

Abstract
Cardiac microvascular dysfunction is associated with cardiac hypertrophy and can eventually lead to heart failure. Dysregulation of long non-coding RNAs (lncRNAs) has recently been recognized as one of the key mechanisms involved in cardiac hypertrophy. However, the potential roles and underlying mechanisms of lncRNAs in cardiac microvascular dysfunction have not been explicitly delineated. Our results confirmed that cardiac microvascular dysfunction was related to cardiac hypertrophy and ferroptosis of cardiac microvascular endothelial cells (CMECs) occurred during cardiac hypertrophy. Using a combination of in vivo and in vitro studies, we identified a lncRNA AABR07017145.1, named as lncRNA AAB for short, and revealed that lncRNA AAB was upregulated in the hearts of cardiac hypertrophy rats as well as in the Ang II-induced CMECs. Importantly, we found that lncRNA AAB sponged and sequestered miR-30b-5p to induce the imbalance of MMP9/TIMP1, which enhanced the activation of transferrin receptor 1 (TFR-1) and then eventually led to the ferroptosis of CMECs. Moreover, we have developed a delivery system based on neutrophil membrane (NM)-camouflaged mesoporous silica nanocomplex (MSN) for inhibition of cardiac hypertrophy, indicating the potential role of silenced lncRNA AAB (si-AAB) and overexpressed miR-30b-5p as the novel therapy for cardiac hypertrophy.
Keywords: cardiac hypertrophy, cardiac microvascular endothelial cell, ferroptosis, lncRNA AABR07017145.1, miR-30b-5p, neutrophil membrane-camouflaged nanocomplex
Graphical abstract
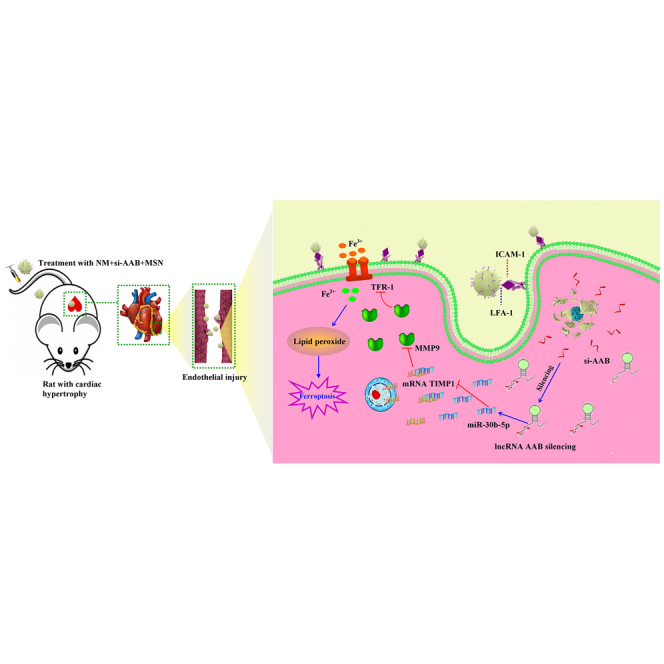
Neutrophil membrane-camouflaged core-shell structure for siRNA of lncRNA AAB inhibits CMEC ferroptosis in cardiac hypertrophy rat NM + si-AAB + MSN targets injured CMECs. Upon the uptake by CMECs, NM + si-AAB + MSN is decomposed and releases siRNA into cytoplasm. si-AAB increases miR-30b-5p, and then miR-30b-5p regulates the MMP9/TIMP1 balance, which leads to suppression of the activation of TFR-1 to reduce iron toxicity, and ultimately inhibits CMEC ferroptosis.
lncRNA AAB sponged and sequestered miR-30b-5p to induce the imbalance of MMP9/TIMP1, which enhanced the activation of TFR-1 and then eventually led to the ferroptosis of CMECs in cardiac hypertrophy. We have developed a nanocomplex that can be targeted to delivery si-AAB or miR-30b-5p to injured CMECs, which inhibits CMEC ferroptosis.
Introduction
Cardiac hypertrophy is a critical pathophysiological feature of heart failure development due to various cardiovascular disorders, including coronary heart disease, valvular disease, and hypertension. However, the molecular mechanisms involved in the hypertrophic reaction have not been clearly elucidated. Currently, the researchers focus on the changes in cardiac microvessels to explore the mechanism of cardiac hypertrophy. There was evidence that endothelial dysfunction was associated with hypertension and cardiac hypertrophy.1
Non-coding RNAs (ncRNAs) are a class of RNAs without protein-coding function and play crucial roles in cellular pathology and physiology.2 A large number of studies have shown that microRNAs (miRNAs), such as miR-20, miR-122, miR-103, and miR-30b-5p, are involved in the process of cardiac hypertrophy.3, 4, 5, 6 More recent reports have demonstrated that lncRNAs through miRNA-mediated regulatory axes by the competing endogenous RNA (ceRNA) mechanism play important roles in the progress of cardiac hypertrophy. For instance, lncRNA CHRF induced cardiac hypertrophy by repressing miR-489 expression.7 Fang et al. found that lncRNA TUG1 alleviated cardiac hypertrophy by targeting miR-34a/DKK1/Wnt-β-catenin signaling pathway.8 The above experimental studies were mainly carried out in cardiomyocytes (CMs). However, many potential mechanisms of myocardial hypertrophy caused by myocardial microvascular injury regulated by ncRNA have not been identified.
Ferroptosis is characterized by excessive accumulation of lipid peroxides, which is an iron-dependent form of regulated cell death.9 Lipid peroxidation and iron are recognized as central mediators of ferroptosis.10 Iron is essential for the survival of almost all organisms because it is a cofactor in many biochemical processes, including oxidative phosphorylation, the enzymatic reactions, and oxygen storage required for cell proliferation.11 However, the level of free iron in cells must be strictly controlled to avoid the production of reactive oxygen species (ROS) via the Fenton reaction.12 TFR-1 and metalloprotein complexes called ferritins are well-known regulators of cellular iron. TFR-1, a transmembrane glycoprotein, is responsible for internalizing the transferrin-bound iron, which is then released into the cytoplasm and stored in a non-toxic form inside ferritins.13 Therefore, the abnormal increase of TFR-1 expression leads to the increase of iron uptake, which results in the accumulation of lipid peroxides in the cell and causes ferroptosis. Ferroptosis is a recently discovered, pharmacologically amenable form of regulated necrosis that is implicated in a myriad of pathophysiologically degenerative conditions, including ischemia/reperfusion injury, acute kidney failure, and cardiomyopathy.14,15 However, the molecular mechanism of ferroptosis in cardiac hypertrophy has rarely been studied. Importantly, ncRNAs are reported to participate in regulating variety of diseases by ferroptosis pathways. For example, Bai et al. found that miRNA-214-3p enhanced erastin-induced ferroptosis by targeting ATF4 in hepatoma cells.16 Studies had shown that a G3BP1-interacting lncRNA promoted ferroptosis and apoptosis in cancer via nuclear sequestration of p53.17 Whether ferroptosis is regulated by ncRNAs in cardiac microvascular endothelial cells (CMECs) during cardiac hypertrophy has not been clearly elucidated.
Currently, the regulatory mechanisms of a series of functional ncRNAs have been widely elucidated, but their clinical application has been greatly hindered due to little attention being paid to the development of effective in vivo delivery strategies to regulate the biological functions. Moreover, compared with the billions of intracellular ncRNAs that may exert their biological functions, the proportion of ncRNAs currently found to be related to cardiac hypertrophy is still very low. Therefore, the regulatory mechanism and effective delivery of more functional ncRNAs remain to be explored. Herein, experimental results in vivo showed that cardiac hypertrophy was accompanied by abnormal cardiac microvascular function. Moreover, we found that the imbalance of MMP9/TIMP1 led to the dysfunction of CMEC. Subsequent research confirmed that lncRNA AAB could induce CMEC ferroptosis by targeting miR-30b-5p, which endogenously bound to the 3′UTR of TIMP1, then activating TFR-1. Based on this regulatory mechanism, a nanoparticle delivery platform was used to transport silenced lncRNA AAB (si-AAB) and overexpressed miR-30b-5p for the reversal of cardiac hypertrophy. This study indicated that manipulating the expression of lncRNA AAB and miR-30b-5p represents a promising therapeutic strategy for cardiac hypertrophy.
Results
Ferroptosis occurs in cardiac microvascular damage during cardiac hypertrophy
We first established 4-week abdominal aorta constriction (AAC) models in rats. Four weeks after surgery, the heart volume (Figure S1A), heart weight/body weight ratio (HW/BW), left ventricle weight/body weight ratio (LVW/BW), heart weight/tibial length ratio (HW/TL), left ventricular posterior wall dimension (LVPWd), left ventricular internal diastolic diameter (LVIDd), and left ventricular internal systolic diameters (LVIDs) were markedly increased in the model group. In contrast, left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF) were significantly decreased (Table S1). Moreover, hematoxylin-eosin (HE) and wheat germ agglutinin (WGA) staining marked an increased CM area in model groups (Figures S1B and S1C). Western blot analysis indicated that cardiac hypertrophy marker proteins (β-MHC and BNP) were remarkably increased in model groups (Figures S1D–S1G). Hemodynamics analysis revealed abnormal parameters in model groups (Table S2). Additionally, the expression of PECAM-1 was decreased to a significantly greater extent by pressure overload (Figures S1H and S1I). As shown in Figure S1J, the AAC-induced heart manifested a linear and irregular filling defect in most microvessels. Similarly, the histological examination of PECAM-1 staining revealed a significant loss of CMECs in model groups (Figure S1K). Furthermore, we detected the protein expression of p-eNOS (Figures S1L and S1M); the content of NO (Figure S1N) and cGMP (Figure S1O); and the expression of Ang-2 and PDGFR-β, considered as the markers of CMEC function7 (Figures S1P and S1Q) in rats with cardiac hypertrophy. Experimental data indicated that cardiac hypertrophy models were established and cardiac microvascular function was abnormal in cardiac hypertrophy.
