

Abstract
Biliary tract cancers (BTCs) consist of a group of highly heterogeneous malignancies that are characterized by genomic differences among tumors from different anatomic sites. The current treatment for BTC includes surgery, chemotherapy, target therapy, and immunotherapy. Although surgery remains the primary option for localized disease, representing the only potential curative treatment, a high risk of recurrence cannot be neglected. Chemotherapy has been considered the standard of care for both advanced and metastatic disease and in adjuvant settings. However, drug resistance is a major obstacle associated with chemotherapy. The development of genetic testing technologies, including next‐generation sequencing, has opened the door for the identification of drug targets and candidate molecules. A series of preclinical studies has demonstrated the role of gene mutations, abnormal signaling pathways, and immunosuppression in the pathogenesis of BTC, laying the foundation for the application of targeted therapy and immunotherapy. A variety of molecularly targeted agents, including pemigatinib, have shown promising survival benefits in patients with advanced disease. The rapidly evolving role of multimodal therapy represents the subject of this review.
Keywords: biliary tract cancers, clinical trials, immunotherapy, molecular characterization, targeted therapy
Biliary tract cancers (BTCs) consist of a group of highly heterogeneous malignancies, which are characterized by genomic differences among tumors from different anatomic sites. A series of preclinical studies have proven the role of gene mutations, abnormal signaling pathways, and immunosuppression in the pathogenesis of BTC, laying the foundation for the application of targeted therapy and immunotherapy. The disappointing response to conventional cytotoxic drugs along with the genetic heterogeneity of BTC led to both immunotherapy and targeted therapy–oriented research based on the genetic characterization of BTC.
1. BACKGROUND
Biliary tract cancers (BTCs) consist of a group of highly heterogeneous malignancies originating from the biliary system, including gallbladder cancer (GBC) and cholangiocarcinoma (CCA). BTC is a rare but fatal disease whose incidence varies according to geographic location and ethnicity. 1 The diagnosis of BTC often relies on imaging and tumor tissue biopsy, with the latter involving surgical resection or mass puncture. Due to its latent onset, BTC is usually diagnosed in late stage, thereby deprived of the opportunity for surgical resection. 2 The purpose of this review is to discuss the characteristics of BTC subtypes, current multimodal treatments (including surgery and adjuvant therapies for patients with early disease and chemotherapies for those with advanced disease), and recent progress related to targeted therapies and immune therapies.
2. ANATOMICAL AND HISTOPATHOLOGICAL CLASSIFICATION OF BTC
According to anatomic location, BTCs can be classified as GBC, intrahepatic cholangiocarcinoma (ICC), perihilar cholangiocarcinoma (pCCA), or distal cholangiocarcinoma (dCCA). The latter two are collectively referred to as extrahepatic cholangiocarcinoma (ECC). Most cases of GBC (60%) arise from the fundus of the gallbladder, while 30% and 10% arise from the body and neck, respectively. ICC is defined as a CCA located proximally to the secondary bile ducts, while pCCA appears in the right and left hepatic ducts and the insertion of the cystic duct into the common bile duct. 3 dCCA is confined to the area between the start of the common bile duct and the ampulla of Vater.
ICC has three main subtypes: the mixed subtype (derived from cuboidal mucin‐negative cholangiocytes in small bile ducts), the mucinous subtype (derived from cylindrical mucous cholangiocytes in larger bile ducts), and the cholangiolocellular subtype (derived from hepatic progenitor cells [HPCs] in the most peripheral biliary branches). 4 Some studies suggest that hepatitis B virus and hepatitis C virus (HCV) infections play a role in the development of mixed‐ICC. In Japan, the risk of ICC in patients with liver cirrhosis caused by HCV is approximately 1000 times higher than that in the general population. 5 Mixed‐ICCs and cholangiolocellular‐ICCs (CLCs) display almost mass‐forming growth patterns, while muc‐ICCs exhibit mass‐forming patterns or a combination of periductal infiltration and mass formation. 6 In addition to conventional ICC, CLC is a rare variant of ICC originating from bipotential HPCs. 7 Studies have found that mice with genetic alterations that affect the Hippo pathway within the liver exhibit HPC expansion and subsequently develop CLC. 8 In this case, a single tumor consisting of both differentiations develops, rather than one containing a mixture of hepatocytes and cholangiocytes. Although it shares biliary features with ICC, studies to date have shown that its radiological, clinicopathological, and molecular characteristics are different from those of ICC and hepatocellular carcinoma (HCC). Combined HCC may be more accurate terminology. 8 In addition, recent research indicates that the combination of activated Notch signaling and AKT overexpression may lead to the rapid formation of ICCs from hepatocytes. 9 The molecular characteristics still need to be further elucidated.
ECC is believed to derive from cholangiocytes or pluripotent stem cells, which originate from the peribiliary glands located at the branch points of the biliary tree (eg, portal and ampulla regions). 5 Conventional ECCs are mucin‐producing adenocarcinomas. They frequently appear as flat or poorly defined nodular sclerosing tumors with a periductal infiltrating growth pattern (>80%) and are less common as intraductal polypoid or papillary tumors. 6 , 10
3. MOLECULAR CHARACTERISTICS OF BTC
Several studies have reported the molecular characteristics of BTC, highlighting the genomic differences among tumors from different anatomic sites. Their findings may contribute to a more precise classification of BTC and have potential therapeutic significance (Figure 1).
FIGURE 1.
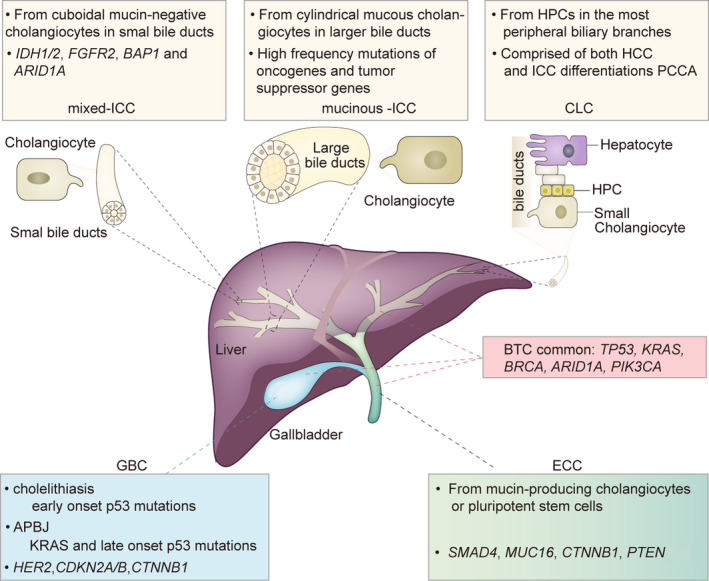
Molecular characterization of BTC (biliary tract cancer). APBJ, abnormalities of the pancreatic bile‐duct junction; ARID1A, AT‐rich interaction domain 1A; BAP1, BRCA1‐associated protein 1; CDKN2A/B, cyclin‐dependent kinase inhibitor 2A and B; CLC, cholangiolocellular‐ICC; CTNNB1, catenin, cadherin‐associated protein, beta 1; ECC, extrahepatic cholangiocarcinoma; FGFR2, fibroblast growth factor receptor 2; GBC, gallbladder cancer; HCC, hepatocellular carcinoma; HER2, human epidermal growth factor receptor 2; HPC, hepatic progenitor cell; ICC, intrahepatic cholangiocarcinoma; IDH1/2, isocitrate dehydrogenase 1 and 2; KRAS, Kirsten rat sarcoma viral oncogene homolog; MUC16, mucoprotein 16; PCCA, perihilar cholangiocarcinoma; PIK3CA, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha; PTEN, gene of phosphate and tension homology deleted on chromosome ten; ; SMAD4, SMAD family member 4; TP53, tumor protein 53
3.1. Gallbladder cancer
Two key pathways for the onset of GBC have been identified. The most common is gallstones and chronic inflammation of the gallbladder, which tend to occur in female patients over 65 years of age. The second, less frequent pathway is related to congenital abnormalities of the pancreatic bile duct junction (APBJ), which are especially common in Japanese patients and are more likely to occur in young patients. 11
