has been only ~6 months. Pathological diagnosis of this tumor is challenging because of the morphological features with poor differentiation that often mimic undifferentiated carcinoma or carcinoma of unknown primary.
SMARCA4-DTS has a unique biologic signature: co-loss of SMARCA4 and SMARCA2 with overexpression of SOX2. SMARCA4 loss by IHC varies in studies from 30% to 100% of cases. Durable responses to the ICI pembrolizumab have been documented.(50) Patients do not generally have evidence of germline transmission.(31)
SMARCA4-deficient uterine sarcoma (24):
SMARCA4-deficient uterine sarcomas (subset of malignant rhabdoid tumor of the uterus) are rare uterine malignant neoplasms with IHC, morphologic, and genomic similarities to SCCOHT. Sheets of large atypical epithelioid cells with prominent rhabdoid morphology, indistinguishable from the large cell variant of SCCOHT are observed. Median age of onset is about 51 years old. These cancers are aggressive, with a median survival of only ~7 months.
At the molecular level, the tumors have SMARCA4 loss by IHC due to SMARCA4 gene mutations. Germline SMARCA4 variants have been reported in SMARCA4-deficient uterine sarcomas.(51)
Hereditary Syndromes with SMARCA4 alterations
Since the SWI/SNF complex plays a central role in gene expression, it is not surprising that there is an association between variants in the SWI/SNF complex, malignant neoplasms and developmental disorders. Germline variants in the SWI/SNF complex have now been shown to be a driving force for several types of cancers and other disorders.(52)
Germline variants in SMARCA4 have been found in RTPS2, and Coffin-Siris syndrome (Table 2)(32–37). Moreover, ~35% of rhabdoid tumors are associated with germline SWI/SNF variants (mostly in SMARCB1 and less commonly in SMARCA4), and almost half of women with SCCOHT have germline SMARCA4 variants.(46)
The cancer risk in individuals with germline SMARCA4 pathogenic variants remains unclear, but it is likely high. For instance, only one publication reports a female with a SMARCA4 germline variant who remained cancer-free past her sixth decade(53); however, there is also a publication bias for cancer-positive cases that confounds risk assessment.
Most rhabdoid tumors have been associated with SMARCB1 loss-of-function mutations, but they can also be caused by SMARCA4 loss-of-function mutations (leading to non-expression of their respective proteins). In general, prognosis is worse for patients with SMARCA4-related malignancies. To differentiate between the two variant types, rhabdoid-neoplasm related hereditary disorders have been separated into two categories:
-
→
RTPS1, for patients who carry a germline variant in SMARCB1;
-
→
RTPS2, for patients who carry a germline variant in SMARCA4.
Germline carriers are more at risk of developing second primary tumors. Females that carry the SMARCA4 germline variant have a higher risk of developing SCCOHT. This has led to the suggestion that SCOOHT be considered part of the RTPS family of tumors.
Rhabdoid tumor predisposition syndrome 2 (RTPS2)(32,54,55):
RTPS’s main feature is a high risk of developing rhabdoid tumors, which are lethal cancers presenting mostly in infants and preschool children. The diagnosis of RTPS2 is established by one or more features: (i) a germline heterozygous deleterious variant in SMARCA4; (ii) multiple SMARCA4-deficient malignancies; (iii) a proband with a rhabdoid tumor; and/or (iv) a family history of rhabdoid malignant neoplasm.
Rhabdoid tumors most commonly afflict the central nervous system (i.e., atypical teratoid/rhabdoid tumor [AT/RT]), with more than half arising in the cerebellum. These cancers can also arise as extracranial extrarenal malignant rhabdoid tumors, such as rhabdoid tumors of the heart, bladder, liver, retroperitoneum, head and neck, paravertebral muscles, mediastinum, pelvis, rhabdoid tumor of the kidney, and SCCOHT (also known as malignant rhabdoid tumor of the ovary).
Because RTPS2 is ultra-rare, the standard of care for management is not firmly established. However, intensive surveillance from birth is needed. Most patients are treated with aggressive chemotherapy, surgery, and radiotherapy. Preventative bilateral ovary removal may be suggested, perhaps after childbearing or earlier.
Individuals diagnosed with SMARCA4-related RTPS2 generally inherited a deleterious variant from an unaffected parent. Since RTPS2 can be passed on in an autosomal dominant fashion, each offspring of an individual with a germline SMARCA4 pathogenic variant has a 50% chance of inheriting the pathogenic variant.(32) Even so, penetrance appears to be incomplete, and the RTPS-related tumors can differ even within the same family. Pre-implantation genetic testing/prenatal screening are feasible if the deleterious family variant has been identified.
Coffin–Siris syndrome (CSS):
Coffin-Siris is an ultra-rare and clinically heterogeneous congenital disorder with the main characteristics being developmental disability, microcephaly, hypoplastic nails of the fifth fingers or toes, and distinct facial features.