Different forms of cell death, such as apoptosis, autophagy, and necroptosis, have been shown to be implicated in cardiac hypertrophy.18, 19, 20 These findings indicated that several forms of cell death could collectively contribute to cardiac hypertrophy, and the probability of an alternating new form of cell death cannot be ignored. Ferroptosis is an iron-dependent, nonapoptotic form of cell death that mediates its effects in part by promoting the accumulation of lethal lipid ROS.21 Furthermore, ferroptosis is involved in cardiac injury.22,23 Based on the above research, we detected the biological indicators of apoptosis, autophagy, and necroptosis in hypertrophic heart tissue, and western blot results showed that the protein expressions of LC3II, caspase-3, RIP1, and RIP3 were significantly upregulated, while the protein level of Bcl-2 was significantly decreased in vivo (Figures S2A–S2F). In addition, to investigate whether pressure overload induced cardiac damage via an oxidative stress mechanism, biomarkers of oxidative stress were evaluated. As shown in Table S3, our results showed that the model rats exhibited reduced activities of antioxidative enzymes, including superoxide dismutase (SOD), Mn-SOD, Cu/Zn-SOD, glutathione peroxidase (GSH-PX), and catalase (CAT) and glutathione (GSH) level, and increased the level of malondialdehyde (MDA) in comparison with the sham group. The following experiment was performed to determine the effects of ferrostatin-1 (Ferr-1) on the cardiac microvascular function in cardiac hypertrophy rats. First, we found that Ferr-1 reduced the increase in lipid peroxides in the hearts of the model group by detecting MDA level (Figure 1A) and the ptgs2 considered as a marker of ferroptosis24 mRNA level (Figure 1B). Moreover, the expression of H2AX was decreased by Ferr-1 (Figures 1C and 1D). In addition, an electron microscope was used to observe the ultrastructural alterations of microvessels with Ferr-1 treatment. As a result, our analysis revealed that the cardiac microvascular endothelial mitochondria in AAC-treated rats were severely distorted and atrophied, which were regarded as the main characteristic of ferroptosis,25 and these AAC-induced effects were rescued by Ferr-1 treatment (Figure 1E). Gelatin-ink staining showed that Ferr-1 markedly increased cardiac microvascular density (Figure 1F). In addition, we detected the effects of Ferr-1 on the protein expression of p-eNOS, the content of NO, and cGMP (Figures 1G–1J) in rats with cardiac hypertrophy. Experimental data suggested that Ferr-1 could improve cardiac microvascular function. Ang II could induce hypertension and vascular injury.26 Moreover, damage of microvessel integrity and barrier function is considered the key step in microcirculatory dysfunction.27 Hence, we examined LC3II, caspase-3, Bcl-2, RIP1, and RIP3. Interestingly, CMEC underwent apoptosis, necroptosis, without a significant degree of autophagy after treatment with Ang II (Figures S2G–S2L). More importantly, we found that Ang II increased the ROS level; reduced the levels of SOD, Mn-SOD, Cu/Zn-SOD, GSH-PX, CAT, and GSH; and increased the level of MDA in comparison with the control group (Table S4). Further study indicated that Ferr-1 reduced the increase in ROS, lipid peroxides, and the mRNA level of ptgs2 (Figures 1K–1M), and increased the cell viability of Ang II-induced CMECs (Figures 1N and 1O). Immunofluorescence results revealed that PECAM-1 was significantly lacking in Ang II-treated CMECs. However, Ferr-1 increased PECAM-1 expression significantly (Figure 1P). Furthermore, the migration ability that was decreased by Ang II and Ferr-1 reversed this situation (Figure 1Q). Western blot analysis showed that the expression of p-eNOS was significantly increased compared with the Ang II group after treatment with Ferr-1 (Figures 1R–1S). The detection of NO content suggested that Ferr-1 increased the content of NO in CMEC exposed to Ang II (Figure 1T). These findings indicated that ferroptosis occurred in cardiac microvascular damage, suggesting that the protection of Ferr-1 on hypertrophic heart may involve improving CMEC function by suppressing ferroptosis.
Figure 1.

Inhibition of ferroptosis improves the function of CMECs
(A) Changes of MDA content in heart tissue after AAC treatment for 4 weeks and Ferr-1 intervention for 2 weeks. (B) The expression of ptgs2 mRNA in myocardial tissue was detected by qRT-PCR. (C and D) Effect of treatment with Ferr-1 for 2 weeks on H2AX protein expression in hypertrophic myocardial tissue. (E) Electron microscopy was used to observe morphological changes of mitochondria in CMECs. (F) Gelatin ink was injected into hypertrophic hearts after treatment with Ferr-1 for 2 weeks, and samples were observed via microscope. (G-H) Effect of treatment with Ferr-1 for 2 weeks on the expression of p-eNOS in cardiac tissue. (I) Statistical graph for NO content in the cardiac tissue after treatment with Ferr-1 for 2 weeks. (J) Effect of treatment with Ferr-1 for 2 weeks on the level of cGMP. Ferr-1, ferrostatin-1; AAC, abdominal aorta constriction. Model group: rats were treated by AAC for 4 weeks. M + Ferr-1, rats of model group induced by AAC 2 weeks were injected with Ferr-1 at a dose of 2 mg/kg/day for another 2 weeks. Data were represented by means ± SEM (n = 6). ##p < 0.01 versus sham group; ∗p < 0.05 and ∗∗p < 0.01 versus model group. (K) Immunofluorescence results of ROS in CMECs induced by Ang II for 24 h following by treatment with 2 μΜ Ferr-1 for 48 h. (L) Changes of MDA content in CMECs. (M) The effect of treatment with Ferr-1 for 48 h on the mRNA expression of ptgs2 in CMECs. (N and O) The effect of Ferr-1 on the viability of CMECs. (P) Representative image of immunofluorescence of PECAM-1 in CMECs. (Q) The scratch test was used to detect the effect of the treatment with 2 μΜ Ferr-1 for 48 h on the function of CMECs. (R and S) Western blot analysis of p-eNOS expression in CMECs. (T) Statistical graph for NO content in CMECs. Ang II group: CMECs were treated by Ang II for 24 h. Ferr-1 group: CMECs were given 100 nM Ang II for 24 h followed by treatment with 2 μΜ Ferr-1 for 48 h. Data were represented by means ± SEM (n = 6). ##p < 0.01 versus control group; ∗∗p < 0.01 versus Ang II group. Scale bars, (E) 1.0 μm; (F) 100 μm (×100); 100 μm (×200); (O and Q) 10 μm; (K and P) 50 μm.
Imbalance of MMP9/TIMP1 ratio leads to dysfunction of CMECs in cardiac hypertrophy
Angiogenesis requires invasion by endothelial cells and localized proteolytic modification of the extracellular matrix. Proteolysis of the extracellular matrix allows cell migration and may also release stored signaling molecules. Matrix metalloproteinases (MMPs) are implicated in these processes owing to their ability to cleave extracellular matrix components.20 MMP9 is one of the two major gelatinases in the MMP family. In addition to effectively and rapidly cutting unfolded collagen, MMP9 has been reported to be able to cut other matrix and non-matrix components, and then participate in angiogenesis.28 Moreover, TIMP1 is an endogenous specific inhibitor of MMP9.29 Therefore, the imbalance of MMP9/TIMP1 is involved in the occurrence and development of a variety of diseases. Ang II was found to upregulate TIMP1 gene expression in rat heart endothelia cells in a dose- and time-dependent manner.30 According to the above studies, we detected the protein expression of MMP9 and TIMP1 in rats with cardiac hypertrophy induced by pressure overload and CMECs exposed to Ang II by western blot. The results indicated that the imbalance of MMP9/TIMP1 occurred both in cardiac hypertrophy rat hearts and CMECs (Figures S3A–S3H). Further studies showed that the expression of MMP9 was significantly increased after TIMP1 silencing (Figures 2A–2D). In addition, silencing TIMP1 increased the cell viability of Ang II-induced CMECs (Figures 2E and 2F). To investigate the effect of imbalance of MMP9/TIMP1 on the function of CMECs, the expression of PECAM-1 was detected by immunofluorescence and western blot. Immunofluorescence results revealed that PECAM-1 was remarkably lacking in Ang II-treated CMECs, but TIMP1 silencing significantly increased PECAM-1 expression and integrity (Figure 2G). The western blot results showed a similar tendency (Figures 2H and 2I). Furthermore, we investigated the effects of TIMP1 silencing on CMEC migration and tube formation. The results indicated that Ang II suppressed cell migration and tube formation ability. However, the migration distances and tube formation in the TIMP1 silencing-treated groups were increased obviously (Figures 2J and 2K). In addition, the expression of p-eNOS and NO content was decreased after treatment with Ang II, and TIMP1 silencing reversed the effects of Ang II (Figures 2L–2N). Furthermore, we constructed the full-length plasmid of TIMP1 and transfected it into CMECs. We found that overexpression of TIMP1 significantly induced the dysfunction of CMECs (Figures S3I–S3N). These results indicated that the imbalance of MMP9/TIMP1 leads to the dysfunction of CMECs.