Cases of GBC that develop in the context of an APBJ are consistently associated with KRAS (50%‐83%) and late‐onset p53 mutations. On the other hand, cholecystitis‐associated GBCs rarely exhibit KRAS mutations and are characterized by early p53 mutations. 12 TP53 is the most frequently altered gene in patients with GBC. In general, the estimated frequency of p53 mutations in patients with GBC ranges from 21% to 39%. 13 , 14 Studies investigating the PI3K pathway have also identified activating mutations in PIK3CA in cases of GBC (12%‐13%). 1 HER2/neu amplification or protein overexpression has been detected in 12%‐16% of GBC cases, a pattern unique to GBC. 15 High‐level microsatellite instability (MSI) has been reported in 5% of GBC cases, 16 occurring more frequently in patients with APBJ. 1 A previous study reported that the incidence of breast cancer susceptibility gene (BRCA) mutations is higher in patients with MSI‐H/deficient mismatch repair (dMMR) than in those without (19.5% vs. 1.7%, P < .0001). 17 However, there is no significant difference in the frequency of BRCA mutations among tumor sites. In GBC and ICC, BRCA2 mutations (4.0% and 2.7%) are more frequent than BRCA1 mutations (0.3% and 0.4%, P < .05), although the observed frequency of the two mutation types is similar for ECC (BRCA2: 2.6%; BRCA1: 2.1%). 17
3.2. ICC
Several independent studies have defined two distinctive molecular groups of ICCs: the proliferation molecular subclass with shorter survival and the inflammation subclass with better prognosis. 12 From the perspective of anatomic location and growth patterns, the former corresponds to the mucinous subtype to some extent. They both activate oncogenic signaling pathways, including KRAS (8.6%‐25%), BRAF, SMAD4, and genes from the RAS‐MAPK and MET signaling networks. 3 , 10 , 14 Unfortunately, the latter do not have much in common with the mixed subtype, although they have both been observed in the setting of chronic liver disease. 14 The inflammation subclass is distinguished by overexpression of cytokines and IL‐6‐STAT3 signaling, while mixed‐ICC displays a typically high rate of missense mutations in the isocitrate dehydrogenase 1 and 2 (IDH1/2) genes (10%‐23%), 18 fibroblast growth factor receptor 2 (FGFR2) fusions (14%), and BRCA1‐associated protein 1 (BAP1) (15%). 10 , 12 , 13 Interestingly, KRAS and IDH1 mutations are mutually exclusive. FGFR2 aberrations appear to be mutually exclusive with other mutations such as IDH1/2, KRAS, and BRAF. 19 , 20 MSI status is observed in 10% of patients with ICC and is more frequent than GBC (5%) and ECC (5%). 16
IDH1/2 mutation is a molecular feature of intrahepatic bile duct origin and therefore occurs exclusively in ICC and rarely in ECC and GBC. 7 , 21 The correlation between IDH mutation status and prognosis remains controversial. In the Fudan cohort of 252 patients with ICC with follow‐up data, the mutation status of IDH genes was associated with longer overall survival (OS) and a lower probability of recurrence, 18 which is contrary to the findings of another study with a small sample size (n = 34). 13 Data have also shown that IDH1/2 mutation predicts a good prognosis in mixed‐ICC and a poor prognosis in muc‐iCC. 22 Notably, FGFR2 fusions that lead to ligand‐independent activation of the receptor tyrosine kinase have been identified almost only in patients with ICC. 21 , 23 , 24 FGFR2 aberrations in ICC have been associated with an apparent female predominance and an apparent tendency for a better prognosis. 24 Epidermal growth factor receptor (EGFR) expression is an independent prognostic factor in ICC, whereas vascular endothelial growth factor (VEGF) expression is associated with intrahepatic metastasis. 25
3.3. ECC
ECC, ICC, and GBC exhibit partial overlap in terms of molecular expressions, albeit at different frequencies. These include TP53, KRAS, CDKN2A/B, ARID1A, and PTEN. KRAS mutations are consistently more prevalent in ECC than in ICC and GBC. 6 HER2 is present in 9% of ECCs. However, unlike in cases of GBC, these alterations are mostly base substitution and insertion mutations rather than amplifications. 26
In recent years, Japanese scholars have reported a type of occupational CCA caused by long‐term exposure to chlorinated organic solvents, including 1,2‐dichloropropane (DCP) and dichloromethane (DCM). 27 Although the genetic mutations of occupational CCA is similar to that of conventional BTC, its tumor mutation burden (TMB) is about 30 times higher than that of the latter. 28
4. STATE‐OF‐THE‐ART TREATMENT FOR BTC
BTC treatment varies according to the stage defined by the Bismuth‐Corlette and TNM classifications. Surgery with negative margins including lymphadenectomy remains the primary treatment option for localized disease, representing the only potential curative treatment. Chemotherapy has also been applied in adjuvant and metastatic/locally advanced settings (Figure 2).
FIGURE 2.
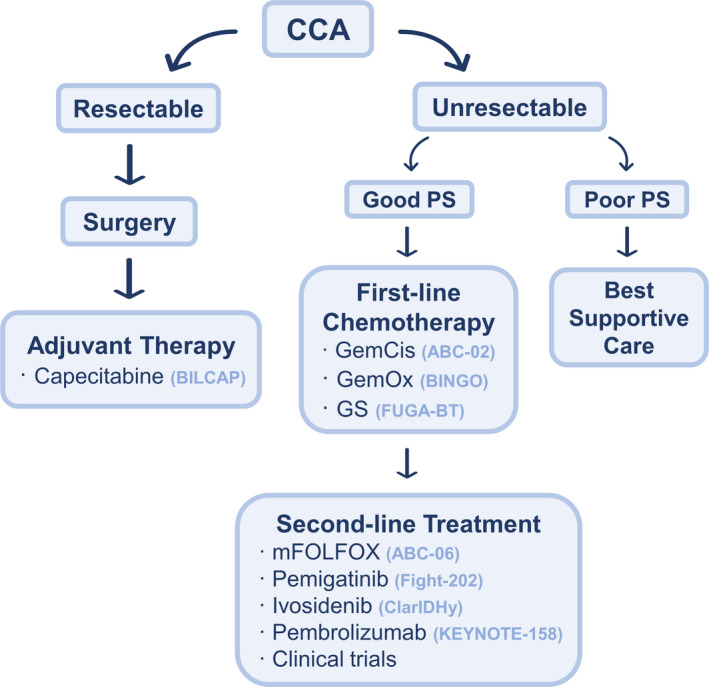
Therapeutic strategy in cholangiocarcinoma (CCA). GemCis, gemcitabine + cisplatin; GemOx, gemcitabine + oxaliplatin; GS, gemcitabine + S‐1; mFOLFOX, modified 5‐fluorouracil + oxaliplatin; PS, performance status
4.1. Neoadjuvant therapy
Due to atypical symptoms in the early stages of the disease, only about 20%‐50% of patients with BTC have the opportunity to undergo surgery. 2 , 29 Several retrospective studies indicated that neoadjuvant therapies, including preoperative radiotherapy and chemotherapy, could improve the resectability of locally advanced CCA and prolong the survival time. 30 , 31 , 32 Previous research provided information that in patients with localized node‐negative pCCA, compared with traditional resection, neoadjuvant chemoradiation combined with liver transplantation achieved a better 5‐year survival rate (82% versus 21%, P = .022) and less recurrence rate. 33 A prospective case series showed that ICC with pretransplant stable disease (SD) in gemcitabine‐based neoadjuvant chemotherapy may benefit from liver transplantation. 34 The application of neoadjuvant therapy in BTC requires further exploration in prospective trials.
4.2. Adjuvant therapy
Even with R0 resection, the risk of 5‐year recurrence is 64% for GBC and 53.5% for ICC. 35 , 36 The reported 5‐year survival rates for patients with CCA after surgery range from 22% to 44%. 2 , 36 After surgery, patients are advised to undergo adjuvant therapy to reduce the likelihood of recurrence, especially those with positive lymph nodes or resection margins. 37 For occupational BTC, aggressive treatment including secondary surgical resection of the recurrent lesion appears to be effective. 38
Adjuvant chemotherapy is based on the results of the BILCAP study, 39 which was a randomized, controlled phase III study that compared capecitabine with observation in patients with resected BTC. The primary endpoint was not met in the intention‐to‐treat population, and the authors reported a median OS of 51.1 months in the capecitabine group and 36.4 months in the observation group (hazard ratio [HR]: 0.81, P = .97). In a protocol‐specified analysis adjusted for minimization factor, nodal status, grade, and sex, the authors reported a statistically significant survival advantage in the capecitabine group (HR: 0.75, P = .028, respectively). Nevertheless, in the same setting, the PRODIGE‐12 40 and BCAT 41 phase III studies reported no benefit from the combination of gemcitabine and oxaliplatin (GemOx) or from the single agent gemcitabine, respectively.