It is caused by germline variants in different subunits of the ATP-dependent SWI/SNF chromatin remodeling complex--ARID1A, ARID1B, SMARCA4, SMARCA2, SMARCB1 or SMARCE1 as well as SOX11.(33–36) SMARCA4 is mutated in ~7–11% of patients. Most SMARCA4 germline alterations that have been reported in Coffin-Siris syndrome patients are non-truncating (either missense or small in-frame deletions) clustered within the highly conserved ATPase/helicase domain, thus suggesting dominant-negative or gain-of-function effects. In contrast, most SCCOHTs are due to biallelic germline and/or somatic inactivating (nonsense or frameshift) alterations causing complete loss of SMARCA4 expression. A case report of a patient with mild Coffin-Siris syndrome who developed SCCOHT as a teenager showed an inactivating SMARCA4 variant, which is, as mentioned, non-traditional for this syndrome.(33)
Coffin-Siris syndrome appears to follow an autosomal dominant pattern of inheritance. However, the condition is not usually inherited from an affected parent, but rather occurs from de novo mutations that likely take place during the embryonic period.
Potential Therapeutic Targeting
Because tumor suppressor loss is not directly druggable, investigators have pursued therapeutic vulnerabilities that exploit concomitant changes in gene expression and signaling pathways. In the case of SMARCA4-alterated cancers, immunotherapy with ICIs has emerged as a promising treatment modality (Table 3)(18,48–50,56–61). SMARCA4 alterations result in abnormal SWI/SNF complexes; such aberrant chromatin remodeling complexes can influence the transcription of interferon-stimulated genes important for immune responsiveness, as well as the differentiation, activation and recruitment of several immune cell types.(62) Multiple other signals have also been investigated for potential pharmacologic intervention.
Table 3:
Examples of SMARCA4 alterations in cancer treated with immunotherapy (immune checkpoint inhibitors)
| Disease | Report type | Immunotherapy | Outcome | Comment | References |
|---|---|---|---|---|---|
| POSITIVE OUTCOME | |||||
| SMARCA4-deficient thoracic sarcoma | Case report | Pembrolizumab | PR after first dose | PD-L1 positive on tumor cells = 60% | (56) |
| SMARCA4-deficient thoracic sarcoma | Case report | Pembrolizumab | PR, ongoing at 11+ months | PD-L1 negative, TMB low | (50) |
| SMARCA4-altered NSCLC | Retrospective review of patients treated at Sloan Kettering | ICIs | ICI treatment correlated with improved outcomes (P = 0.01) Class 1 mutations (truncating mutations, fusions, and homozygous deletion—associated with protein loss) had the best response |
SMARCA4 alterations found in ~10% of NSCLC and associated with poor prognosis | (18) |
| SMARCA4-mutated NSCLC (lung adenocarcinoma) | Case report | Nivolumab | PR for 14+ months | TMB very high at 396 mutations/mb; PD-L1 negative by IHC Patient’s tumor had failed to respond to three prior lines of therapy |
(57) |
| SCCOHT | Case series of four patients | ICIs | All patients responded to ICI
|
8 of 11 cases demonstrated PD-L1 expression (IHC) with strong associated T-cell infiltration | (48,49) |
| NEGATIVE OUTCOME | |||||
| SMARCA4-deficient small-cell lung carcinoma | Case report | Nivolumab | Hyperprogressive disease | (58–61) | |
Abbreviations: CR = complete remission; ICI= immune checkpoint inhibitor; NSCLC = non-small cell lung cancer; PR = partial response; SCCOHT = small cell carcinoma ovary hypercalcemic type; TMB = tumor mutation burden
Immunotherapy:
Although the low mutation burden of SCCOHT would not predict responsiveness to ICI responsiveness, programmed cell death protein [ligand] 1 (PD-[L]1) inhibitors such as pembrolizumab have shown exceptional and durable responses in patients with relapsed SCCOHT.(48,49) Case reports of remarkable responses in aggressive SMARCA4-deficient thoracic sarcoma and in SMARCA4-altered NSCLC have also been published.(50,56,57) Although the number of patients reported above is small, it should be noted that the hallmark of the above cancers is SMARCA4 alterations. Furthermore, ICI treatment correlated with significantly improved outcomes overall in SMARCA4-aberrant NSCLC.(18) Some of these ICI responses were seen in patients with high tumor mutation burden (TMB) and/or high PDL1 expression by IHC, each of which are markers for immunotherapy response, but other patients had low TMB and were PD-L1 negative and still responded (Table 3)(63–67). Unfortunately, SMARCA4-deficient small-cell lung carcinoma has been reported to have had a negative outcome on ICI, as reflected by hyperprogressive disease (accelerated progression, which is sometimes observed in patients treated with immunotherapy).(58–61)
Bromodomain/BET inhibitors:
BET inhibitors are a class of drugs that reversibly bind the bromodomains of Bromodomain and Extra-Terminal motif (BET) proteins BRD2–4, and thwart protein-protein interaction between BET proteins and acetylated histones and transcription factors. BET inhibitors have been investigated in SCCOHT models, based on the reliance of SMARCA4-mutant esophageal cancer models for BET protein BRD4 and the co-regulation of an oncogenic network by BRD4 and SMARCA4. SCCOHT cells in orthotopic xenograft models showed sensitivity to BET inhibitors.(68)