Figure 2.

Silencing TIMP1 improves the dysfunction of CMECs induced by Ang II
(A and B) The efficiency and specificity of siRNA directed against TIMP1 in CMECs treated with siTIMP1 for 24 h. (C and D) Detection of MMP9 protein expression in CMECs by western blot analysis after treatment with siTIMP1 for 24 h and then exposed to Ang II for 24 h. (E and F) Cell viability of different groups was detected in CMECs. (G) Representative image of immunofluorescence of PECAM-1 in CMECs. (H and I) Western blotting was performed to quantify the expression of PECAM-1 in CMECs. (J) The scratch test was used to detect the migration ability of CMECs. (K) Tube formation of CMECs was examined in three-dimensional collagen gels. (L and M) Western blot analysis of p-eNOS expression in CMECs. (N) Statistical graph for NO content in CMECs. siTIMP1: TIMP1 was silenced. Ang II group: CMECs were treated by Ang II for 24 h. Ang II + siTIMP1 group: CMECs were exposed to siTIMP1 for 24 h and then treated by Ang II for 24 h. Ang II + NC-siTIMP1 group: CMECs were treated with negative control of siTIMP1 for 24 h and then exposed to Ang II for 24 h. Data are represented by means ± SEM (n = 6). ##p < 0.01 versus control group; ∗p < 0.05 and ∗∗p < 0.01 versus Ang II group. Scale bars, (E, J, and K) 10 μm; (G) 50 μm.
Imbalance of MMP9/TIMP1 ratio induces CMEC ferroptosis by activating TFR-1
Previous studies have confirmed that MMP9 is an angiogenic protease involved in the mobilization of vascular endothelial growth factor (VEGF).31 Our above studies have confirmed that silencing TIMP1 can significantly improve the CMEC function. However, the specific protective mechanism remains unclear and needs further study. Therefore, we silenced TIMP1 and detected the protein expression of caspase-3, Bcl-2, RIP1, and RIP3, and the results indicated that silencing TIMP1 could not inhibit cell apoptosis and necroptosis induced by Ang II in CMECs (Figures S4A–S4D). Interestingly, we also examined the expression of ferroptosis-related proteins, including SLC7A11, GPx4, TFR-1, and Fpn1.32, 33, 34, 35 The results showed that silencing TIMP1 significantly inhibited the increase of TFR-1 expression induced by Ang II but had no effect on SLC7A11, GPx4, and Fpn1 protein expression (Figures 3A and 3B). Further studies showed that silencing TIMP1 markedly reduced the iron ion level, the content of ROS and MDA, and the ptgs2 mRNA expression (Figures 3C–3F). Figure 3G suggested that silencing TIMP1 inhibited the cell death of CMECs. These findings indicated that the decrease in MMP9/TIMP1 ratio leads to ferroptosis of CMEC by activation of TFR-1.
Figure 3.

Silencing TIMP1 inhibits CMEC ferroptosis by downregulation of TFR-1
(A and B) Western blot analysis of the expression of SLC7A11, GPx4, TFR-1, and Fpn1 in CMECs treated by siTIMP1 for 24 h and then treated by Ang II for 24 h. (C) Statistical analysis chart of Fe2+ content in CMECs. (D) Immunofluorescence results of ROS in CMECs. (E) Changes of MDA content in CMECs. (F) The mRNA expression of ptgs2 in CMECs. (G) Statistical analysis of CMEC mortality. siTIMP1: TIMP1 was silenced. Ang II group: CMECs were treated with Ang II for 24 h. Ang II + siTIMP1 group: CMECs were exposed to siTIMP1 for 24 h and then treated by Ang II for 24 h. Ang II + NC-siTIMP1 group: CMECs were treated with negative control of siTIMP1 for 24 h and then exposed to Ang II for 24 h. Data are represented by means ± SEM (n = 6). #p < 0.05 and ##p < 0.01 versus control group; ∗∗p < 0.01 versus Ang II group. Scale bars, 50 μm.
MiR-30b-5p, the potential target of lncRNA AAB, inhibits ferroptosis by binding TIMP1
miRNAs negatively regulate gene expression by promoting mRNA degradation or inhibiting mRNA translation.36 Interestingly, a complementary binding site between miR-30b-5p and TIMP1 was predicted using the database of miRNA targets (http://www.targetscan.org/). Then, we analyzed the 3ʹUTR region of TIMP1 by RNAhybrid and noticed that miR-30b-5p has a complementary sequence with TIMP1 3ʹUTR (Figure 4A). Besides, luciferase assay revealed that miR-30b-5p was able to suppress the luciferase activity of wild-type (WT) TIMP1. However, the mutated form of TIMP1 3ʹUTR demonstrated lesser response to miR-30b-5p (Figure 4B). Moreover, the researchers found that miR-30b-5p may act as a hypertrophic suppressor.6 Therefore, we detected and analyzed miR-30b-5p expression in vivo and vitro, and found that miR-30b-5p was decreased in myocardial tissue of cardiac hypertrophy rat and CMECs induced by Ang II (Figures 4C and 4D). The next experiment was to determine whether miR-30b-5p was linked to CMEC ferroptosis by regulating the balance of MMP9/TIMP1. We transfected the miR-30b-5p mimic and AMO-miR-30b-5p into CMECs to detect the effect of miR-30b-5p on ferroptosis of CMECs (Figure 4E). Our data indicated that, in Ang II-induced CMECs, enforced expression of miR-30b-5p leads to a significant reduction of the protein expression of TIMP1 and TFR-1, ROS and MDA content, iron ion concentration, the mRNA level of ptgs2, and a remarkable increase in the cell viability and the protein expression of MMP9, which could be reversed by AMO-miR-30b-5p (Figures 4F–4M). Overexpression of miR-30b-5p markedly inhibited the ferroptosis of CMECs exposed to Ang II. However, AMO-miR-30b-5p suppressed the protective effect of miR-30b-5p on CMECs (Figure 4N). Recently, the ceRNA mechanism of lncRNAs has attracted a lot of research interest. The concept of ceRNAs demonstrates that the RNA molecules containing the binding sites (sequence complementarity) to a particular miRNA can competitively bind to this individual miRNA to reduce its functional availability.37 lncRNAs often act as ceRNAs, which can sponge and sequester miRNAs, indirectly regulate the target genes of miRNAs, or specifically release the target genes from repression by the targeted miRNA.38 Bioinformatics tools (integrating the miRanda algorithm) were used to analyze the lncRNAs targeting miR-30b-5p (Table S5). We found that 11 lncRNAs shared a highly conserved binding site with miR-30b-5p. Next, we detected the expression changes of these 11 lncRNAs with a high score predicted by bioinformatics methods in cardiac tissue of rats with cardiac hypertrophy, and the results showed that the mRNA expressions of lncRNA AC115159.2, lncRNA AABR07017145.1 (lncRNA AAB), and lncRNA AABR07030334.1 were significantly upregulated (Figure 4O). Then, we performed quantitative real-time PCR (qRT-PCR) to detect these three lncRNAs levels in response to Ang II treatment in CMECs. Among the lncRNAs, AABR07017145.1, which was named lncRNA AAB, was substantially elevated (Figure 4P). Moreover, we compared the sequence of lncRNA AAB with that of miR-30b-5p using RNAhybrid and noticed that lncRNA AAB contained a target site of miR-30b-5p (Figure 4Q). Therefore, we produced a luciferase construct of TIMP1. Luciferase assay revealed that miR-30b-5p could suppress the luciferase activity of TIMP1, but it could be reversed by lncRNA AAB and a mutated form (AAB-mut) had less effect on the luciferase activity of TIMP1 (Figure 4R). To investigate the effect of lncRNA AAB on the ferroptosis of CMECs via inhibition of miR-30b-5p, co-transfection of lncRNA AAB and miR-30b-5p was performed in CMECs. Enforced expression of lncRNA AAB markedly increased the protein expression of TIMP1 and TFR-1, and remarkably decreased the protein expression of MMP9, but these effects were reversed by treatment with miR-30b-5p (Figures S5A and S5B). Moreover, overexpression of lncRNA AAB increased the level of Fe2+ in CMECs and promoted the ferroptosis of CMECs, but miR-30b-5p suppressed the effect of lncRNA AAB on ferroptosis (Figures S5C and S5D). These results revealed that lncRNA AAB interacted with miR-30b-5p by this putative binding site and then regulated the ferroptosis of CMECs.
Figure 4.