Survival data from a phase II randomized prospective study (KHBO 1208) in Japan indicate that S‐1 adjuvant therapy may be better than gemcitabine adjuvant therapy after major hepatectomy in patients with BTC. 42 A phase III prospective randomized trial (ACTICCA‐1) is assessing the clinical performance of gemcitabine + cisplatin (GemCis) in patients after curative intent resection of BTC. 43
4.3. First‐line chemotherapy
Systemic chemotherapy is the standard of care for advanced or metastatic diseases. GemCis should be the preferred choice in patients with good Eastern Cooperative Oncology Group (ECOG) performance status (PS) and normal renal function based on the results of the ABC‐02 trial. 44 This Phase III clinical trial demonstrated the superiority of GemCis compared with gemcitabine monotherapy, with an OS of 11.7 months vs 8.1 months, respectively (HR: 0.64 P < .001). GemOx is generally preferred if renal function is a concern. 45 In patients with ECOG PS 2, gemcitabine alone should be considered. In Asia, the combination of gemcitabine + S‐1 (GS) is considered a new option for advanced BTC thanks to the phase III trial FUGA‐BT, which demonstrated the noninferiority of GS to GemCis in terms of OS (GemCis: 11.2 months vs. GS: 13 months; HR: 0.94, P = .046). 46 Another phase III clinical trial revealed that capecitabine plus oxaliplatin (XELOX) exhibited similar efficacy to GemOx in terms of 6‐month progression‐free survival (PFS), highlighting its potential as an alternative first‐line treatment. 47
It is worth noting that there are new therapeutic candidates with improved survival rates. A phase III clinical trial in Japan showed that the clinical efficacy of gemcitabine + cisplatin + S‐1 (GCS) is significantly better than that of GemCis. The former was associated with a longer median PFS (mPFS; 7.4 months vs. 5.5 months, HR: 0.75; P = .0015), and there were no significant differences in adverse events (AEs), except for those related to S‐1. Such therapy may thus become a new standard for the treatment of advanced BTC. 48 A phase II clinical trial has shown that adding nab‐paclitaxel to first‐line treatment with GemCis may prolong survival, with an mPFS of 11.8 months and an mOS of 19.2 months. 49
4.4. Second‐line treatment
Second‐line chemotherapy has been widely utilized despite limited evidence to support a benefit. The results of the randomized phase III ABC 06 trial demonstrated an OS advantage for modified 5‐fluorouracil + oxaliplatin (mFOLFOX) compared with best supportive care (6.2 vs. 5.3 months, HR: 0.69, P = .031), suggesting that mFOLFOX is the treatment of choice in patients with ECOG PS 0‐1 previously treated with GemCis first‐line chemotherapy. 50 However, considering the limited survival benefit, studies have explored the possibility of other options in the second‐line treatment of BTC. Retrospective data for 87 patients with patients receiving second‐line therapy indicated that the capecitabine and irinotecan (XELIRI) regimen is safe and effective, with a disease control rate (DCR) of 41.3% and an OS of 8 months. 51 A phase II trial conducted in 60 patients demonstrated that, compared with irinotecan monotherapy, the XELIRI regimen has significant advantages in terms of prolonging PFS and improving 9‐month OS rates (3.7 vs. 2.4, P = .036, and 60.0% vs. 32.9%, P = .045, respectively). 52
For those who have used GemOx as first‐line treatment, 5‐fluorouracil‐irinotecan (FOLFIRI)‐based chemotherapy may be a good choice. And the sensitivity to this chemotherapy regimen is correlated with sensitivity to the first‐line GemOx regimen (P = .007). 53 However, FOLFIRI is not superior to FOLFOX in patients with BTC after failure of first‐line GemCis treatment. 54
Regorafenib is a potent inhibitor of angiogenic and oncogenic kinases and has been approved for the treatment of patients with metastatic colorectal cancer refractory to standard chemotherapy. An earlier study showed that, among 43 patients with chemotherapy‐refractory advanced/metastatic BTC in whom the response to regorafenib treatment could be assessed, the mPFS was 15.6 weeks (90% confidence interval [CI]: 12.9‐24.7 weeks). 55 The randomized phase II study REACHIN showed that, although regorafenib doubled the mPFS compared with placebo (3.0 months to 1.5 months; HR: 0.49; 95% CI: 0.29‐0.81; P = .004), there was no significant difference in OS. 56
Based on the results of the Fight‐202 trial, pemigatinib (a potent and selective oral inhibitor of FGFR 1‐3 and VEGFR2) has been approved by the Food and Drug Administration (FDA) for the treatment of previously treated patients with CCA exhibiting FGFR2 rearrangement or fusions, with an overall response rate (ORR) of 35.5% and an overall median follow‐up time of 17.8 months. 57 Based on the results of the phase III ClarIDHy study, ivosidenib has also been recommended as a second‐line treatment for patients with IDH1‐mutant CCA, which reduces the risk of death by 63% compared with placebo. 58 In addition, in 2018, the FDA included pembrolizumab as a treatment option for patients with unresectable or metastatic MSI‐H/dMMR BTC. 59
5. TARGETED THERAPY IN CCA
With the development of gene detection technologies, including next‐generation sequencing, studies have elucidated the role of gene mutations and abnormal signaling pathways in the pathogenesis of BTC, leading to the discovery of new therapies for molecular targets. Liquid biopsy could overcome the limitations of tissue biopsy (limited tissue, tumor heterogeneity), aid in the analysis of biomarkers of drug resistance, and assist with longitudinal monitoring. 60 In the prospective clinical study MOSCATO‐01, patients with BTC who received targeted therapy based on identified molecules exhibited an encouraging improvement in mOS compared with those who received unselected therapy (17 months vs. 5 months; P = .008). 61 The important targets involved in this pathway include FGFR, IDH, ERBB, c‐MET, BRAF, NTRK, and anti‐VEGF therapy (Figure 3).
FIGURE 3.
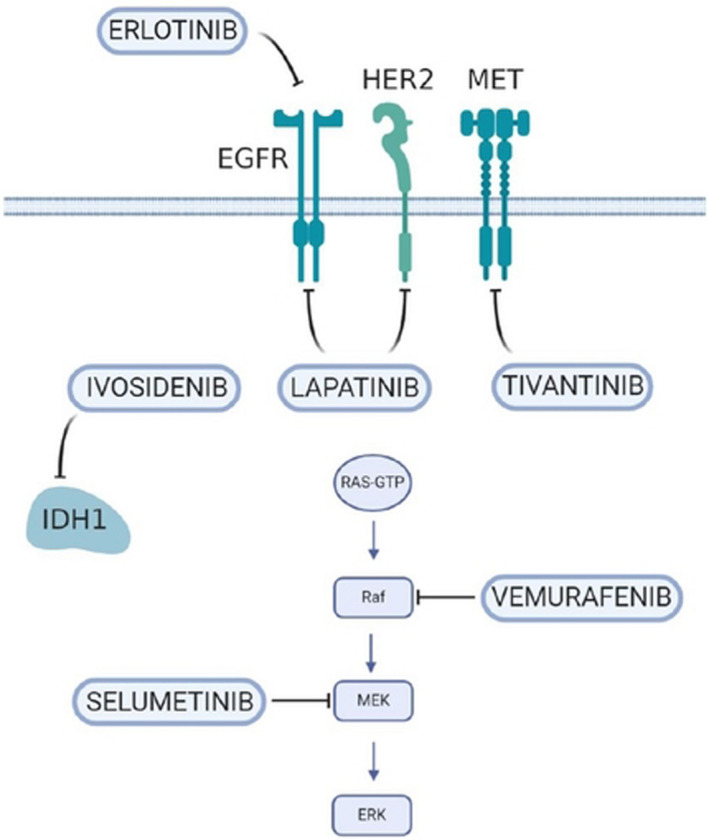
New therapeutic target in cholangiocarcinoma (CCA). EGFR, epidermal growth factor receptor; IDH1, isocitrate dehydrogenase 1; MEK, mitogen‐activated protein kinase; MET, tyrosine protein kinase MET; RAF, serin‐threonine protein)
5.1. FGFR‐targeted therapy
FGFR2 is one of the most promising therapeutic targets identified in CCA in recent years. Derazantinib (ARQ087), a potent orally bioavailable multikinase inhibitor with pan‐FGFR activity, has shown encouraging antitumor activity and a manageable safety profile in patients with chemotherapy‐refractory ICC harboring FGFR2 fusion. 62 A pivotal phase II study of derazantinib is currently underway in patients with inoperable or advanced ICC (NCT03230318). A multicenter, open‐label, phase II study demonstrated the acceptable toxicity and potential efficacy of infigratinib (BGJ398)—an orally bioavailable, selective pan‐FGFR kinase inhibitor—in 61 patients with FGFR‐altered advanced CCA. 63 The ORR was 14.8%, and the DCR was 75.4%, with an mPFS of 5.8 months (95% CI: 4.3 to 7.6 months). A phase III study compared the efficacy of infigratinib and GemCis as first‐line treatment for patients with unresectable locally advanced or metastatic ICC with FGFR2 gene fusions/translocations (NCT03773302). Following the recent FDA approval of pemigatinib and its appearance in the National Comprehensive Cancer Network (NCCN) Guidelines, the European Medicine Agency's Committee for Medicinal Products for Human Use (CHMP) also recommended it for patients with locally advanced and previously treated CCA and for those with metastatic CCA harboring an FGFR2 gene fusion or rearrangement. However, the rapid emergence of acquired resistance has often been observed. Recent studies have shown that the third‐generation irreversible FGFR inhibitor futibatinib (TAS‐120) provides clinical benefits for patients with ICC who have acquired resistance to infigratinib. 64 In a phase II study (FOENIX‐CCA2), 67 patients with ICC harboring FGFR2 fusions/other rearrangements received futibatinib and had an ORR of 37.3%, a median duration of response (DOR) of 8.3 months, and a DCR of 82%. 65 Clinical trials of other selective FGFR inhibitors are underway (NCT04238715, NCT04526106, NCT04565275).
5.2. IDH‐targeted therapy
IDH gene mutations affect the proliferation and differentiation of HPCs by inhibiting HNF‐4α (a primary regulator of hepatocyte identity and quiescence), which plays an important role in the pathogenesis of CCA. 66 The phase III ClarIDHy study showed that ivosidenib improved PFS relative to placebo treatment: The mPFS was 2.7 months in the 126 patients treated with ivosidenib, while it was 1.4 months in the 61 patients receiving placebo (HR: 0.37, P < .001). The median OS for ivosidenib was 10.3 months, compared with 7.5 months for placebo (HR: 0.79; 95% CI: 0.56‐1.12). 58 However, given the limited survival benefits and high costs, it is necessary to consider the patients’ financial situation and ensure proper communication. A study of high‐throughput drug screening in a large panel of cancer cell lines found that IDH‐mutant ICC cells are highly sensitive to the multi‐kinase inhibitor dasatinib. 67 Furthermore, a phase II clinical trial is exploring the safety and effectiveness of dasatinib in patients with IDH‐mutant advanced ICC (NCT02428855).
5.3. ERBB‐targeted therapy
The receptor tyrosine protein kinase ERBB‐2, also known as HER2, is a member of the EGFR family of receptor tyrosine kinases. In a retrospective analysis, eight patients with GBC with HER2/neu amplification or overexpression achieved a 100% DCR (1 CR, 4 PR, and 3 SD) after receiving HER2‐directed therapy (trastuzumab, lapatinib, or pertuzumab). 15 In the SUMMIT global basket study, treatment with neratinib (an irreversible inhibitor of HER1, HER2, and HER4) resulted in an ORR of 22% in nine patients with BTC exhibiting HER2 mutations. 68 Despite the limited number of patients involved, the results of these studies provide insights into the treatment of patients with HER2‐positive BTC. However, two phase II trials demonstrated that lapatinib, a dual‐target tyrosine kinase inhibitor (TKI) of HER1 and HER2, showed no benefits in patients with advanced BTC when administered as monotherapy, 69 , 70 although it improved the clinical outcome of patients with breast cancer with HER2 amplification or overexpression.