EZH2 inhibitors:
A potential therapeutic target for SMARCA4 aberrations comes from studies demonstrating that SWI/SNF loss leads to elevated PRC2.(48) Indeed, SMARCA4-deficient cancer cells display sensitivity to suppression of the methyltransferase known as enhancer of zeste homolog 2 (EZH2), which serves as the catalytic subunit of PRC2. In SCCOHT cells, EZH2 inhibitors potently suppress growth of SCCOHT cell line xenografts. The only trial (NCT02601950) to include patients with SCCOHT investigated the EZH2 inhibitor tazemetostat, and early results were reported for 10 patients with SCCOHT, one of whom achieved a partial response (PR).(69)
Histone deacetylase inhibitors (HDAC):
Targeting histone modification complexes has also shown promise for treatment of patients with SCCOHT. HDAC inhibitors in SCCOHT result in re-expression of SMARCA2, which strongly suppresses proliferation of SCCOHT cells including in in vivo xenograft models of SCCOHT cells, which were responsive to the HDAC inhibitor, quisinostat.(70) However, a single clinical case report did not find efficacy with this approach.(71)
Cyclin inhibitors:
SCCOHT cells are also sensitive to cyclin-dependent kinase 4/6 (CDK4/6) inhibition. SMARCA4 loss causes downregulation of cyclin D1, limiting CDK4/6 kinase activity in SCCOHT cells and leading to in vitro and in vivo susceptibility to CDK4/6 inhibitors.(72) A similar synthetic lethal interaction between SMARCA4-loss and CDK4/6 inhibition was noted in SMARCA4-deficient NSCLC. (73)
Kinase inhibitors:
Exploiting an arrayed kinome-focused siRNA screen, Lang and colleagues showed sensitivity of SCCOHT cell lines in culture and in xenograft models to the multi-targeted tyrosine kinase inhibitor ponatinib (approved in the USA for BCR-ABL-positive leukemia treatment).(74) A reliance upon FGFR signaling as the primary mechanism for this sensitivity was implicated (keeping in mind that ponatinib is a potent FGFR inhibitor (in addition to a BCR-ABL kinase inhibitor)). These results are in agreement with observations in rhabdoid tumors where re-expression of SMARCB1 resulted in decreased expression of FGFR1 and FGFR2, as well as the relative in vitro and in vivo sensitivity of rhabdoid tumor cell lines ponatinib.(75,76) Ponatinib warrants further investigation in SCCOHT and other rhabdoid tumors.
DNA repair:
SMARCA4 binds BRCA1(8), a gene product key to DNA damage repair. Furthermore, preclinical data suggests that SMARCA4-deficient lung cancer cells showed enhanced replication stress. Exposure to ATR inhibitors (which impair DNA repair) resulted in replication catastrophe in these cells.(77) Similarly, low expression of SMARCA4 is significantly associated with platinum-based chemotherapy responsiveness in NSCLC, probably because platinum induces extensive DNA damage.(78)
Other therapeutic targets:
In the preclinical setting, Wang et al(79) have demonstrated killing of SMARCA4-deficient tumors of gynecologic origin with mitochondrial oxidative phosphorylation inhibitors. These agents may be effective because SMARCA4 loss attenuates glucose transport leading to decreased glycolysis and increased dependence on mitochondria respiration. This type of synthetic lethality has also been reported for lung cancer cell lines.(80)
AURKA activity has also been identified as essential for survival and proliferation in NSCLC cells lacking SMARCA4. In these cells, RNAi-mediated depletion or chemical inhibition of AURKA using VX-680 induces cell death in vitro and in xenograft murine models. This effect may be because AURKA-dependent, centrosome-independent mitotic spindle assembly, is vital for the survival of SMARCA4-mutant but not of SMARCA4 wild-type cells. Therefore, AURKA inhibitors may exploit a synthetic lethal vulnerability for NSCLCs carrying SMARCA4-inactivating alterations.(81)
CONCLUSIONS
The protein encoded by SMARCA4 is an ATPase member of the SWI/SNF chromatin remodeling protein family. It provides the energy machinery for remodeling nucleosomes and thereby regulating the transcriptional activation of genes normally repressed by chromatin. Approximately 5–7% of cancers have aberrations in SMARCA4. SMARCA4 anomalies are the molecular hallmark of several ultra-rare aggressive cancers (Table 1): SCCOHT and SMARCA4-deficient thoracic and uterine sarcomas. SMARCA4 abnormalities are also discerned in a small subgroup of more common tumors including, but not limited to lung, colon, bladder, and breast carcinomas. Germline variants in the SMARCA4 gene lead to various hereditary conditions (Table 2): RTPS2 due to loss-of-function SMARCA4 abnormalities, and presenting with lethal rhabdoid tumors in infants and young children; and Coffin-Siris syndrome, a subset of which is due to dominant negative/gain-of function SMARCA4 alterations, and presenting with craniofacial differences, short fifth fingers and toes with underdeveloped or absent nails, feeding difficulties, hypotonia, and intellectual disability.