MiR-30b-5p inhibits ferroptosis in the Ang II-induced CMEC
(A) MiR-30b-5p targeting site in TIMP1 3ʹUTR. (B) Luciferase assay. Cells were transfected with miR-30b-5p and then with luciferase constructs of WT TIMP1 3ʹUTR (TIMP1-wt) or mutated TIMP1 3ʹUTR (TIMP1-mut). Luciferase activity was analyzed. ##p < 0.01 versus TIMP1-wt group. (C) The mRNA expression of miR-30b-5p in cardiac tissue of rats induced by AAC for 4 weeks. Data are represented by means ± SEM (n = 6). ##p < 0.01 versus sham group. (D) The mRNA expression of miR-30b-5p in CMECs exposed to Ang II for 24 h. (E) The mRNA expression of miR-30b-5p in CMECs. (F and G) Cell viability of CMECs was detected. (H and I) Western blot analysis of TIMP1, MMP9, and TFR-1 in CEMCs. (J) Statistical analysis chart of Fe2+ content. (K) Immunofluorescence results of ROS in CMECs. (L) Changes of MDA content in CMECs. (M) The mRNA expression of ptgs2 in different groups. (N) Statistical analysis of CMEC mortality. AMO: AMO-miR-30b-5p. NC: negative control. Ang II group: CMECs were exposed to Ang II for 24 h. Ang II + miR-30b-5p group: CMECs were exposed to Ang II for 24 h and then treated with miR-30b-5p mimics for 24 h. Ang II + miR-30b-5p + AMO group: CMECs were exposed to Ang II for 24 h and then treated with miR-30b-5p mimics and AMO-miR-30b-5p for 24 h. Ang II + NC-miR-30b-5p group: CMECs were exposed to Ang II for 24 h and then treated with negative control of miR-30b-5p. Ang II + NC-AMO group: CMECs were exposed to Ang II for 24 h and then treated with negative control of AMO-miR-30b-5p. Data are represented by means ± SEM (n = 6). ##p < 0.01 versus control group; ∗∗p < 0.01 versus Ang II group. (O) qRT-PCR analysis of lncRNAs in the hearts of cardiac hypertrophy rats induced by AAC. AABR07017145.1: lncRNA AAB. Data are represented by means ± SEM (n = 6). #P < 0.05 and ##p < 0.01 versus sham group. (P) The expression of the three lncRNAs in CMECs exposed to Ang II for 24 h. Data are represented by means ± SEM (n = 6). ##p < 0.01 versus control group. (Q) lncRNA AAB contained a site complementary to miR-30b-5p. (R) Luciferase activity analyses in cells co-transfected with TIMP1-wt and luciferase reporters containing miR-30b-5p mimic or lncRNA AAB. Data are represented by means ± SEM (n = 6). ##p < 0.01 versus TIMP1-wt group; ∗∗p < 0.01 versus TIMP1-wt and miR-30b-5p mimic group. Scale bars, (G) 10 μm; (K) 50 μm.
Preparation, characterization, and cellular uptake of nanocomplex
Hence, if we can effectively deliver small interfering RNA (siRNA) of lncRNA AAB via nanocomplex, it may be possible to inhibit ferroptosis in CMECs. Next, our results showed the preparation and characterization of nanocomplex. As displayed in Figure 5A, neutrophil membrane (NM) + si-AAB + MSN clearly demonstrated a eukaryotic-cell-like structure, with an outermost slightly gray soft layer thickness of ∼10 nm, which was consistent with the thickness of the living cellular membrane. The obtained MSNs were uniform mesoporous nanospheres with a mean particle size of ∼100 nm, as shown in the scanning electron microscopy image (Figures 5B and 5C). The pore size was tuned to a suitable size for siRNA loading and the surface area ensured the high loading capacity of siRNA. Furthermore, the MSN was biodegradable within 24 h when incubated in fetal bovine serum, which was important for efficient in vivo drug release and reducing the body’s immune side reaction and accumulation.39 The zeta potential was measured to investigate the surface modification and charge changes of the MSN. When MSN was loaded with siRNA, the zeta potential decreased to −14.6 ± 1.06 mV, which was attributed to the negative charge of siRNA (Figure 5D).40 To further confirm the mesoporous structure of the MSNs, the surface area and pore diameter of MSN nanoparticles were detected. The values were 519 m2 g−1 and a well-aligned mesoporous around 2.4 nm (inset of Figure 5E), respectively, which could satisfy the subsequent siRNA loading. The X-ray diffraction (XRD) pattern showed that the MSNs had no prominent peaks but a broad diffraction peak at 2θ of about 22°, because the MSNs are amorphous (Figure 5F). The existence of membrane protein LFA-1 and β2 Integrin of cell membrane on NM + si-AAB + MSN was further identified by western blotting analysis. In the samples of cell membrane and NM + si-AAB + MSN, LFA-1 and β2 Integrin bands were observed on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel (Figure 5G). The existence of LFA-1 and β2 Integrin on NM + si-AAB + MSN would ensure effective targeting of NM + si-AAB + MSN toward the injured CMECs. To demonstrate that Ang II could induce CMECs injury and inflammation, we determined ICAM-1 levels by western blot. The protein expression of ICAM-1 in injured CMECs was significantly more increased than that in normal CMECs (Figures 5H and 5I). To demonstrate specific recognition and targeted delivery of the NM + si-AAB + MSN toward CMECs, Ang II was used to induce the injury of CMs and CMECs. Then, after incubation with NM + si-AAB + MSN, the nuclei of cells were stained with DAPI (blue), and NM + si-AAB + MSN was labeled with fluorescein isothiocyanate (FITC) (green). Weak signal of FITC was observed in CMs, indicating low uptake efficiency of NM + si-AAB + MSN. However, in CMECs, the intracellular FITC signal was 3.8-fold that in CMs (Figures 5J and 5K). Besides, cellular uptake results of si-AAB + MSN and NM + si-AAB + MSN by CMECs were evaluated using flow cytometric analysis in Figures 5L and 5M. The fluorescence intensity of NM + si-AAB + MSN in cells was obviously higher than that of PBS and si-AAB + MSN group. It demonstrated that NM-modified MSNs could be taken in more by CMECs. Endosomal escape of nanocomplex was essential for efficient siRNA delivery, which avoided intracellular degradation of siRNA in lysosomes.41 The lysosomal escape of NM + si-AAB + MSN in CMECs was tracked via fluorescent colocalization by confocal microscopy. NM + si-AAB + MSN, cell nuclei, and lysosome were labeled with FITC (green), DAPI (blue), and lysosome marker LysoTracker (red), respectively. CMECs were incubated with NM + si-AAB + MSN for varied time duration (i.e., 1 h, 2 h, 4 h, and 24 h). Upon 1-h incubation, the signals of FITC and LysoTracker were separated, representing the initial stage of NM + si-AAB + MSN taken up by CMECs; after 2-h incubation, the signals of FITC and LysoTracker overlaid, reflecting the entrance of NM + si-AAB + MSN in lysosome; after 4 h, signals of FITC and LysoTracker partially separated, indicating that NM + si-AAB + MSN began to escape from lysosome; after 24 h, most signals of FITC and LysoTracker separated, showing the successful lysosomal escape of NM + si-AAB + MSN (Figure 5N). These results demonstrated that we had successfully synthesized neutrophil membrane-coated siRNA of lncRNA AAB nanoparticles.
Figure 5.

Preparation and characterization of nanocomplex
(A) Transmission electron microscopy images of MSN and NM + si-AAB + MSN. Insets: the magnified images of individual particles. (B and C) Size distribution. (D) Surface charge of MSN, NM + MSN, and NM + si-AAB + MSN was measured by dynamic light scattering and zeta potential. (E) N2 adsorption-desorption isotherms. (F) XRD pattern. (G) LFA-1 and β2 Integrin protein level on NM and NM + si-AAB + MSN were detected by western blotting. (H and I) Western blot analysis of ICAM-1 expression in CMECs induced by Ang II for 24 h. Data are represented by means ± SEM (n = 6). ##p < 0.01 versus control group. (J and K) Cellular uptake of NM + si-AAB + MSN by CMs and CMECs was observed by fluorescence microscopy after they were exposed to Ang II for 24 h and then treated by NM + si-AAB + MSN, respectively. Data are represented by means ± SEM (n = 6). ##p < 0.01 versus CM group. (L and M) Cellular uptake of si-AAB + MSN and NM + si-AAB + MSN by CMECs after treatment with Ang II for 24 h. (N) Confocal microscopy images of the intracellular localization of NM + si-AAB + MSN within CMECs at different time points. NM + si-AAB + MSN is labeled with FITC (green), lysosomes are labeled with LysoTracker (red), and nuclei are stained with DAPI (blue). Yellow color represented colocalization of fluorescent signals from NM + si-AAB + MSN and lysosomes. si-AAB: siRNA of lncRNA AAB. MSN, mesoporous silica nanocomplex. Scale bars, (A and B) 200 μm; (J and N) 50 μm.