5.4. KRAS‐BRAF‐MEK‐ERK pathway
A phase II basket study of vemurafenib, a BRAF inhibitor, has reported a potential benefit in patients with CCA harboring BRAF V600 mutations. 71 In another basket trial (ROAR) the strategy of simultaneously inhibiting BRAF (dabrafenib) and MEK (trametinib) was tested in solid tumors. In the subgroup of 32 evaluable patients with BTC, the ORR was 41% (13/32; 95% CI: 24‐59%), mPFS was 7.2 months (95% CI: 4.6‐10.1), and the mOS was 11.3 months (95% CI: 7.3‐17.6). 72 Based on these studies, routine screening for BRAF V600e mutations is recommended in patients with CCA. A phase II study of the MEK1/2 inhibitor selumetinib has reported an mPFS of 3.7 months and an mOS of 9.8 months in patients with advanced BTC. 73
5.5. Neurotropic tyrosine receptor kinase (NTRK)‐targeted therapy
The binding of NTRK to the tropomyosin‐related kinase (TRK) receptor leads to the activation of the Ras/MAPK pathway, which leads to increased conduction and cell growth through ERK signaling, thus playing a carcinogenic role. 74 As a pioneer of broad‐spectrum anticancer drugs, NTRK inhibitors may play a role in a small proportion of patients with BTC (0.67%). 75 In a previous study, 54 patients with advanced or metastatic NTRK fusion‐positive solid tumors (one patient with CCA) treated with entrectinib achieved an ORR of 57%. 76 Larotrectinib (another highly selective NTRK inhibitor) also showed excellent antitumor activity in patients with NTRK fusion‐positive tumors, with an ORR of 93% (14 of 15 patients). 77
5.6. Anti‐VEGF therapy
Two phase II clinical trials have revealed that regorafenib has promising efficacy in patients with BTC after failure of GemCis first‐line treatment. 55 , 56 However, treatment with vandetanib (an orally available, dual VEGFR2 and EGFR TKI) did not improve PFS in patients with advanced BTC, according to the VanGogh Phase II multicenter study. 72 The ABC‐03 study also showed that adding cediranib (an oral VEGFR inhibitor) to GemCis could not slow the growth of cancer. 78 The role of VEGF inhibition in patients with advanced BTC is still under investigation.
5.7. c‐MET targeted therapy
The activity of tinvatinib, a c‐MET kinase inhibitor, has been investigated both in cell lines and in a phase I study (ARQ 197), showing antineoplastic activity. 79 , 80 A phase II trial evaluated the activity of cabozantinib, an inhibitor of the tyrosine kinases c‐Met and VEGFR2, in patients with advanced CCA after progression on first‐line or second‐line systemic therapy, which demonstrated limited clinical activity and significant toxicity 81 : In the 19 patients enrolled, the mPFS was 1.8 months, and the mOS was 5.2 months (95% CI: 2.7‐10.5 months). A phase II study is now assessing the efficacy and safety of crizotinib in patients harboring alterations in ALK, MET, or ROS1 (NCT02034981).
5.8. Poly (ADP‐ribose) polymerase inhibitors (PARPi)
PARP1 is an abundant ribozyme that can be activated when DNA is notched or broken, playing an important role in maintaining genomic stability. Abnormal functioning of BRCA1 or BRCA2 makes cells significantly sensitive to the inhibition of PARP enzyme activity, leading to chromosomal instability, cell cycle arrest, and subsequent apoptosis. 82 Ongoing trials are investigating the use of niraparib in patients with BAP1‐mutant tumors (NCT03207347). In addition, in vitro and in vivo IDH mutant tumor models show sensitivity to PARPi, 83 and a phase II trial involving patients with CCA is underway to test this concept (NCT03212274).
5.9. Anti‐Dickkopf‐1 (Dkk1) antibody
Dkk1 is an inhibitor of oncogenic beta‐catenin–dependent Wnt signaling through negative feedback. 84 But DKK1 can also target β‐catenin in myeloid‐derived suppressor cells (MDSC), which may lead to immune escape and promote the proliferation of cancer cells. 85 DKN‐01, an anti‐DKK1 antibody, has proven safety in studies of various malignancies and showed potential efficacy in patients with Wnt activating mutations (such as CTNNB1 and APC mutations) or high expression of Dkk1. 84 , 86 A phase II trial of the combination of DKN‐01 and nivolumab is ongoing in previously treated patients with advanced BTC (NCT04057365).
6. IMMUNOTHERAPY IN BTC
Many cancers are able to evade the immune system, mainly by overexpressing inhibitory ligands to suppress T cell attack. It is now evident that immune checkpoint modulation plays a relevant role in gastrointestinal cancers as well. Therefore, an increasing number of research groups aim to enhance T cell–mediated antitumor immune responses through checkpoint blockade and adoptive cell therapy (ACT).
6.1. Immune checkpoint inhibition
The clinical benefits of immune checkpoint inhibitors (CPIs) have been demonstrated in tumors with MSI‐H/dMMR and TMB‐H (TMB ≥10 mutations/megabase) status. 59 Higher PD‐L1 expression in tumors is correlated with venous invasion and advanced tumor staging, leading to poor prognosis of BTC. 87 , 88 According to a previous study of 652 patients with BTC, 8.6% were programmed cell death protein 1 ligand (PD‐L1) positive with the following distribution: GBC 12.3%, ICC 7.3%, and ECC 5.2%. 89 It is worth stating that compared with conventional CCA, higher PD‐L1 overexpression and TMB was observed in occupational CCA (P < .01). 90 The number of inhibitory ligand–positive tumor‐infiltrating cells in the tumor microenvironment of occupational CCA was also significantly increased, which may be promising targets for mobilizing immune cells to attack the tumors. 91 Taken together, these studies provide the basis for treating patients with BTC using CPIs.
Early data from KEYNOTE‐028 investigating outcomes in PD‐L1–positive patients with advanced BTC pretreated with blockers reported that four (17%) of the 24 patients had a PR, while another four achieved SD. 92 A phase II study using another programmed cell death protein 1 (PD‐1) antibody, nivolumab, was conducted in 54 patients with advanced BTC after progression despite standard‐line therapy. The ORR was 22%, and the associated mPFS and mOS were 3.9 and 14.2 months, respectively. 89 It is also worth mentioning that, in KEYNOTE‐158, pembrolizumab achieved an ORR of 40.9% and an mPFS of 4.2 months in patients with BTC presenting with MSI‐H/dMMR status. 59
Cytotoxic T lymphocyte–associated protein 4 (CTLA‐4) and PD‐1/PD‐L1 suppress T cell–mediated immune responses at different stages and locations; thus, they may act synergistically to activate antitumor immune responses. 93 A phase II clinical trial evaluated the efficacy and safety of combining nivolumab and ipilimumab (an anti‐CTLA‐4 inhibitor) in patients with advanced BTC, with an ORR of 23% and DCR of 44%. 94 There are other ongoing clinical trials evaluating the effects of dual immune checkpoint suppression therapy in patients with BTC (Table 1b).
TABLE 1.