A small subset of patients with rhabdoid tumors and ~43% of women with SCCOHT have a germline SMARCA4 alteration. Emerging literature suggests that, despite its role as a tumor suppressor, SMARCA4 alterations, especially those that result in loss of function, are actionable, with immune checkpoint blockade demonstrating responses in a small number of published patients with SCCOHT or SMARCA-4 deficient thoracic or uterine sarcomas, as well as in SMARCA4-altered NSCLC (Table 3). Targeted therapies with BET, EZH2, HDAC, CDK4/6, FGFR inhibitors, and inhibitors of DNA damage repair are mechanistically sound as they have shown activity in preclinical models and/or have reasonable biologic rationale. Whether or not some of these compounds could also alleviate the congenital manifestations of disease (aside from the cancer itself) may merit investigation.(82) More trials with a biomarker-driven approach are needed for patients whose malignancies harbor SMARCA4-alterations.(83,84)
Acknowledgements:
Funded in part by the Joan and Irwin Jacobs Fund, and by National Cancer Institute grants P30 CA023100 (RK).
KM and JJA have no disclosures.
GPB receives speaker’s fees from Natera and serves as a consultant to TumorGen Inc. and CEND Therapeutics.
SK serves as a consultant for Foundation Medicine. He receives speaker’s fee from Roche and advisory board for Pfizer. He has research funding from ACT Genomics, Sysmex, Konica Minolta and OmniSeq.
RK has research funding from Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, Boehringer Ingelheim, and Konica Minolta, as well as consultant and/or speaker fees from LOXO, X-Biotech, Actuate Therapeutics, Genentech, Pfizer, Roche, TD2, Biologic Dynamics, Neogenomics, and NeoMed. RK has an equity interest in IDbyDNA and Curematch, Inc and is Board member of CureMatch and CureMetrix and co-founder of CureMatch.
REFERENCES
- 1.Mittal P, Roberts CWM. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol 2020;17(7):435–48 doi 10.1038/s41571-020-0357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Haswell JR, Roberts CW. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer--mechanisms and potential therapeutic insights. Clin Cancer Res 2014;20(1):21–7 doi 10.1158/1078-0432.CCR-13-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oike T, Ogiwara H, Nakano T, Yokota J, Kohno T. Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn J Clin Oncol 2013;43(9):849–55 doi 10.1093/jjco/hyt101. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida A, Kobayashi E, Kubo T, Kodaira M, Motoi T, Motoi N, et al. Clinicopathological and molecular characterization of SMARCA4-deficient thoracic sarcomas with comparison to potentially related entities. Mod Pathol 2017;30(6):797–809 doi 10.1038/modpathol.2017.11. [DOI] [PubMed] [Google Scholar]
- 5.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013;45(6):592–601 doi 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One 2013;8(1):e55119 doi 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helming KC, Wang X, Roberts CWM. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014;26(3):309–17 doi 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell 2000;102(2):257–65 doi 10.1016/s0092-8674(00)00030-1. [DOI] [PubMed] [Google Scholar]
- 9.Strobeck MW, DeCristofaro MF, Banine F, Weissman BE, Sherman LS, Knudsen ES. The BRG-1 subunit of the SWI/SNF complex regulates CD44 expression. J Biol Chem 2001;276(12):9273–8 doi 10.1074/jbc.M009747200. [DOI] [PubMed] [Google Scholar]
- 10.Agaimy A, Daum O, Markl B, Lichtmannegger I, Michal M, Hartmann A. SWI/SNF Complex-deficient Undifferentiated/Rhabdoid Carcinomas of the Gastrointestinal Tract: A Series of 13 Cases Highlighting Mutually Exclusive Loss of SMARCA4 and SMARCA2 and Frequent Co-inactivation of SMARCB1 and SMARCA2. Am J Surg Pathol 2016;40(4):544–53 doi 10.1097/PAS.0000000000000554. [DOI] [PubMed] [Google Scholar]
- 11.Huang SC, Ng KF, Yeh TS, Cheng CT, Chen MC, Chao YC, et al. The clinicopathological and molecular analysis of gastric cancer with altered SMARCA4 expression. Histopathology 2020;77(2):250–61 doi 10.1111/his.14117. [DOI] [PubMed] [Google Scholar]
- 12.Herpel E, Rieker RJ, Dienemann H, Muley T, Meister M, Hartmann A, et al. SMARCA4 and SMARCA2 deficiency in non-small cell lung cancer: immunohistochemical survey of 316 consecutive specimens. Ann Diagn Pathol 2017;26:47–51 doi 10.1016/j.anndiagpath.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez-Nieto S, Canada A, Pros E, Pinto AI, Torres-Lanzas J, Lopez-Rios F, et al. Massive parallel DNA pyrosequencing analysis of the tumor suppressor BRG1/SMARCA4 in lung primary tumors. Hum Mutat 2011;32(2):E1999–2017 doi 10.1002/humu.21415. [DOI] [PubMed] [Google Scholar]