NM + si-AAB + MSN inhibits CMEC ferroptosis by upregulation of miR-30b-5p
Figure 6A shows the cellular internalization and intracellular deconstruction of nanocomplex, including (1) targeted recognition of NM + si-AAB + MSN with injured CMECs via specific binding between LFA-1, β2 Integrin, and ICAM-1, (2) cellular uptake of NM + si-AAB + MSN within CMECs, (3) intracellular cell membrane dissociation, and (4) MSN degradation and siRNA release. With this favorable reduction response, the NM + si-AAB + MSN could efficiently silence lncRNA AAB expression in CMECs and more than 70% of lncRNA AAB expression could be suppressed at a siRNA dose of 30 nM (Figure 6B). To further study the mechanism of ferroptosis inhibition, we treated Ang II-induced CMECs with si-AAB, si-AAB + MSN, and NM + si-AAB + MSN. The results showed that cell viability of si-AAB + MSN and NM + si-AAB + MSN groups were increased significantly compared with that in the Ang II group (Figures 6C and 6D). Moreover, to investigate the effect of si-AAB, si-AAB + MSN, and NM + si-AAB + MSN on the function of CMECs, we detected the expression of PECAM-1 by immunofluorescence. The data indicated that the PECAM-1 of CMECs treated by Ang II was incomplete. However, after the treatment with si-AAB + MSN and NM + si-AAB + MSN, it showed uniform peripheral staining (Figure 6E). The results of cell migration, the expression of p-eNOS, and NO production showed that si-AAB + MSN and NM + si-AAB + MSN significantly improved the function of CMECs (Figures S6A–S6D). Further study found that si-AAB + MSN and NM + si-AAB + MSN significantly upregulated the mRNA expression of miR-30b-5p and the protein expression of MMP9, and downregulated the protein expression of TIMP1 and TFR-1 (Figures 6F–6H). Compared with the Ang II group, the iron ion, ROS, MDA, ptgs2 levels, and cell death of the si-AAB group almost did not change, while those of si-AAB + MSN and NM + si-AAB + MSN groups were reduced significantly. CMECs treated with NM + si-AAB + MSN showed the lowest level of iron ion, ROS, MDA, ptgs2, and cell death, demonstrating the effective suppression of ferroptosis by NM + si-AAB + MSN (Figures 6I–6M). All these results strongly showed that NM + si-AAB + MSN was the most effective in inhibiting the ferroptosis of CMECs exposed to Ang II by upregulation of miR-30b-5p.
Figure 6.

Silencing lncRNA AAB by NM + si-AAB + MSN reverses the ferroptosis of CMEC
(A) Illustration of the targeted recognition of NM + si-AAB + MSN, cellular uptake within a CMEC, intracellular cell membrane dissociation, MSN degradation, and siRNA release. (B) lncRNA AAB expression level was determined by qRT-PCR in CMECs treated with the NM + si-AAB + MSN for 48 h at different siRNA doses. Data are represented by means ± SEM (n = 6). #p < 0.05 and ##p < 0.01 versus 0 nM. (C and D) Cell viability of CMECs induced by Ang II for 24 h was detected after treatment with si-AAB, si-AAB + MSN, and NM + si-AAB + MSN, respectively. (E) Immunofluorescence results of PECAM-1 in CMECs. (F) The miR-30b-5p mRNA level in CMECs. (G and H) Western blot analysis of TIMP1, MMP9, and TFR-1 in CEMCs. (I) Statistical analysis chart of iron ions content. (J) Immunofluorescence results of ROS in CMECs of different groups. (K) Statistical analysis chart of MDA content. (L) qRT-PCR analysis of the expression level of ptgs2 mRNA in CMECs. (M) Statistical analysis of CMEC mortality. Ang II group: CMECs were treated with 100 nM Ang II for 24 h following by treatment with PBS for 48 h. si-AAB group: CMECs were given 100 nM Ang II for 24 h following by treatment with si-AAB for 48 h. si-AAB + MSN group: CMECs were given 100 nM Ang II for 24 h following by treatment with si-AAB + MSN for 48 h. NM + si-AAB + MSN group: CMECs were given 100 nM Ang II for 24 h following by treatment with NM + si-AAB + MSN for 48 h. Data are represented by means ± SEM (n = 6). ##p < 0.01 versus control group; ∗p < 0.05 and ∗∗p < 0.01 versus Ang II group. Scale bars, (D) 10 μm; (E and J) 50 μm.
NM + si-AAB + MSN repairs cardiac microvascular endothelial injury in cardiac hypertrophy
The animal model of cardiac hypertrophy was created following AAC, and the rats with cardiac hypertrophy were randomly divided into four groups. After 24 h, the rats with cardiac hypertrophy were respectively treated with PBS, si-AAB, si-AAB + MSN, and NM + si-AAB + MSN via tail vein injection once every 2 days for 4 weeks (Figure 7A). To ensure that NM + si-AAB + MSN was always able to target the hearts, we measured the protein level of ICAM-1 in myocardial tissue on day 3, day 9, and day 27 after AAC. The results showed that the protein expression of ICAM-1 at these different time points was significantly higher than that in the sham group (Figures 7B and 7C). Then, we studied the in vivo distribution of the nanocomplex via bioimaging by quantification of the MSN fluorescent signals in heart. In the si-AAB + MSN group, slight fluorescent signal appeared in the heart; in the NM + si-AAB + MSN group, significant signal was observed in the heart, indicating good target specificity of the nanocomplex toward damaged endothelium (Figure 7D). In addition, we studied in vivo targeting of the nanocomplex via qRT-PCR. In the model group treated with PBS, the expression of lncRNA AAB was markedly increased, the treatment of si-AAB and si-AAB + MSN had no effect on the expression of lncRNA AAB, while NM + si-AAB + MSN remarkably decreased the lncRNA AAB expression (Figure S7A). Moreover, we detected the expression of miR-30b-5p, and found that only treatment with NM + si-AAB + MSN could reverse the reduction in the expression of miR-30b-5p (Figure S7B). Western blot analysis showed that NM + si-AAB + MSN remarkably decreased the TIMP1 and TFR-1 expression, and markedly increased the MMP9 expression (Figures S7C and S7D). The ferroptosis of cardiac microvessel in the hypertrophic hearts was analyzed to evaluate the therapeutic effect of NM + si-AAB + MSN in vivo. As indicated by ELISA, qRT-PCR, and electron microscopy, the model group treated with PBS exhibited high levels of Fe2+, MDA and ptgs2 (Figures S7E–S7G), and the atrophic mitochondria of CMECs (Figure 7E). The ferroptosis levels showed negligible change in the si-AAB and si-AAB + MSN group, while that in the NM + si-AAB + MSN groups remarkably dropped, revealing the good anti-ferroptosis therapeutic effect of NM + si-AAB + MSN on cardiac microvessels. To verify the effect of NM + si-AAB + MSN on cardiac microvascular function, we further detected the cardiac microvessel density, the protein expression of PECAM-1 and p-eNOS, NO production capacity, and cGMP content. The results suggested that NM + si-AAB + MSN increased cardiac microvessel density (Figure 7F) and improved cardiac microvascular function in cardiac hypertrophy rats (Figures S8A–S8F). Furthermore, to investigate whether NM + si-AAB + MSN could inhibit cardiac hypertrophy by preventing ferroptosis of CMECs, the therapeutic effect was evaluated via echocardiography measurements, histopathological examination, and western blot. Representative indicators of cardiac function and cardiac hypertrophy were evaluated. In comparison with the PBS, bare si-AAB, and si-AAB + MSN groups, the NM + si-AAB + MSN group performed much better in restoring LVEF and LVFS (Figures 7G–7I), exhibiting remarkable therapeutic efficiency of NM + si-AAB + MSN to improve the functions of hypertrophic myocardium. Moreover, NM + si-AAB + MSN markedly reduced the values of HW/BW, LVW/BW, HW/TL, LVPWd, LVIDd, and LVIDs, showing the inhibiting of cardiac hypertrophy (Table S6). HE staining of heart tissue sections and WGA staining were further performed to investigate the histopathological morphology of ventricles. In comparison with the model group treated with PBS, only the NM + si-AAB + MSN group produced the smaller cross-sectional area, exhibiting significant heart morphology protective effect (Figures 7J and 7K). To verify the siRNA-mediated in vivo cardiac hypertrophy inhibition, we further detected the related regulatory factors by western blotting analysis. The protein levels of BNP and β-MHC in the si-AAB and si-AAB + MSN groups showed negligible change compared with the model group, while the NM + si-AAB + MSN group showed significant decrease, demonstrating the effective suppression of cardiac hypertrophy by NM + si-AAB + MSN in vivo (Figures 7L and 7M). In addition, to investigate the effect of miR-30b-5p on cardiac hypertrophy rats, we also developed a miR-30b-5p delivery system based on NM-MSN (NM + miR-30b-5p + MSN). Our results showed that NM + miR-30b-5p + MSN markedly improved cardiac microvascular function and reversed cardiac hypertrophy the same as NM + si-AAB + MSN (Figures S9A–S9J). In general, the above experimental data confirmed that NM + si-AAB + MSN and NM + miR-30b-5p + MSN could target the damaged cardiac microvascular endothelium and effectively inhibit ferroptosis, thus playing a protective role in the myocardium against cardiac hypertrophy.
Figure 7.