Selected ongoing clinical trials with immunotherapy in BTC
| Therapeutic | Target | Phase | Status | Design | Trail NCT | Endpoints |
|---|---|---|---|---|---|---|
| (a) Checkpoint inhibitor monotherapy | ||||||
| Pembrolizumab (MK‐3475) | PD‐1 | Ib | Active, not recruiting | Single Group Assignment | NCT02054806 | ORR/PFS |
| Pembrolizumab (MK‐3475) | PD‐1 | II | Recruiting | Single Group Assignment | ORR/PFS; ORR | |
| Nivolumab | PD‐1 | II | Active, not recruiting | Single Group Assignment | NCT02829918 | ORR |
| STI‐3031 | PD‐L1 | II | Not yet recruiting | Single Group Assignment | NCT03999658 | ORR/DOR/PFS |
| M7824 | PD‐L1 | II | Recruiting | Single Group Assignment | NCT03833661 | OR/DOR |
| Nivolumab or pembrolizumab | PD‐1 | ‐ | Recruiting | Prospective Cohort Study | NCT03695952 | ORR |
| (b) Dual checkpoint inhibition | ||||||
|
MEDI4736 Tremelimumab |
PD‐L1 monotherapy/with CTLA‐4 | I | Active, not recruiting | Parallel Assignment | NCT01938612 | Safety and tolerability |
|
Nivolumab Ipilimumab |
PD‐1 CTLA‐4 |
II | Recruiting | Parallel Assignment | NCT02834013 | ORR |
| Gemcitabine + Cisplatin + Nivolumab; Nivolumab + Ipilimumab |
PD‐1 PD‐1+ CTLA‐4 |
II | Active, not recruiting | Parallel Assignment (Randomized) | NCT03101566 | PFS |
| (c) Checkpoint inhibition plus myeloid cell immunosuppression | ||||||
|
Pembrolizumab Sargramostim |
PD‐1 GM‐CSF |
II | Active, not recruiting | Single Group Assignment | NCT02703714 | ORR |
|
Nivolumab Nivolumab + Cabrilizumab |
PD‐1 CSF‐1R |
‐ | Recruiting | Parallel Assignment (Randomized) | NCT03768531 | Safety and tolerability |
| (d) Adoptive cell therapy | ||||||
|
IL‐2 HER2Bi‐Armed T Cells |
I | Unknown | Single Group Assignment | NCT02662348 | Safety and tolerability | |
| CART‐HER‐2 | I | Unknown | Single Group Assignment | NCT01935843 | Safety and tolerability | |
| CART‐EGFR | I | Unknown | Single Group Assignment | NCT01869166 | Safety and tolerability | |
| Cytokine induced killer cells | I | Recruiting | Single Group Assignment | NCT01868490 | Tumor size and CIK cell–homing | |
|
TC‐210 T Cells TC‐210 + fludarabine + cyclophosphamide TC‐210+ fludarabine + cyclophosphamide + anti‐PD1 |
I/II | Recruiting | Single Group Assignment | NCT03907852 | Safety and tolerability | |
| Tumor‐Infiltrating Lymphocytes (TIL) | II | Recruiting | Single Group Assignment | NCT03801083 | ORR/CRR/DOR | |
| MUC‐1 CART cell | ‐ | Recruiting | Single Group Assignment | NCT03633773 | DCR/ORR | |
| (e) Adoptive cell therapy plus checkpoint inhibition | ||||||
|
CD8+ T Cell Aldesleukin Cyclophosphamide Pembrolizumab |
I | Active, not recruiting | Single Group Assignment | NCT02757391 | Safety and tolerability | |
|
'SMT‐NK' Inj Pembrolizumab |
I/IIa | Recruiting | Single Group Assignment | NCT03937895 |
Phase I: RP2D Phase IIa: ORR |
|
| (f) Checkpoint inhibition plus targeting | ||||||
|
Ramucirumab Pembrolizumab |
VEGFR‐2 PD‐1 |
I | Active, not recruiting | Parallel Assignment | NCT02443324 | Number of participants who experienced DLTs |
|
Guadecitabine Durvalumab |
DNMT PD‐L1 |
Ib | Recruiting | Single Group Assignment | NCT03257761 | Incidence of TEAEs, RP2D |
|
Anlotinib TQB245 |
VEGFR, PDGFR, FGFR, c‐Kit PD‐L1 |
Ib | Recruiting | Single Group Assignment | NCT03996408 | DLT/MTD/RP2D/ORR |
|
Regorafenib Avelumab |
VEGFR1‐3, PDGFR‐β, Kit, RET, Raf‐1 PD‐L1 |
I/II | Recruiting | Sequential Assignment | NCT03475953 |
Phase I: RP2D Phase II: ORR |
|
FT‐2102 Nivolumab |
IDH1 PD‐1 |
Ib/II | Recruiting | Parallel Assignment | NCT03684811 |
Phase Ib: DLT/RP2D Phase II: ORR |
|
Lenvatinib Pembrolizumab |
VEGFR1‐3, FGFR1‐4, PDGFR‐β, RET, KIT PD‐1 |
II | Recruiting | Single Group Assignment | NCT03895970 | ORR/DCR/PFS |
|
Lenvatinib Pembrolizumab |
VEGFR1‐3, FGFR1‐4, PDGFR‐β, RET, KIT PD‐1 |
II | Active, not recruiting | Single Group Assignment | NCT03797326 | ORR, Incidence of TEAEs |
|
Rucaparib Nivolumab |
PARP PD‐1 |
II | Recruiting | Single Group Assignment | NCT03639935 | PFS |
|
Axitinib Toripalimab |
VEGFR1‐3, PDGFR‐β, c‐Kit PD‐1 |
II | Not yet recruiting | Single Group Assignment | NCT04010071 | ORR/PFS |
|
Olaparib Durvalumab |
PARP PD‐L1 |
II | Not yet recruiting | Parallel Assignment | NCT03991832 | ORR/DCR |
|
DKN‐01 Nivolumab |
DKK1 PD‐1 |
II | Recruiting | Parallel Assignment | NCT04057365 | ORR |
|
SHR‐1210 + Apatinib SHR‐1210 + FOLFOX4/GEMOX regimen |
PD‐1, VEGFR‐2 PD‐1+ chemotherapy |
II | Recruiting | Parallel Assignment | NCT03092895 | Safety and tolerability |
|
Entinostat Nivolumab |
HDAC1, HDAC3 PD‐1 |
II | Recruiting | Parallel Assignment | NCT03250273 | ORR |
|
Atezolizumab Atezolizumab + Cobimetinib |
PD‐L1 MEK |
II | Active, not recruiting | Parallel Assignment (Randomized) | NCT03201458 | PFS |
| (g) Checkpoint inhibition plus chemotherapy | ||||||
|
Nivolumab Gemcitabine Cisplatin |
PD‐1 | I/II | Unknown | Single Group Assignment | NCT03311789 | 6‐month PFS rate, mOS |
|
INT230‐6 Pembrolizumab or anti‐CTLA‐4 antibody |
PD‐L1 CTLA‐4 |
I/II | Recruiting | Sequential Assignment | NCT03058289 | Safety and tolerability |
|
Manganese chloride Nab‐paclitaxel Gemcitabine Anti‐PD‐1 antibody |
PD‐1 | I/II | Recruiting | Single Group Assignment | NCT04004234 | Incidence of TEAEs, PFS |
|
Nivolumab Nanoliposomal‐Irinotecan 5‐Fluorouracil Leucovorin |
PD‐1 | Ib/II | Recruiting | Single Group Assignment | NCT03785873 |
Phase Ib: Incidence of DLTs Phase II: mPFS |
|
Nivolumab TS‐1 Gemcitabine |
PD‐1 | II | Recruiting | Single Group Assignment | NCT04172402 | ORR |
|
Toripalimab Gemcitabine 5‐ fluorine pyrimidine |
PD‐1 | II | Recruiting | Single Group Assignment | NCT03982680 | 6‐month PFS rate, mPFS, toxic side effects |
|
Toripalimab S1 Albumin Paclitaxel |
PD‐1 | II | Recruiting | Single Group Assignment | NCT04027764 | ORR/PFS/DCR/OS |
|
SHR‐1210 GEMOX |
PD‐1 | II | Recruiting | Single Group Assignment | NCT03486678 | 6‐month PFS rate, incidence of TEAEs |
|
Pembrolizumab Oxaliplatin Capecitabine |
PD‐1 | II | Recruiting | Single Group Assignment | NCT03111732 | 5‐month PFS rate, safety, ORR, OS |
|
Pembrolizumab Cisplatin Gemcitabine |
PD‐1 | II | Recruiting | Single Group Assignment | NCT03260712 | 6‐month PFS rate, ORR, toxicity |
|
GS Toripalimab |
PD‐1 | II | Recruiting | Single Group Assignment | NCT03796429 | PFS/OS/ORR |
|
Durvalumab Tremelimumab Gemcitabine/ Cisplatin |
PD‐L1 CTLA‐4 |
II | Recruiting | Single Group Assignment | NCT03046862 | ORR/DCR/PFS/OS |
|
Durvalumab + Tremelimumab + Gemcitabine Durvalumab + Tremelimumab + Gemcitabine + Cisplatin Gemcitabine + Cisplatin |
PD‐L1 CTLA‐4 |
II | Recruiting | Parallel Assignment (Randomized) | NCT03473574 | ORR, OS, incidence of TEAEs |
|
M7824 + Gemcitabine + Cisplatin Placebo + Gemcitabine + Cisplatin |
PD‐L1 | II/III | Recruiting | Parallel Assignment (Randomized) | NCT04066491 | Number of participants who experienced DLTs, OS |
|
Pembrolizumab + Gemcitabine + Cisplatin Placebo + Gemcitabine + Cisplatin |
PD‐1 | III | Recruiting | Parallel Assignment (Randomized) | NCT04003636 | PFS/OS/ORR |
|
Durvalumab + Gemcitabine + Cisplatin Placebo + Gemcitabine + Cisplatin |
PD‐L1 | III | Recruiting | Parallel Assignment (Randomized) | NCT03875235 | OS/PFS/ORR |
|
KN035 + Gemcitabine + oxaliplatin Gemcitabine + oxaliplatin |
PD‐L1 | III | Recruiting | Parallel Assignment (Randomized) | NCT03478488 | OS/PFS/ORR |
| (h) Checkpoint inhibition plus targeting and chemotherapy | ||||||
|
Oxaliplatin Gemcitabine Lenvatinib JS001 |
VEGFR1‐3, FGFR1‐4, PDGFRα, RET, KIT PD‐1 |
II | Active, not recruiting | Single Group Assignment | NCT03951597 | ORR/OS/PFS |
| (i) Checkpoint inhibition plus ablative local therapy | ||||||
|
Tremelimumab Durvalumab Radiation |
CTLA‐4 PD‐L1 |
II | Recruiting | Single Group Assignment | NCT03482102 | ORR, incidence of TEAEs, OS, DCR, PFS |
|
Durvalumab + Tremelimumab Durvalumab + Tremelimumab + TACE Durvalumab + Tremelimumab + RFA Durvalumab + Tremelimumab + Cryoablation |
PD‐L1 CTLA‐4 |
II | Recruiting | Parallel Assignment | NCT02821754 | PFS, incidence of TEAEs |
|
Radiotherapy + Camrelizumab Gemcitabine + Cisplatin |
PD‐1 | II | Recruiting | Parallel Assignment (Randomized) | NCT03898895 | PFS, OS, incidence of TEAEs |
|
Nivolumab + RA Nivolumab + Ipilimumab + RA |
PD‐1 CTLA‐4 |
II | Recruiting | Parallel Assignment (Randomized) | NCT02866383 | CBR, incidence of TEAEs, ORR, PFS, OS |
Abbreviations: CTLA‐4, cytotoxic T lymphocyte–associated protein 4; DCR, disease control rate; DLT, dose‐limiting toxicity; DOR, duration of response; GM‐CSF, human granulocyte‐macrophage colony‐stimulating factor; HDAC, histone deacetylases; MEK, methyl ethyl ketone; mOS, median OS; mPFS, median PFS; ORR, objective response rate; OS, overall survival; PDGFR‐β, platelet‐derived growth factor receptor beta; PD‐1, programmed cell death protein 1; PD‐L1, programmed cell death 1 ligand 1; PFS, progression‐free survival; RP2D, recommended phase 2 dose; NCT, national clinical trial; MTD, maximum tolerated dose; CIK, cytokine‐induced killer; TEAE, treatment emergent adverse event; CBR clinical benefit rate.