- 14.Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res 2003;63(3):560–6. [PubMed] [Google Scholar]
- 15.Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat 2008;29(5):617–22 doi 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 16.Oike T, Ogiwara H, Tominaga Y, Ito K, Ando O, Tsuta K, et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res 2013;73(17):5508–18 doi 10.1158/0008-5472.CAN-12-4593. [DOI] [PubMed] [Google Scholar]
- 17.Matsubara D, Kishaba Y, Ishikawa S, Sakatani T, Oguni S, Tamura T, et al. Lung cancer with loss of BRG1/BRM, shows epithelial mesenchymal transition phenotype and distinct histologic and genetic features. Cancer Sci 2013;104(2):266–73 doi 10.1111/cas.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoenfeld AJ, Bandlamudi C, Lavery JA, Montecalvo J, Namakydoust A, Rizvi H, et al. The Genomic Landscape of SMARCA4 Alterations and Associations with Outcomes in Patients with Lung Cancer. Clin Cancer Res 2020;26(21):5701–8 doi 10.1158/1078-0432.CCR-20-1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karnezis AN, Wang Y, Ramos P, Hendricks WP, Oliva E, D’Angelo E, et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J Pathol 2016;238(3):389–400 doi 10.1002/path.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jelinic P, Mueller JJ, Olvera N, Dao F, Scott SN, Shah R, et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 2014;46(5):424–6 doi 10.1038/ng.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramos P, Karnezis AN, Craig DW, Sekulic A, Russell ML, Hendricks WP, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet 2014;46(5):427–9 doi 10.1038/ng.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perret R, Chalabreysse L, Watson S, Serre I, Garcia S, Forest F, et al. SMARCA4-deficient Thoracic Sarcomas: Clinicopathologic Study of 30 Cases With an Emphasis on Their Nosology and Differential Diagnoses. Am J Surg Pathol 2019;43(4):455–65 doi 10.1097/PAS.0000000000001188. [DOI] [PubMed] [Google Scholar]
- 23.Sauter JL, Graham RP, Larsen BT, Jenkins SM, Roden AC, Boland JM. SMARCA4-deficient thoracic sarcoma: a distinctive clinicopathological entity with undifferentiated rhabdoid morphology and aggressive behavior. Mod Pathol 2017;30(10):1422–32 doi 10.1038/modpathol.2017.61. [DOI] [PubMed] [Google Scholar]
- 24.Kolin DL, Dong F, Baltay M, Lindeman N, MacConaill L, Nucci MR, et al. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): a clinicopathologic entity distinct from undifferentiated carcinoma. Mod Pathol 2018;31(9):1442–56 doi 10.1038/s41379-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 25.Fernando TM, Piskol R, Bainer R, Sokol ES, Trabucco SE, Zhang Q, et al. Functional characterization of SMARCA4 variants identified by targeted exome-sequencing of 131,668 cancer patients. Nat Commun 2020;11(1):5551 doi 10.1038/s41467-020-19402-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Hamel N, Tomiak E, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 2014;46(5):438–43 doi 10.1038/ng.2931. [DOI] [PubMed] [Google Scholar]
- 27.Karanian-Philippe M, Velasco V, Longy M, Floquet A, Arnould L, Coindre JM, et al. SMARCA4 (BRG1) loss of expression is a useful marker for the diagnosis of ovarian small cell carcinoma of the hypercalcemic type (ovarian rhabdoid tumor): a comprehensive analysis of 116 rare gynecologic tumors, 9 soft tissue tumors, and 9 melanomas. Am J Surg Pathol 2015;39(9):1197–205 doi 10.1097/PAS.0000000000000475. [DOI] [PubMed] [Google Scholar]
- 28.Clarke BA, Witkowski L, Ton Nu TN, Shaw PA, Gilks CB, Huntsman D, et al. Loss of SMARCA4 (BRG1) protein expression as determined by immunohistochemistry in small-cell carcinoma of the ovary, hypercalcaemic type distinguishes these tumours from their mimics. Histopathology 2016;69(5):727–38 doi 10.1111/his.12988. [DOI] [PubMed] [Google Scholar]
- 29.Conlon N, Silva A, Guerra E, Jelinic P, Schlappe BA, Olvera N, et al. Loss of SMARCA4 Expression Is Both Sensitive and Specific for the Diagnosis of Small Cell Carcinoma of Ovary, Hypercalcemic Type. Am J Surg Pathol 2016;40(3):395–403 doi 10.1097/PAS.0000000000000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kupryjanczyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J, Plisiecka-Halasa J, Szafron L, Podgorska A, et al. Ovarian small cell carcinoma of hypercalcemic type - evidence of germline origin and SMARCA4 gene inactivation. a pilot study. Pol J Pathol 2013;64(4):238–46 doi 10.5114/pjp.2013.39331. [DOI] [PubMed] [Google Scholar]