Silencing lncRNA AAB by NM + si-AAB + MSN inhibits cardiac hypertrophy
(A) Scheme of time axis showing the design of animal study. (B and C) Detection of ICAM-1 protein expression by western blot analysis at different time points of AAC. (D) Images of hearts with fluorescent signals of MSN after tail vein injection of PBS, si-AAB, si-AAB+MSN, and NM+si-AAB+MSN, respectively. (E) Electron microscopy was used to observe morphological changes of mitochondria in CMECs. (F) Gelatin ink was injected into hearts after tail vein injection of PBS, si-AAB, si-AAB+MSN, and NM+si-AAB+MSN for 4 weeks, respectively, and samples were observed via microscope. (G) Typical images of the echocardiography in different groups. (H–I) The echocardiographic parameters including LVFS and LVEF were measured. (J and K) Representative sections of hearts stained for HE and WGA staining. (L and M) Detection of BNP and β-MHC protein expression by western blot analysis in cardiac tissue of rats. Model group: rats were subjected to AAC and received vehicle (PBS, caudal vein injection) 24 h later once every 2 days for 4 weeks. si-AAB group: rats were subjected to AAC and treated with si-AAB via tail vein injection 24 h later every 2 days for 4 weeks. si-AAB+MSN group: rats were subjected to AAC and treated with si-AAB+MSN via tail vein injection 24 h later every 2 days for 4 weeks. NM+si-AAB+MSN group: rats were subjected to AAC and treated with NM + si-AAB + MSN via tail vein injection 24 h later every 2 days for 4 weeks. Data are represented by means ± SEM (n=6). ##p <0.01 versus sham group; ∗∗p <0.01 versus model group. Scale bars, (E) 1.0 μm; (F) 200 μm (×100); 100 μm (×200); (J) 50 μm.
Discussion
Cardiac hypertrophy is a common response to multiple physiological and pathological stimuli, ultimately leading to heart failure. Heart failure is one of the most fatal cardiovascular diseases worldwide.42 It is important to identify effective therapeutic targets that inhibit maladaptive hypertrophy and subsequent heart failure. In the present study, we found that lncRNA AAB was remarkably upregulated in cardiac hypertrophy rats and this upregulation promoted ferroptosis. The pro-ferroptotic action of lncRNA AAB was mediated by the imbalance of MMP9/TIMP1 via sponging miR-30b-5p. In contrast, silencing lncRNA AAB and overexpressing miR-30b-5p alleviated CMEC ferroptosis and improved the function of CMEC treated by Ang II. In this study, the nanocomplex was rationally designed to realize efficient si-AAB loading and cell membrane cloaking, and thus to effectively perform its function of precise repair toward endothelial injury in cardiac hypertrophy (Figure 8). The synthesis of the nanocomplex included three steps (Figure 8A): (1) synthesis of mesoporous silica nanoparticle (MSN) with si-AAB (si-AAB + MSN), (2) neutrophil membrane (NM) isolation from neutrophil, and (3) construction of cell membrane-camouflaged si-AAB + MSN (NM + si-AAB + MSN) complex. Moreover, the synthesis of NM + miR-30b-5p + MSN was the same as NM + si-AAB + MSN. Silica nanoparticles have been proved to be ideal nucleic acid vehicles for loading and delivery.43,44 The targeted delivery of NM + si-AAB + MSN was achieved through the antibody LFA-1 and β2 Integrin on the neutrophil membrane, which specifically bound to ICAM-1 on the injured endothelial cells45,46 (Figure 8B), considering the high expression of ICAM-1 in endothelial cells of hypertrophic hearts.47 In addition, our results indicated that the protein expression of ICAM-1 was markedly increased in CMECs induced by Ang II and myocardial tissue of cardiac hypertrophy rats. Upon uptake by CMECs, NM + si-AAB + MSN was triggered to decompose in CMECs, and si-AAB was consequently released into the cytoplasm for efficient silencing of lncRNA AAB. Therefore, administration of NM + si-AAB + MSN in a rat model of cardiac hypertrophy can effectively inhibit CMEC ferroptosis, leading to preservation of viable CMECs and augmentation of cardiac functions.
Figure 8.

The mechanism of NM + si-AAB + MSN regulating cardiac hypertrophy via inhibiting the ferroptosis of CMEC
(A) The synthesis of NM + si-AAB+ MSN. (B) The targeted delivery of NM + si-AAB + MSN on the injured endothelial cells.
Recent studies on miRNAs have renewed our understanding about the regulation of cardiac hypertrophy. Various studies, including ours, have provided strong evidence that miRNAs play important roles in cardiac hypertrophy.48, 49, 50 Notably, the expression of miR-30b-5p was significantly downregulated in the rat model of cardiac hypertrophy after AAC. In addition, miR-30b-5p in CMECs was resistant to Ang II-induced damage under pathological stimulation. Furthermore, more importantly, we found that forced expression of miR-30b-5p attenuated CMEC ferroptosis and improved the function of CMEC treated with Ang II. Our study elucidated the anti-ferroptotic effect and mechanisms of miR-30b-5p in cardiac hypertrophy, and indicated that the exogenous application of miR-30b-5p might be a novel therapeutic strategy for cardiac hypertrophy. In the present study, enforced expression of miR-30b-5p resulted in a reduction of TIMP1 in CMECs. We also verified the interaction of TIMP1 and miR-30b-5p by luciferase assay. This indicated that the interaction between miR-30b-5p and TIMP1 3ʹUTR actually existed in vitro. Our present work used siRNA TIMP1 to study its role in CMEC and demonstrated that TIMP1 was a prerequisite for Ang II to initiate injury of CMECs.
Mammalian genomes encode a large number of lncRNAs.51,52 lncRNAs are defined as having important functions in specific cell types, tissues, and developmental conditions, such as structural scaffolding,53 RNA processing,54 chromatin modification,55 and the regulation of apoptosis and invasion.56 Although lncRNAs are biologically important, the role of lncRNAs in the regulation of myocardial hypertrophy is not fully understood. Previous studies have shown that lncRNAs may interact with miRNA as endogenous sponge RNA and affect the expression of miRNA target genes. In addition, we found other differentially expressed lncRNAs that contained miR-30b-5p binding sites in myocardial hypertrophy rats. We know that a single miRNA can regulate multiple targets, and a single target can be co-regulated by multiple miRNAs. Besides, one lncRNA may regulate multiple miRNAs and many lncRNAs form a complex lncRNA-miRNA-target regulatory network to participate in the progression of cardiac hypertrophy. In the future, the lncRNA-miRNA-target regulatory network should be systematically investigated to reveal the mechanism of cardiac hypertrophy. For example, Song et al found that inhibition of lncRNA Gm15834 attenuated autophagy-mediated myocardial hypertrophy via miR-30b-3p/ULK1 axis in mice.57 Other studies have shown that lncRNA NEAT1 promoted cardiac hypertrophy through the microRNA-19a-3p/SMYD2 axis.58 Our present work revealed a novel function of lncRNA AAB in regulating cardiac hypertrophy. lncRNA AAB served as a sponge of miR-30b-5p to regulate the imbalance of MMP9/TIMP1, which activated hypertrophic responses. Our results may provide new clues for understanding the cellular events controlled by lncRNAs. The discovery of lncRNA in cardiac hypertrophy may provide a new way to explore the complex molecular mechanism of cardiac hypertrophy.
Currently, many functional lncRNAs are known, and their regulatory mechanisms have been widely elucidated. However, few efforts have been made to develop effective in vivo regulatory strategies to improve treatment outcomes. RNA interference technology is the most convenient method because it has a strong ability to silence the expression of target genes. However, due to the polyanion and biomacromolecule characteristics of RNA interference therapies such as siRNA, specific delivery vehicles are required to facilitate siRNA delivery in vivo.59,60 Nanotechnology has shown great promise for improvement of in vivo siRNA delivery, and several RNA interference nanoparticle platforms have been marketed or entered into early phase clinical trials for the treatment of various diseases, especially liver diseases.61 However, effective and safe systemic delivery of siRNAs into heart in vivo remains challenging, due to the complexities of cardiovascular disease.
In this study, we report a self-assembled neutrophil membrane-camouflaged nanocomplex with a mesoporous core for siRNA-mediated repair of endothelial injury in cardiac hypertrophy. Mesoporous silica nanoparticle core possesses high siRNA loading capacity and effectively protects NM + si-AAB + MSN from degradation in body fluid. The si-AAB released long term at the endothelial injury site inhibits ferroptosis of CMECs, and therefore improves cardiac microvascular function. More importantly, considering the potential clinical application of this study, we compared the lncRNA AAB sequence with the human homologous sequence and found that the two sequences were well conserved (Figure S10). Furthermore, to compare the NM nanoparticles and lipidosome for cell uptake, we developed a delivery system based on the standard lipid-vesicle-coated MSN, and then the cell uptake was evaluated via immunofluorescence. The results indicated the NM nanoparticles were better than the lipidosome for cell uptake (Figure S11). Therefore, our study may provide a new therapeutic strategy for the clinical treatment of cardiac hypertrophy.