6.2. ACT
ACT uses host cells that have been genetically engineered with antitumor T cell receptors (TCRs) or chimeric antigen receptors (CARs), as well as natural host cells such as natural killer (NK) cells , to induce antitumor reactivity.
Recently, a phase I trial of 19 patients with EGFR‐positive advanced BTC reported that one patient achieved complete response (CR) and 10 patients achieved SD after CAR T‐EGFR cell immunotherapy, demonstrating the safety and feasibility of this therapy. 95 Another phase I trial of 11 patients with HER2‐positive (>50%) BTC reported an mPFS of 4.8 months (range: 1.5‐8.3 months), suggesting encouraging clinical activity. 96
Other studies have indicated that ex vivo–expanded and activated NK cells exhibit effective antitumor activity both in vitro and in vivo. A phase I/IIa clinical trial is evaluating the safety and efficacy of allogeneic NK cells ("SMT‐NK") in combination with pembrolizumab for patients with gemcitabine‐refractory BTC (NCT03937895).
The successful use of ACT lies in identifying appropriate targets that are selectively expressed in cancers, which could be challenging in solid tumors.
6.3. Immunotherapy in combination with other therapies
The tumor microenvironment plays a vital role in accelerating the formation of new blood vessels, enhancing the proliferation and invasion of cancer cells, and preventing apoptosis of tumor cells. 97 Complementary combinations of immunotherapy are increasingly being explored as strategies to improve efficacy. Multiple clinical trials using CPIs plus targeted or cytotoxic drugs are underway (Table 1).
6.3.1. Immunotherapy in combination with targeted therapy
Preclinical evidence indicates a close correlation between angiogenesis and suppression of the antitumor response. VEGF increases T cell exhaustion by enhancing the expression of inhibitory checkpoints on T cells, while simultaneous blocking of VEGFR and PD‐1/PD‐L1 can induce cumulative antitumor effects. 98 Lenvatinib is a multiple receptor TKI that mainly targets VEGFR and FGFR. Flow cytometric analysis has revealed that lenvatinib enhances the ability to induce CD8+ T cells via the interferon (IFN) signaling pathway when combined with PD‐1 blockade. 99 The efficacy and safety of this strategy are being evaluated in patients with CCA (NCT03797326, NCT03895970). However, only one patient achieved an objective response in the trial of ramucirumab (an antagonist of VEGFR2) and pembrolizumab, in which 46.2% (11/26) of patients were PD‐L1 positive. 98 PARPi can upregulate PD‐L1 expression in breast cancer cells, which may lead to increased sensitivity to CPI therapy. 100 A phase II trial of this combination therapy is expected to start recruiting patients to assess the efficacy of IDH‐mutated CCAs (NCT03991832).
6.3.2. Immunotherapy in combination with chemotherapy
There is growing evidence that cytotoxic drugs can also strengthen the immune system by increasing the ratio of cytotoxic lymphocytes to regulatory T cells and the number of antigen‐presenting cells. In vitro, CCA cells treated with gemcitabine can induce the mRNA expression of PD‐L1. 101 These encouraging experiments highlight the potential and feasibility of this treatment strategy, and larger clinical trials are thus underway. A phase II study demonstrated tolerability and good response durability for GemCis combined with durvalumab (D) ± tremelimumab (T) in 121 first‐line patients with advanced BTC, with an ORR of 70% and a DCR of 95%. 102 The efficacy of GemCis combined with durvalumab is being further studied in the phase III TOPAZ‐1 trial.
6.3.3. Immunotherapy in combination with cancer vaccine
A phase I trial of 36 patients with ICC confirmed that adoptive T cell transfer combined with a dendritic cell vaccine may magnify tumor destruction. The mPFS and mOS were 18.3 and 31.9 months in patients receiving adjuvant immunotherapy and 7.7 and 17.4 months in patients undergoing surgery alone (P = .005 and .022, respectively). 103
7. CONCLUSION
Given the heterogeneity of BTC, molecular characterization may represent the key to improving clinical outcomes through personalized therapy. Targeted therapies have shown remarkable results in patients with BTC harboring identified actionable mutations (such as FGFR2 fusions and IDH gene mutations) and have been successively approved as the standard treatment. Therefore, clinical research should focus on developing targeted therapies. Immunotherapy represents a promising approach for patients with BTC with MSI‐H/dMMR disease. Nevertheless, identifying the characteristics of specific patients likely to benefit from each treatment remains a challenge that must be overcome to guide the precise treatment of BTC. Longitudinal liquid biopsies such as ctDNA analyses may aid in promoting the development of precision medicine and improve therapeutic benefits in some patients. To date, chemotherapy remains the standard of care for advanced diseases. As we wait for new randomized clinical trials to shed light on the current dilemma, the possible options for patients with BTC should be discussed by a multidisciplinary team to ensure selection of the most appropriate treatment for each patient.
CONFLICT OF INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
Zhang R, Puzzoni M, Mariani S, et al. Emerging treatment evolutions and integrated molecular characteristics of biliary tract cancers. Cancer Sci. 2021;112:4819–4833. doi: 10.1111/cas.15139
Zhang and Puzzoni contributed equally as first authors.
Funding information
This paper is funded by the NationalNational Major Scientific and Technological Special Project for Significant New Drugs Development during the Thirteenth Five‐Year Plan Period 2020ZX 09201‐003. Major Scientific and Technological Special Project for Significant New Drugs Development during the Thirteenth Five‐Year Plan Period 2020ZX 09201‐003.
Contributor Information
Weijia Fang, Email: weijiafang@zju.edu.cn.
Mario Scartozzi, Email: marioscartozzi@gmail.com.
REFERENCE
- 1. Valle JW, Kelley RK, Nervi B, Oh D‐Y, Zhu AX. Biliary tract cancer. Lancet. 2021;397(10272):428‐444. [DOI] [PubMed] [Google Scholar]
- 2. Gunasekaran G, Bekki Y, Lourdusamy V, Schwartz M. Surgical Treatments of Hepatobiliary Cancers. Hepatology. 2021;73:128‐136. [DOI] [PubMed] [Google Scholar]
- 3. Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935):2168‐2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim H, Park C, Han KH, et al. Primary liver carcinoma of intermediate (hepatocyte‐cholangiocyte) phenotype. J Hepatol. 2004;40(2):298‐304. [DOI] [PubMed] [Google Scholar]
- 5. Komuta M, Govaere O, Vandecaveye V, et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology. 2012;55(6):1876‐1888. [DOI] [PubMed] [Google Scholar]
- 6. Banales JM, Cardinale V, Carpino G, et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS‐CCA). Nat Rev Gastroenterol Hepatol. 2016;13(5):261‐280. [DOI] [PubMed] [Google Scholar]
- 7. Jain A, Kwong LN, Javle M. Genomic profiling of biliary tract cancers and implications for clinical practice. Curr Treat Options Oncol. 2016;17(11):58. [DOI] [PubMed] [Google Scholar]
- 8. Brunt E, Aishima S, Clavien PA, et al. cHCC‐CCA: Consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology. 2018;68(1):113‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J, Dong M, Xu Z, et al. Notch2 controls hepatocyte‐derived cholangiocarcinoma formation in mice. Oncogene. 2018;37(24):3229‐3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kendall T, Verheij J, Gaudio E, et al. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019;39(Suppl 1):7‐18. [DOI] [PubMed] [Google Scholar]
- 11. Wistuba II, Gazdar AF. Gallbladder cancer: lessons from a rare tumour. Nat Rev Cancer. 2004;4(9):695‐706. [DOI] [PubMed] [Google Scholar]
- 12. Marcano‐Bonilla L, Mohamed EA, Mounajjed T, Roberts LR. Biliary tract cancers: epidemiology, molecular pathogenesis and genetic risk associations. Chin Clin Oncol. 2016;5(5):61. [DOI] [PubMed] [Google Scholar]
- 13. Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45(12):1470‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roos E, Soer EC, Klompmaker S, et al. Crossing borders: A systematic review with quantitative analysis of genetic mutations of carcinomas of the biliary tract. Crit Rev Oncol Hematol. 2019;140:8‐16. [DOI] [PubMed] [Google Scholar]
- 15. Javle M, Churi C, Kang HC, et al. HER2/neu‐directed therapy for biliary tract cancer. J Hematol Oncol. 2015;8:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Silva VWK, Askan G, Daniel TD, et al. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chinese Clin Oncol. 2016;5(5):62. [DOI] [PubMed] [Google Scholar]
- 17. Spizzo G, Puccini A, Xiu J, et al. Molecular profile of BRCA‐mutated biliary tract cancers. Esmo Open. 2020;5(3):e000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32(25):3091‐3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Akita M, Sofue K, Fujikura K, et al. Histological and molecular characterization of intrahepatic bile duct cancers suggests an expanded definition of perihilar cholangiocarcinoma. HPB. 2019;21(2):226‐234. [DOI] [PubMed] [Google Scholar]
- 20. Mahipal A, Tella SH, Kommalapati A, Anaya D, Kim R. FGFR2 genomic aberrations: Achilles heel in the management of advanced cholangiocarcinoma. Cancer Treat Rev. 2019;78:1‐7. [DOI] [PubMed] [Google Scholar]