- 31.Rekhtman N, Montecalvo J, Chang JC, Alex D, Ptashkin RN, Ai N, et al. SMARCA4-Deficient Thoracic Sarcomatoid Tumors Represent Primarily Smoking-Related Undifferentiated Carcinomas Rather Than Primary Thoracic Sarcomas. J Thorac Oncol 2020;15(2):231–47 doi 10.1016/j.jtho.2019.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemes K, Bens S, Bourdeaut F, Hasselblatt M, Kool M, Johann P, et al. Rhabdoid Tumor Predisposition Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al. , editors. GeneReviews((R)). Seattle (WA)1993. [Google Scholar]
- 33.Errichiello E, Mustafa N, Vetro A, Notarangelo LD, de Jonge H, Rinaldi B, et al. SMARCA4 inactivating mutations cause concomitant Coffin-Siris syndrome, microphthalmia and small-cell carcinoma of the ovary hypercalcaemic type. J Pathol 2017;243(1):9–15 doi 10.1002/path.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsurusaki Y, Koshimizu E, Ohashi H, Phadke S, Kou I, Shiina M, et al. De novo SOX11 mutations cause Coffin-Siris syndrome. Nat Commun 2014;5:4011 doi 10.1038/ncomms5011. [DOI] [PubMed] [Google Scholar]
- 35.Dsouza NR, Zimmermann MT, Geddes GC. A case of Coffin-Siris syndrome with severe congenital heart disease and a novel SMARCA4 variant. Cold Spring Harb Mol Case Stud 2019;5(3) doi 10.1101/mcs.a003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kosho T, Okamoto N, Coffin-Siris Syndrome International C. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet 2014;166C(3):262–75 doi 10.1002/ajmg.c.31407. [DOI] [PubMed] [Google Scholar]
- 37.Podwika SE, Jenkins TM, Khokhar JK, Erickson SH, Modesitt SC. Optimal age for genetic cancer predisposition testing in hereditary SMARCA4 Ovarian Cancer Families: How young is too young? Gynecol Oncol Rep 2020;32:100569 doi 10.1016/j.gore.2020.100569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magnani L, Cabot RA. Manipulation of SMARCA2 and SMARCA4 transcript levels in porcine embryos differentially alters development and expression of SMARCA1, SOX2, NANOG, and EIF1. Reproduction 2009;137(1):23–33 doi 10.1530/REP-08-0335. [DOI] [PubMed] [Google Scholar]
- 39.Zhou J, Zhang M, Fang H, El-Mounayri O, Rodenberg JM, Imbalzano AN, et al. The SWI/SNF chromatin remodeling complex regulates myocardin-induced smooth muscle-specific gene expression. Arterioscler Thromb Vasc Biol 2009;29(6):921–8 doi 10.1161/ATVBAHA.109.187229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi X, Wang Q, Gu J, Xuan Z, Wu JI. SMARCA4/Brg1 coordinates genetic and epigenetic networks underlying Shh-type medulloblastoma development. Oncogene 2016;35(44):5746–58 doi 10.1038/onc.2016.108. [DOI] [PubMed] [Google Scholar]
- 41.Hodges HC, Stanton BZ, Cermakova K, Chang CY, Miller EL, Kirkland JG, et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat Struct Mol Biol 2018;25(1):61–72 doi 10.1038/s41594-017-0007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romero OA, Setien F, John S, Gimenez-Xavier P, Gomez-Lopez G, Pisano D, et al. The tumour suppressor and chromatin-remodelling factor BRG1 antagonizes Myc activity and promotes cell differentiation in human cancer. EMBO Mol Med 2012;4(7):603–16 doi 10.1002/emmm.201200236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodges C, Kirkland JG, Crabtree GR. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med 2016;6(8) doi 10.1101/cshperspect.a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7(8):818–31 doi 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mashtalir N, Suzuki H, Farrell DP, Sankar A, Luo J, Filipovski M, et al. A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 2020;183(3):802–17 e24 doi 10.1016/j.cell.2020.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bourdeaut F, Lequin D, Brugieres L, Reynaud S, Dufour C, Doz F, et al. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 2011;17(1):31–8 doi 10.1158/1078-0432.CCR-10-1795. [DOI] [PubMed] [Google Scholar]