In summary, our present work indicated that the MMP9/TIMP1 imbalance resulted in CMEC ferroptosis and, further, induced the dysfunction of CMECs. MiR-30b-5p, an anti-hypertrophic miRNA, showed an inhibitory effect on CMEC ferroptosis by targeting TIMP1. Moreover, we have identified and revealed the important biological function of lncRNA AAB in cardiac hypertrophy. This lncRNA could downregulate miR-30b-5p and thus induce the imbalance of MMP9/TIMP1, leading to ferroptosis of CMECs. Using NM + si-AAB + MSN and NM + miR-30b-5p + MSN to transport si-AAB and miR-30b-5p could efficiently silence lncRNA AAB and overexpress miR-30b-5p, and then inhibit the ferroptosis of CMECs in vivo. The new strategy developed in this work showed great potential for treatment of patients with cardiac hypertrophy.
Materials and methods
Ethics statement
The procedures for the use of animals (male Wistar rat; 200–250 g) in this work were in accordance with the regulations of the Ethic Committees of Harbin Medical University-Daqing (ethics approval number: HMU20210801001) and conformed to the National Research Council (NRC) Guide for the Care and Use of Laboratory Animals (2011, 8th edition).
Isolation of neutrophils
The neutrophils were isolated by Percoll gradient method. In brief, blood samples were collected in a test tube containing 3.8% sodium citrate, purified by centrifugation, and then resuspended in 2 mL of PBS. The cells were carefully added into three layers of Percoll gradient solution diluted at 78%, 69%, and 52% (v/v) in PBS and centrifuged at 2,000 rpm for 20 min. Neutrophils were recovered from interfaces 69% and 78% and from the upper layer of 78%. The high-purity neutrophils were obtained by lysing the residual erythrocytes with 4°C lytic buffer.
Isolation of NM
The frozen neutrophils were homogenized after thawing. The homogenate was extracted, centrifuged (3,500 rpm, 5 min) at 4°C, and the nuclei and intact cells were removed. The supernatant was centrifuged (20,000 rpm, 25 min) at 4°C to remove the precipitate and mitochondria. The supernatant was then centrifuged (50,000 rpm, 1 h) at 4°C. The NM vesicles containing precipitation were collected and washed with 0.2 mM EDTA Na2 twice. The NM vesicles were extruded on 400-nm and 100-nm polycarbonate membranes for several times continuously. After 5 min of ultrasound, the membranes were stored at −80°C for later use. The membrane content was determined by Pierce BCA protein detection kit (Beyotime Biotechnology).
Preparation and characterization of MSN nanoparticles covered by NM
MSN nanoparticles (0.5 mg) were dispersed in siRNA of lncRNA AAB or miR-30b-5p solution for 12 h under magnetic stirring. For membrane coating, the NM vesicles underwent fusion with an equal volume of MSN by ultrasound for 5 min, and the resulting specimens were filtered 20 times using porous syringe filters with a membrane pore size of 200 nm. After centrifugation (2,500 rpm, 10 min), excess neutrophil membrane vesicles were removed, and NM + si-AAB + MSN or NM + miR-30b-5p + MSN was obtained.
Images were captured by FEI Magellan 400 scanning electron microscope (United States). N2 adsorption-desorption isotherm was measured on a Micrometitics Tristar 3000 system. Dynamic Light Scattering (DLS) measurement and zeta potential were conducted on a zeta sizer Nano series instrument (Nano ZS90). Protein analysis was achieved by SDS-PAGE and western blot analysis.
Establishing cardiac hypertrophy model and detecting cardiac function in vivo
AAC or sham surgery was performed on male Wistar rat as previously described.62 The rats were randomly divided into five groups as follows: (1) sham group: the rats underwent sham operation and received vehicle (PBS, caudal vein injection). (2) Model group: the rats were subjected to AAC and received vehicle (PBS, caudal vein injection). (3–5) The treatment groups: the rats were subjected to AAC and after 24 h treated with si-AAB, si-AAB + MSN, or NM + si-AAB + MSN (50 nM siRNA dose per rat every 2 days for 4 weeks, caudal vein injection). To investigate the effect of NM + miR-30b-5p + MSN on cardiac hypertrophy rats, the rats were subjected to AAC and treated with miR-30b-5p, miR-30b-5p + MSN, or NM + miR-30b-5p + MSN. In addition, after 14 days of surgery, eight rats were randomly selected from the model group and intraperitoneally injected with ferrostatin-1 (Ferr-1; ferroptosis inhibitor; Xcess Biosciences) at a dose of 2 mg/kg. In this study, rats were subjected to AAC and treated with the above interventions randomly. Efficacy was evaluated based on morphology, transthoracic echocardiography, and biomarker analyses in a blinded fashion. Finally, in the condition of anesthetization with pentobarbital sodium (40 mg/kg), the rat was sacrificed by cervical dislocation and the heart was removed rapidly after the last treatment. Transthoracic echocardiography was performed according to manufacturer's guide for small animal echocardiography (Visual Sonica, Toronto, Ontario, Canada). The functional indicators included LVPWd, LVIDd, LVIDs, LVFS, and LVEF. In addition, left ventricular function was continuously recorded with BL-420N organism function experiment system (Cheng Du Tai Meng, China). The indicators included left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), and maximum rate of left ventricular pressure rise and fall (+dp/dtmax and −dp/dtmax).
Histological analysis
The hearts of AAC rats or sham rats were quickly dissected and immersed in 4% paraformaldehyde for 24 h and stained with HE or FITC-labeled WGA staining for histopathology. Photographs of HE staining were taken with a microscope and left ventricular cardiac myocyte membranes were observed by fluorescent microscopy.
Isolation and culture of CMECs
The CMECs were isolated from Wistar rats (100–120 g) as previously described.7 In brief, the left ventricles of male rats were harvested and minced into 1-mm3 pieces after the epicardial coronaries and endocardial endothelium were removed. The pieces of cardiac tissue were dissociated by collagenase II and subsequently cultured in DMEM/F12 supplemented with 1% VEGF, 1% endothelial cell growth supplement (ECGS), 10% fetal bovine serum (FBS), and 100 μg/mL penicillin/streptomycin. Ang II (Sigma, St Louis, MO) was used to establish the damage model of CMEC. CMECs were randomly divided into five groups as follows: (1) control group: the cell was incubated under normal conditions throughout the experiments. (2) Model group: the cell was treated with 100 nM Ang II for 24 h following by treatment with PBS for 48 h. (3–5) si-AAB groups: the cells were given 100 nM Ang II for 24 h followed by treatment with si-AAB, si-AAB + MSN, and NM + si-AAB + MSN for 48 h, respectively. Moreover, the cells in the Ferr-1 group were given 100 nM Ang II for 24 h following by treatment with 2 μΜ Ferr-1 for 48 h.
Western blot
Protein from frozen CMECs and cardiac tissues was extracted. After denaturation, proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane. After blocking, the membranes were incubated with primary antibodies and secondary antibodies. Primary antibodies included GAPDH (1:2,000, Sigma, St Louis, United States), BNP (1:1,000, Sigma, St Louis, United States), β-MHC (1:2,000, Sigma, St Louis, United States), PECAM-1 (1:300, Abcam, Cambridge, MA), H2AX (1:500, Cell Signaling Technology, Danvers, MA), MMP9 (1:1,000, Cell Signaling Technology, Beverly, MA), TIMP1 (1:500, Abcam, Cambridge, MA), SLC7A11 (1:500, Abcam, Cambridge, MA), GPx4 (1:1,000, Abcam, Cambridge, MA), ferroportin 1 (Fpn1, 1:1,000, Abcam, Cambridge, MA), TFR-1 (1:1,000, Abcam, Cambridge, MA), p-eNOS (Ser1177) (1:1,000, Cell Signaling Technology, Beverly, MA), eNOS (1:1,000, Cell Signaling Technology, Beverly, MA), LC3II (1:2,000, Cell Signaling Technology, Beverly, MA), Caspase-3 (1:300, Sigma, St Louis, United States), Bcl-2 (1:1,000, Cell Signaling Technology, Beverly, MA), RIP1 (1:1,000, Cell Signaling Technology, Beverly, MA), RIP3 (1:1,000, Cell Signaling Technology, Beverly, MA), β2 Integrin (1:20,000, Abcam, Cambridge, MA), LFA-1 (0.1 μg/mL, Abcam, Cambridge, MA), and ICAM-1 (1:5,000, Abcam, Cambridge, MA). The nitrocellulose membranes were treated with ECL reagents prior to visualization using an Imaging System (LI-COR Biosciences). The specific protein expression levels were normalized to the levels of GAPDH on the same nitrocellulose membrane.
Electron microscopy
To detect the changes of mitochondria in CMECs during cardiac hypertrophy, electron microscopy was conducted as described previously.63 Heart tissues were fixed at 4°C with 2% glutaraldehyde in a 0.1 M sodium cacodylate buffer and postfixed for 1 h on ice with 1% osmium tetroxide. The slices were stained with uranyl acetate and observed under an electron microscope (HITACHI HT7650, Japan).
Gelatin-ink staining
To observe the patency of the microvasculature, gelatin ink (3% gelatin and ink) was injected into the heart via the jugular vein at a room temperature of 30°C. Then, the hearts were cut and maintained at 4°C for 1 h. Cryosections was performed after 4% paraformaldehyde fixation and the samples were observed under a microscope.64
Iron measurements
The cardiac tissue and CMECs were pretreated in PBS. The supernatant was collected after centrifugation. The iron level was determined by the Iron Assay Kit (ab83366, Abcam) according to the manufacturer's instructions.