- 21. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47(9):1003‐1010. [DOI] [PubMed] [Google Scholar]
- 22. Ma BQ, Meng HJ, Tian Y, et al. Distinct clinical and prognostic implication of IDH1/2 mutation and other most frequent mutations in large duct and small duct subtypes of intrahepatic cholangiocarcinoma. BMC Cancer. 2020;20(1):318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2‐PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nature Communications. 2015;6(6078). [DOI] [PubMed] [Google Scholar]
- 24. Graham RP, Barr Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45(8):1630‐1638. [DOI] [PubMed] [Google Scholar]
- 25. Yoshikawa D, Ojima H, Iwasaki M, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer. 2008;98(2):418‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee H, Wang K, Johnson A, et al. Comprehensive genomic profiling of extrahepatic cholangiocarcinoma reveals a long tail of therapeutic targets. J Clin Pathol. 2016;69(5):403‐408. [DOI] [PubMed] [Google Scholar]
- 27. Kubo S, Kinoshita M, Takemura S, et al. Characteristics of printing company workers newly diagnosed with occupational cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2014;21(11):809‐817. [DOI] [PubMed] [Google Scholar]
- 28. Mimaki S, Totsuka Y, Suzuki Y, et al. Hypermutation and unique mutational signatures of occupational cholangiocarcinoma in printing workers exposed to haloalkanes. Carcinogenesis. 2016;37(8):817‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu L, Tsilimigras DI, Paredes AZ, et al. Trends in the Incidence, Treatment and Outcomes of Patients with Intrahepatic Cholangiocarcinoma in the USA: Facility Type is Associated with Margin Status, Use of Lymphadenectomy and Overall Survival. World J Surg. 2019;43(7):1777‐1787. [DOI] [PubMed] [Google Scholar]
- 30. Nelson JW, Ghafoori AP, Willett CG, et al. Concurrent chemoradiotherapy in resected extrahepatic cholangiocarcinoma. Int J Radiat Oncol Biol Phys. 2009;73(1):148‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patkar S, Patel S, Gupta A, Ramaswamy A, Ostwal V, Goel M. Revision Surgery for Incidental Gallbladder Cancer‐Challenging the Dogma: Ideal Timing and Real‐World Applicability. Ann Surg Oncol. 2021;28:6758–6766. [DOI] [PubMed] [Google Scholar]
- 32. Le Roy B, Gelli M, Pittau G, et al. Neoadjuvant chemotherapy for initially unresectable intrahepatic cholangiocarcinoma. Br J Surg. 2018;105(7):839‐847. [DOI] [PubMed] [Google Scholar]
- 33. Rea DJ, Heimbach JK, Rosen CB, et al. Liver transplantation with neoadjuvant chemoradiation is more effective than resection for hilar cholangiocarcinoma. Ann Surg. 2005;242(3):451‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lunsford KE, Javle M, Heyne K. Liver transplantation for locally advanced intrahepatic cholangiocarcinoma treated with neoadjuvant therapy: a prospective case‐series. Lancet Gastroenterol Hepatol. 2018;3(5):337–348. [DOI] [PubMed] [Google Scholar]
- 35. Creasy JM, Goldman DA, Gonen M, et al. Evolution of surgical management of gallbladder carcinoma and impact on outcome: results from two decades at a single‐institution. Hpb. 2019;21(11):1541‐1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hyder O, Hatzaras I, Sotiropoulos GC, et al. Recurrence after operative management of intrahepatic cholangiocarcinoma. Surgery. 2013;153(6):811‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horgan AM, Amir E, Walter T, Knox JJ. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta‐analysis. J Clin Oncol. 2012;30(16):1934‐1940. [DOI] [PubMed] [Google Scholar]
- 38. Kubo S, Takemura S, Tanaka S, et al. Outcomes after resection of occupational cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2016;23(9):556‐564. [DOI] [PubMed] [Google Scholar]
- 39. Primrose JN, Fox RP, Palmer DH, et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): a randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 2019;20(5):663‐673. [DOI] [PubMed] [Google Scholar]
- 40. Edeline J, Benabdelghani M, Bertaut A, et al. Gemcitabine and Oxaliplatin Chemotherapy or Surveillance in Resected Biliary Tract Cancer (PRODIGE 12‐ACCORD 18‐UNICANCER GI): A Randomized Phase III Study. J Clin Oncol. 2019;37(8):658‐667. [DOI] [PubMed] [Google Scholar]
- 41. Ebata T, Hirano S, Konishi M, et al. Randomized clinical trial of adjuvant gemcitabine chemotherapy versus observation in resected bile duct cancer. Br J Surg. 2018;105(3):192‐202. [DOI] [PubMed] [Google Scholar]
- 42. Kobayashi S, Nagano H, Tomokuni A, et al. A Prospective, Randomized Phase II Study of Adjuvant Gemcitabine Versus S‐1 After Major Hepatectomy for Biliary Tract Cancer (KHBO 1208): Kansai Hepato‐Biliary Oncology Group. Ann Surg. 2019;270(2):230‐237. [DOI] [PubMed] [Google Scholar]
- 43. Stein A, Arnold D, Bridgewater J, et al. Adjuvant chemotherapy with gemcitabine and cisplatin compared to observation after curative intent resection of cholangiocarcinoma and muscle invasive gallbladder carcinoma (ACTICCA‐1 trial) ‐ a randomized, multidisciplinary, multinational phase III trial. BMC Cancer. 2015;15:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273‐1281. [DOI] [PubMed] [Google Scholar]
- 45. Fiteni F, Nguyen T, Vernerey D, et al. Cisplatin/gemcitabine or oxaliplatin/gemcitabine in the treatment of advanced biliary tract cancer: a systematic review. Cancer Med. 2014;3(6):1502‐1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morizane C, Okusaka T, Mizusawa J, et al. Combination gemcitabine plus S‐1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA‐BT (JCOG1113) randomized phase III clinical trial. Ann Oncol. 2019;30(12):1950‐1958. [DOI] [PubMed] [Google Scholar]
- 47. Kim ST, Kang JH, Lee J, et al. Capecitabine plus oxaliplatin versus gemcitabine plus oxaliplatin as first‐line therapy for advanced biliary tract cancers: a multicenter, open‐label, randomized, phase III, noninferiority trial. Ann Oncol. 2019;30(5):788‐795. [DOI] [PubMed] [Google Scholar]
- 48. Ioka T, Kanai M, Kobayashi S, et al. Randomised phase III study of gemcitabine, cisplatin plus S‐1 (GCS) compared with gemcitabine plus cisplatin (GC) for unresectable or recurrent biliary tract cancer (KHBO1401‐MITSUBA). J Clin Oncol. 2015;33(15_suppl):TPS4141. [Google Scholar]
- 49. Shroff RT, Javle MM, Xiao L, et al. Gemcitabine, Cisplatin, and nab‐Paclitaxel for the Treatment of Advanced Biliary Tract Cancers: A Phase 2 Clinical Trial. JAMA Oncol. 2019;5(6):824‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lamarca A, Palmer DH, Wasan HS, et al. ABC‐06 vertical bar A randomised phase III, multi‐centre, open‐label study of active symptom control (ASC) alone or ASC with oxaliplatin / 5‐FU chemotherapy (ASC plus mFOLFOX) for patients (pts) with locally advanced / metastatic biliary tract cancers (ABC) previously‐treated with cisplatin/gemcitabine (CisGem) chemotherapy. J Clin Oncol. 2019;37(15). [Google Scholar]
- 51. Ramaswamy A, Ostwal V, Pande N, et al. Second‐Line Palliative Chemotherapy in Advanced Gall Bladder Cancer, CAP‐IRI: Safe and Effective Option. J Gastrointest Cancer. 2016;47(3):305‐312. [DOI] [PubMed] [Google Scholar]
- 52. Zheng Y, Tu XX, Zhao P, et al. A randomised phase II study of second‐line XELIRI regimen versus irinotecan monotherapy in advanced biliary tract cancer patients progressed on gemcitabine and cisplatin. Br J Cancer. 2018;119(3):291‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sebbagh S, Roux J, Dreyer C, et al. Efficacy of a sequential treatment strategy with GEMOX‐based followed by FOLFIRI‐based chemotherapy in advanced biliary tract cancers. Acta Oncol. 2016;55(9–10):1168‐1174. [DOI] [PubMed] [Google Scholar]
- 54. Kim JW, Suh KJ, Kim J‐W, et al. A randomized phase II study of oxaliplatin/5‐FU (mFOLFOX) versus irinotecan/5‐FU (mFOLFIRI) chemotherapy in locally advanced or metastatic biliary tract cancer refractory to first‐line gemcitabine/cisplatin chemotherapy. J Clin Oncol. 2020;38(15_suppl):4603. [Google Scholar]
- 55. Sun WJ, Patel A, Normolle D, et al. A Phase 2 Trial of Regorafenib as a Single Agent in Patients With Chemotherapy‐Refractory, Advanced, and Metastatic Biliary Tract Adenocarcinoma. Cancer. 2019;125(6):902‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Demols A, Borbath I, Van den Eynde M, et al. Regorafenib after failure of gemcitabine and platinum‐based chemotherapy for locally advanced/metastatic biliary tumors: REACHIN, a randomized, double‐blind, phase II trial. Ann Oncol. 2020;31(9):1169‐1177. [DOI] [PubMed] [Google Scholar]
- 57. Abou‐Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open‐label, phase 2 study. Lancet Oncol. 2020;21(5):671‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhu AX, Macarulla T, Javle MM, et al. Final results from ClarIDHy, a global, phase III, randomized, double‐blind study of ivosidenib (IVO) versus placebo (PBO) in patients (pts) with previously treated cholangiocarcinoma (CCA) and an isocitrate dehydrogenase 1 (IDH1) mutation. J Clin Oncol. 2021;39(3_suppl):266. [Google Scholar]