- 47.Witkowski L, Goudie C, Foulkes WD, McCluggage WG. Small-Cell Carcinoma of the Ovary of Hypercalcemic Type (Malignant Rhabdoid Tumor of the Ovary): A Review with Recent Developments on Pathogenesis. Surg Pathol Clin 2016;9(2):215–26 doi 10.1016/j.path.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 48.Tischkowitz M, Huang S, Banerjee S, Hague J, Hendricks WPD, Huntsman DG, et al. Small-Cell Carcinoma of the Ovary, Hypercalcemic Type-Genetics, New Treatment Targets, and Current Management Guidelines. Clin Cancer Res 2020;26(15):3908–17 doi 10.1158/1078-0432.CCR-19-3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jelinic P, Ricca J, Van Oudenhove E, Olvera N, Merghoub T, Levine DA, et al. Immune-Active Microenvironment in Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Rationale for Immune Checkpoint Blockade. J Natl Cancer Inst 2018;110(7):787–90 doi 10.1093/jnci/djx277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henon C, Blay JY, Massard C, Mir O, Bahleda R, Dumont S, et al. Long lasting major response to pembrolizumab in a thoracic malignant rhabdoid-like SMARCA4-deficient tumor. Ann Oncol 2019;30(8):1401–3 doi 10.1093/annonc/mdz160. [DOI] [PubMed] [Google Scholar]
- 51.Lin DI, Allen JM, Hecht JL, Killian JK, Ngo NT, Edgerly C, et al. SMARCA4 inactivation defines a subset of undifferentiated uterine sarcomas with rhabdoid and small cell features and germline mutation association. Mod Pathol 2019;32(11):1675–87 doi 10.1038/s41379-019-0303-z. [DOI] [PubMed] [Google Scholar]
- 52.Agaimy A, Foulkes WD. Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol 2018;35(3):193–8 doi 10.1053/j.semdp.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 53.Witkowski L, Goudie C, Ramos P, Boshari T, Brunet JS, Karnezis AN, et al. The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecologic oncology 2016;141(3):454–60 doi 10.1016/j.ygyno.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneppenheim R, Fruhwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 2010;86(2):279–84 doi 10.1016/j.ajhg.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Longy M, Toulouse C, Mage P, Chauvergne J, Trojani M. Familial cluster of ovarian small cell carcinoma: a new mendelian entity? J Med Genet 1996;33(4):333–5 doi 10.1136/jmg.33.4.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takada K, Sugita S, Murase K, Kikuchi T, Oomori G, Ito R, et al. Exceptionally rapid response to pembrolizumab in a SMARCA4-deficient thoracic sarcoma overexpressing PD-L1: A case report. Thorac Cancer 2019;10(12):2312–5 doi 10.1111/1759-7714.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naito T, Umemura S, Nakamura H, Zenke Y, Udagawa H, Kirita K, et al. Successful treatment with nivolumab for SMARCA4-deficient non-small cell lung carcinoma with a high tumor mutation burden: A case report. Thorac Cancer 2019;10(5):1285–8 doi 10.1111/1759-7714.13070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiba Y, Kawanami T, Yamasaki K, Uchimura K, Matsuyama A, Yatera K. Hyperprogressive disease after immune checkpoint inhibitor in SMARCA4-deficient small-cell lung carcinoma. Respirol Case Rep 2020;8(8):e00667 doi 10.1002/rcr2.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adashek JJ, Kato S, Ferrara R, Lo Russo G, Kurzrock R. Hyperprogression and Immune Checkpoint Inhibitors: Hype or Progress? Oncologist 2019. doi 10.1634/theoncologist.2019-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin Cancer Res 2017;23(15):4242–50 doi 10.1158/1078-0432.CCR-16-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adashek JJ, Subbiah IM, Matos I, Garralda E, Menta AK, Ganeshan DM, et al. Hyperprogression and Immunotherapy: Fact, Fiction, or Alternative Fact? Trends Cancer 2020;6(3):181–91 doi 10.1016/j.trecan.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu B, Lin JZ, Yang XB, Sang XT. The roles of mutated SWI/SNF complexes in the initiation and development of hepatocellular carcinoma and its regulatory effect on the immune system: A review. Cell Prolif 2020;53(4):e12791 doi 10.1111/cpr.12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther 2017;16(11):2598–608 doi 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther 2015;14(4):847–56 doi 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 65.Khagi Y, Kurzrock R, Patel SP. Next generation predictive biomarkers for immune checkpoint inhibition. Cancer Metastasis Rev 2017;36(1):179–90 doi 10.1007/s10555-016-9652-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goodman AM, Sokol ES, Frampton GM, Lippman SM, Kurzrock R. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. Cancer Immunol Res 2019;7(10):1570–3 doi 10.1158/2326-6066.CIR-19-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jardim DL, Goodman A, de Melo Gagliato D, Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2020. doi 10.1016/j.ccell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shorstova T, Marques M, Su J, Johnston J, Kleinman CL, Hamel N, et al. SWI/SNF-Compromised Cancers Are Susceptible to Bromodomain Inhibitors. Cancer Res 2019;79(10):2761–74 doi 10.1158/0008-5472.CAN-18-1545. [DOI] [PubMed] [Google Scholar]