- 59. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair‐Deficient Cancer: Results From the Phase II KEYNOTE‐158 Study. J Clin Oncol. 2019;38(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parikh AR, Leshchiner I, Elagina L, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med. 2019;25(9):1415‐1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Verlingue L, Malka D, Allorant A, et al. Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO‐01 trial. Eur J Cancer. 2017;87:122‐130. [DOI] [PubMed] [Google Scholar]
- 62. Mazzaferro V, El‐Rayes BF, Busset MDD, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion‐positive intrahepatic cholangiocarcinoma. Br J Cancer. 2019;120(2):165‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Javle M, Lowery M, Shroff RT, et al. Phase II Study of BGJ398 in Patients With FGFR‐Altered Advanced Cholangiocarcinoma. J Clin Oncol. 2018;36(3):276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Goyal L, Shi L, Liu LY, et al. TAS‐120 Overcomes Resistance to ATP‐Competitive FGFR Inhibitors in Patients with FGFR2 Fusion‐Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019;9(8):1064‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bridgewater J, Meric‐Bernstam F, Hollebecque A, et al. Efficacy and safety of futibatinib in intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 fusions/other rearrangements: Subgroup analyses of a phase II study (FOENIX‐CCA2). Ann Oncol. 2020;31:S261‐S262. [Google Scholar]
- 66. Saha SK, Parachoniak CA, Ghanta KS, et al. Mutant IDH inhibits HNF‐4alpha to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Saha SK, Gordan JD, Kleinstiver BP, et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2016;6(7):727‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Profiling differential responses to Pan‐HER inhibition. Cancer Discov. 2017;7(6):OF12. [DOI] [PubMed] [Google Scholar]
- 69. Peck J, Wei L, Zalupski M, O’Neil B, Villalona Calero M, Bekaii‐Saab T. HER2/neu May Not Be an Interesting Target in Biliary Cancers: Results of an Early Phase II Study with Lapatinib. Oncology. 2012;82(3):175‐179. [DOI] [PubMed] [Google Scholar]
- 70. Ramanathan RK, Belani CP, Singh DA, et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother Pharmacol. 2009;64(4):777‐783. [DOI] [PubMed] [Google Scholar]
- 71. Hyman DM, Blay JY, Chau I, et al. VE‐BASKET, a first‐in‐kind, phase II, histology‐independent "basket" study of vemurafenib (VEM) in nonmelanoma solid tumors harboring BRAF V600 mutations (V600m). J Clin Oncol. 2014;32(15_suppl):2533. [Google Scholar]
- 72. Wainberg ZA, Lassen UN, Elez E, et al. Efficacy and safety of dabrafenib (D) and trametinib (T) in patients (pts) with BRAF V600E–mutated biliary tract cancer (BTC): A cohort of the ROAR basket trial. J Clin Oncol. 2019;37(4_suppl):187. [Google Scholar]
- 73. Bekaii‐Saab T, Phelps MA, Li XB, et al. Multi‐Institutional Phase II Study of Selumetinib in Patients With Metastatic Biliary Cancers. J Clin Oncol. 2011;29(17):2357‐2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Amatu A, Sartore‐Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. Esmo Open. 2016;1(2):e000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Demols A, Perez‐Casanova L, Rocq L, et al. O‐4 NTRK gene fusions in bilio‐pancreatic cancers. Ann Oncol. 2020;31:233. [Google Scholar]
- 76. Doebele RC, Drilon A, Paz‐Ares L, et al . Entrectinib in patients with advanced or metastatic NTRK fusion ‐ positive solid tumours: integrated analysis of three phase 1‐2 trials. Lancet Oncol. 2020;21(2):271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Laetsch TW, DuBois SG, Mascarenhas L, et al . Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open‐label, phase 1/2 study. Lancet Oncol. 2018;19(5):705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Valle JW, Wasan H, Lopes A, et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC‐03): a randomised phase 2 trial. Lancet Oncol. 2015;16(8):967‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wei K, Li M, Zoller M, Wang M, Mehrabi A, Hoffmann K. Targeting c‐MET by Tivantinib through synergistic activation of JNK/c‐jun pathway in cholangiocarcinoma. Cell Death Dis. 2019;10(3):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pant S, Saleh M, Bendell J, et al. A phase I dose escalation study of oral c‐MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann Oncol. 2014;25(7):1416‐1421. [DOI] [PubMed] [Google Scholar]
- 81. Goyal L, Zheng H, Yurgelun MB, et al. A phase 2 and biomarker study of cabozantinib in patients with advanced cholangiocarcinoma. Cancer. 2017;123(11):1979‐1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 83. Wang YX, Wild AT, Turcan S, et al. Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH‐mutant gliomas and cholangiocarcinomas. Science. Advances. 2020;6(17):eaaz3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wall JA, Klempner SJ, Arend RC. The anti‐DKK1 antibody DKN‐01 as an immunomodulatory combination partner for the treatment of cancer. Expert Opin Investig Drugs. 2020;29(7):639‐644. [DOI] [PubMed] [Google Scholar]
- 85. D'Amico L, Mahajan S, Capietto AH, et al. Dickkopf‐related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J Exp Med. 2016;213(5):827‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Arend RC, Castro CM, Matulonis UA, et al. Safety and efficacy of a DKK1 inhibitor (DKN‐01) as monotherapy or in combination with paclitaxel in patients with Wnt activated recurrent gynecologic malignancies. Gynecol Oncol. 2019;154:34. [Google Scholar]
- 87. Gani F, Nagarajan N, Kim Y, et al. Program Death 1 Immune Checkpoint and Tumor Microenvironment: Implications for Patients With Intrahepatic Cholangiocarcinoma. Ann Surg Oncol. 2016;23(8):2610‐2617. [DOI] [PubMed] [Google Scholar]
- 88. Walter D, Herrmann E, Schnitzbauer AA, et al. PD‐L1 expression in extrahepatic cholangiocarcinoma. Histopathology. 2017;71(3):383‐392. [DOI] [PubMed] [Google Scholar]
- 89. Mody K, Starr J, Saul M, et al. Patterns and genomic correlates of PD‐L1 expression in patients with biliary tract cancers. Journal of. Gastrointestinal Oncol. 2019;10(6):1099‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sato Y, Kinoshita M, Takemura S, et al. The PD‐1/PD‐L1 axis may be aberrantly activated in occupational cholangiocarcinoma. Pathol Int. 2017;67(3):163‐170. [DOI] [PubMed] [Google Scholar]
- 91. Sato Y, Tanaka S, Kinoshita M, et al. Immunosuppressive tumor microenvironment in occupational cholangiocarcinoma: Supportive evidence for the efficacy of immune checkpoint inhibitor therapy. J Hepatobiliary Pancreat Sci. 2020;27(11):860‐869. [DOI] [PubMed] [Google Scholar]
- 92. Bang YJ, Doi T, De Braud F, et al. Safety and efficacy of pembrolizumab (MK‐3475) in patients (pts) with advanced biliary tract cancer: Interim results of KEYNOTE‐028. Eur J Cancer. 2015;51:S112. [Google Scholar]
- 93. Rotte A. Combination of CTLA‐4 and PD‐1 blockers for treatment of cancer. J Exp Clinical Cancer Res. 2019;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Klein O, Kee D, Nagrial A, et al. Evaluation of Combination Nivolumab and Ipilimumab Immunotherapy in Patients With Advanced Biliary Tract Cancers Subgroup Analysis of a Phase 2 Nonrandomized Clinical Trial. Jama Oncology. 2020;6(9):1405‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Guo YL, Feng KC, Liu Y, et al. Phase I Study of Chimeric Antigen Receptor‐Modified T Cells in Patients with EGFR‐Positive Advanced Biliary Tract Cancers. Clin Cancer Res. 2018;24(6):1277‐1286. [DOI] [PubMed] [Google Scholar]
- 96. Feng KC, Liu Y, Guo YL, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2‐positive advanced biliary tract cancers and pancreatic cancers. Protein & Cell. 2018;9(10):838‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rimassa L, Personeni N, Aghemo A, Lleo A. The immune milieu of cholangiocarcinoma: From molecular pathogenesis to precision medicine. J Autoimmun. 2019;100:17‐26. [DOI] [PubMed] [Google Scholar]
- 98. Arkenau HT, Martin‐Liberal J, Calvo E, et al. Ramucirumab Plus Pembrolizumab in Patients with Previously Treated Advanced or Metastatic Biliary Tract Cancer: Nonrandomized, Open‐Label, Phase I Trial (JVDF). Oncologist. 2018;23(12):1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kato Y, Tabata K, Kimura T, et al. Lenvatinib plus anti‐PD‐1 antibody combination treatment activates CD8(+) T cells through reduction of tumor‐associated macrophage and activation of the interferon pathway. PLoS One. 2019;14(2):e0212513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jiao SP, Xia WY, Yamaguchi H, et al. PARP Inhibitor Upregulates PD‐L1 Expression and Enhances Cancer‐Associated Immunosuppression. Clin Cancer Res. 2017;23(14):3711‐3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sun DY, Ma JX, Wang JL, et al. Anti‐PD‐1 therapy combined with chemotherapy in patients with advanced biliary tract cancer. Cancer Immunol Immunother. 2019;68(9):1527‐1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Oh D‐Y, Lee K‐H, Lee D‐W, et al. Phase II study assessing tolerability, efficacy, and biomarkers for durvalumab (D) ± tremelimumab (T) and gemcitabine/cisplatin (GemCis) in chemo‐naïve advanced biliary tract cancer (aBTC). J Clin Oncol. 2020;38(15_suppl):4520. [Google Scholar]
- 103. Shimizu K, Kotera Y, Aruga A, Takeshita N, Takasaki K, Yamamoto M. Clinical utilization of postoperative dendritic cell vaccine plus activated T‐cell transfer in patients with intrahepatic cholangiocarcinoma. J Hepato‐Biliary‐Pancreatic Sci. 2012;19(2):171‐178. [DOI] [PubMed] [Google Scholar]