- 69.Jones RL, Blay JY, Agulnik M, Chugh R, Mir O, Italiano A, et al. A phase II, multicenter study of the EZH2 inhibitor tazemetostat in adults (rhabdoid tumor cohort) (NCT02601950). Annals of Oncology 2018;29:viii580–viii1 doi 10.1093/annonc/mdy299.011. [DOI] [Google Scholar]
- 70.Wang Y, Chen SY, Colborne S, Lambert G, Shin CY, Santos ND, et al. Histone Deacetylase Inhibitors Synergize with Catalytic Inhibitors of EZH2 to Exhibit Antitumor Activity in Small Cell Carcinoma of the Ovary, Hypercalcemic Type. Mol Cancer Ther 2018;17(12):2767–79 doi 10.1158/1535-7163.MCT-18-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rao V, Bauer F, Vredenburgh JJ. Refractory Small Cell Carcinoma of the Ovary - Hypercalcemic Type (SCCOHT) Treated with Romidepsin and Topotecan: A Case Report and Review of the Literature. Conn Med 2016;80(9):529–32. [PubMed] [Google Scholar]
- 72.Xue Y, Meehan B, Macdonald E, Venneti S, Wang XQD, Witkowski L, et al. CDK4/6 inhibitors target SMARCA4-determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat Commun 2019;10(1):558 doi 10.1038/s41467-018-06958-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xue Y, Meehan B, Fu Z, Wang XQD, Fiset PO, Rieker R, et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat Commun 2019;10(1):557 doi 10.1038/s41467-019-08380-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lang JD, Hendricks WPD, Orlando KA, Yin H, Kiefer J, Ramos P, et al. Ponatinib Shows Potent Antitumor Activity in Small Cell Carcinoma of the Ovary Hypercalcemic Type (SCCOHT) through Multikinase Inhibition. Clin Cancer Res 2018;24(8):1932–43 doi 10.1158/1078-0432.CCR-17-1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wohrle S, Weiss A, Ito M, Kauffmann A, Murakami M, Jagani Z, et al. Fibroblast growth factor receptors as novel therapeutic targets in SNF5-deleted malignant rhabdoid tumors. PLoS One 2013;8(10):e77652 doi 10.1371/journal.pone.0077652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wong JP, Todd JR, Finetti MA, McCarthy F, Broncel M, Vyse S, et al. Dual Targeting of PDGFRalpha and FGFR1 Displays Synergistic Efficacy in Malignant Rhabdoid Tumors. Cell Rep 2016;17(5):1265–75 doi 10.1016/j.celrep.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kurashima K, Kashiwagi H, Shimomura I, Suzuki A, Takeshita F, Mazevet M, et al. SMARCA4 deficiency-associated heterochromatin induces intrinsic DNA replication stress and susceptibility to ATR inhibition in lung adenocarcinoma. NAR Cancer 2020;2(2) doi 10.1093/narcan/zcaa005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bell EH, Chakraborty AR, Mo X, Liu Z, Shilo K, Kirste S, et al. SMARCA4/BRG1 Is a Novel Prognostic Biomarker Predictive of Cisplatin-Based Chemotherapy Outcomes in Resected Non-Small Cell Lung Cancer. Clin Cancer Res 2016;22(10):2396–404 doi 10.1158/1078-0432.CCR-15-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Y, Ong D, Chen SY, Li E, Orlando K, Raupach E, et al. Abstract 1459: Selective killing of SMARCA4-deficient gynecologic cancers by mitochondria oxidative phosphorylation inhibitors. Cancer Research 2020;80(16 Supplement):1459 doi 10.1158/1538-7445.AM2020-1459. [DOI] [Google Scholar]
- 80.Lissanu Deribe Y, Sun Y, Terranova C, Khan F, Martinez-Ledesma J, Gay J, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat Med 2018;24(7):1047–57 doi 10.1038/s41591-018-0019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tagal V, Wei S, Zhang W, Brekken RA, Posner BA, Peyton M, et al. SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat Commun 2017;8:14098 doi 10.1038/ncomms14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adashek JJ, Kato S, Lippman SM, Kurzrock R. The paradox of cancer genes in non-malignant conditions: implications for precision medicine. Genome Med 2020;12(1):16 doi 10.1186/s13073-020-0714-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kato S, Kim KH, Lim HJ, Boichard A, Nikanjam M, Weihe E, et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun 2020;11(1):4965 doi 10.1038/s41467-020-18613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adashek JJ, Subbiah V, Kurzrock R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2021;7(1):15–28 doi 10.1016/j.trecan.2020.08.009. [DOI] [PubMed] [Google Scholar]