ROS detection
The cells were inoculated in a six-well plate with cover glass, cultured for 24 h, then 2,7-dichlorodi-hydrofluorescein diacetate (DCFH-DA) serum-free medium with a final concentration of 10 μM was added, and the fluorescent probe was vibrated every 3–5 min to make full contact with the cells. The cells were washed with fresh culture solution three times to fully remove the DCFH-DA that did not enter the cells. Fluorescence microscopy was used to detect fluorescence intensity of ROS in cells under the excitation wavelength of 488 nm and emission wavelength of 525 nm.
Oxidative stress measurements
The levels of MDA and GSH, and the enzymatic activities of SOD, Mn-SOD, Cu/Zn-SOD, GSH-PX, and CAT, were detected with different detection kits according to the manufacturer's instructions (Nanjing Jian Cheng Bioengineering Institute).65
qRT-PCR
Total RNA of cardiac tissues and CMECs was extracted with TRIzol reagent (Invitrogen) and reverse transcribed with RT-PCR kit (Takara, Japan). Quantification of gene expression was performed using NanoDrop One (Thermo Fisher Scientific, Waltham, United States). mRNA levels were detected by qRT-PCR and normalized to the levels of β-actin expression. Primer sequences are listed in Table S7.
NO and cGMP measurement
NO levels were detected by testing the levels of nitrite and nitrate, followed by the stable degradation products of NO (Griess reaction NO assay kit; Calbiochem). Moreover, the level of cGMP was determined by using a cGMP complete ELISA kit (cGMP Assay Kit; Abcam) following the manufacturer's protocol.
Immunohistochemistry staining
Immunohistochemistry assay was performed by using antibody PECAM-1. Staining was performed with DAB Kit (Sangon Biotech; Shanghai) according to the manufacturer's instructions.
Immunofluorescence assay
Immunofluorescence assay was performed by using antibody PECAM-1. Briefly, CMECs were fixed with 4% paraformaldehyde, permeabilized using 0.2% Triton X-100, and incubated with anti-PECAM-1 (1:50, Abcam, Cambridge, MA) at 4°C overnight. Thereafter, the samples were washed three times with PBS and incubated with FITC conjugated species-specific secondary antibody. Then, DAPI (Beyotime Biotechnology, Shanghai, China) was counter-stained for the identification of nuclei. Photographs were acquired by using a fluorescence microscope.
The different uptake capacities of NM + si-AAB + MSN by CMs and CMECs were verified by immunofluorescence. The nuclei of cells were stained with DAPI (blue), and NM + si-AAB + MSN was labeled with FITC (green). Moreover, to verify the successful lysosomal escape of NM + si-AAB + MSN in CMECs, fluorescent colocalization was tracked by confocal microscopy.
Cell viability assay
To investigate the cell viability, MTT assay was performed. In brief, CMECs were seeded into 96-well plates and cultured. Cell counting kit-8 (CCK-8; Dojindo Molecular Technologies) was used to measure cell viability according to the manufacturer's protocol. Absorbance was measured at 450 nm using a microplate reader.
Endothelial cell wound healing migration assay
According to Cerezo's method,66 briefly, when the cells achieved 100% confluence, we made a straight lesion using a sterile 200-μL pipette tip in the center of the monolayer. The wounds were photographed by using phase-contrast microscopy on an inverted microscope (Nikon, Tokyo, Japan). Six independent experiments were carried out.
Tube formation assay
Collagen gels (10 mg/mL) were formed by adding Matrigel (Becton Dickinson) into a 24-well plate according to the manufacturer's instruction. CMECs were seeded on coated plates at 1.5 × 105 cells/well and incubated at 37°C for 1 h. Tube formation was observed and photographed by using a phase-contrast microscope.
Cellular uptake of si-AAB + MSN and NM + si-AAB + MSN
The cellular uptake efficiency of si-AAB + MSN and NM + si-AAB + MSN by CMECs was detected by flow cytometry. Briefly, CMECs were seeded on a 12-well plate with a density of 1.0 × 105 cells per well in 1 mL of endothelial culture medium (ECM) and cultured for 24 h. Next, the cells were incubated with the si-AAB + MSN and NM + si-AAB + MSN respectively for 4 h. Then the cells were washed with PBS three times to completely remove excess si-AAB + MSN and NM + si-AAB + MSN. The harvested cells were treated with trypsin for 2 min, centrifuged at 2,000 rpm for 3 min, and then the cell debris was suspended in 300 μL of PBS. Fluorescence intensity of cellular uptake was measured using an FACS Aria I spectral analyzer (Becton Dickinson, United States).
Vector construction and transfection
The full-length of TIMP1 and lncRNA AAB were synthesized by PCR and inserted into pcDNA3.1 vector. In addition, pcDNA3.1 empty vector was used as a negative control. CMECs were washed with serum-free medium and then incubated in 5 mL of serum-free medium for 4–6 h for transfection. TIMP1, lncRNA AAB, or control vectors (1 μg/mL) and x-treme GENE siRNA (Invitrogen, Carlsbad, CA) were separately mixed with 300 μL of serum-free medium for 5 min. Then, two mixtures were combined and incubated at room temperature for 18 min. Finally, the plasmid mixture and x-treme GENE siRNA were added to the CMECs and incubated at 37°C for 24 h.
miRNA mimic, siRNA construction, and transfection
MiR-30b-5p mimics and siRNA sequences that target to TIMP1 and lncRNA AAB were synthesized by Gene Pharma (Shanghai Gene Pharma, China). Primer sequences are listed in Table S7. CMECs were washed with serum-free medium and then incubated in 5 mL of serum-free medium for 4–6 h for transfection. MiR-30b-5p mimics or control vectors (1 μg/mL) and x-treme GENE siRNA (Invitrogen, Carlsbad, CA) were separately mixed with 300 μL of serum-free medium for 5 min. Then, two mixtures were combined and incubated at room temperature for 18 min. Finally, the mixture and x-treme GENE siRNA were added to the CMECs and incubated at 37°C for 24 h. Cells were divided into six groups: (1) control group; (2) Ang II group; (3) Ang II + miR-30b-5p group, with cells exposed to Ang II for 24 h and then treated with miR-30b-5p mimics for 24 h; (4) Ang II + miR-30b-5p + AMO group, with cells exposed to Ang II for 24 h and then treated with miR-30b-5p mimics and AMO-miR-30b-5p for 24 h; (5) Ang II + NC-miR-30b-5p group, with cells exposed to Ang II for 24 h and then treated with NC-miR-30b-5p for 24 h; (6) Ang II + NC-AMO group, with cells exposed to Ang II for 24 h and then treated with NC-AMO-miR-30b-5p for 24 h. lncRNA AAB was silenced as follows: CMECs were seeded in six-well plates (50,000 cells per well) and incubated in 2 mL of DMEM containing 10% FBS for 24 h. Subsequently, the medium was replaced by fresh medium and then the NM + si-AAB + MSN complexes were added to silence the lncRNA AAB expression. After incubation for 24 h, the cells were washed with PBS solution and further incubated in fresh medium for another 48 h.
Luciferase reporter assays
The TIMP1 3′UTR and lncRNA AAB full-length sequences containing miR-30b-5p binding sites were amplified by PCR. For luciferase assay, cells were seeded in 48-well plates in triplicate, and 40 ng/well luciferase reporter vector, 10 pmol miR-30b-5p mimic, or mimic control were co-transfected into plates using Lipofectamine 2000 (Invitrogen). Cells were lysed at 24 h after transfection, and luciferase activity was determined using the Dual Luciferase Reporter Assay kit (Promega).
Cell death assay
Cell death was determined by trypan blue exclusion, and the numbers of trypan blue-positive and trypan blue-negative cells were counted on a hemocytometer.
Statistical analysis
The data were analyzed by Statistical Product and Service Solutions (SPSS) 19.0 statistical software (SPSS, Chicago, IL) and Origin 9.1. The quantitative data were represented as the mean ± SEM. Analyses of multiple group comparisons were performed with one-way analysis of variance (ANOVA). p < 0.05 was considered as a statistically significant difference.
Acknowledgments
This work was supported by National Natural Science Foundation of China (for H.S., 81872856 and 82173810) and the Fundamental Research Funds for the Provincial Universities (for P.S., JFWLD201902).
Author contributions
S.P.L., L.M.H., and S.C. performed the experimental work, collected and analyzed the data, and drafted the original manuscript. Q.H.P. and B.L.N. revised the manuscript. L.M.H. and C.Y.G. performed the synthesis. Z.M.T., X.Y.W., and R.J. helped with the animal experiments. W.J.B. and R.P. helped with the cell experiments. S.H.L. conceived and designed the study and critically revised the manuscript for important intellectual content. All the authors read and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.10.024.
Supplemental information
References
- 1.Pandey K.N., Vellaichamy E. Regulation of cardiac angiotensin-converting enzyme and angiotensin AT1 receptor gene expression in Npr1 gene-disrupted mice. Clin. Exp. Pharmacol. Physiol. 2010;37:e70–e77. doi: 10.1111/j.1440-1681.2009.05315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen G., Huang S., Song F., Zhou Y., He X. Lnc-Ang362 is a pro-fibrotic long non-coding RNA promoting cardiac fibrosis after myocardial infarction by suppressing Smad7. Arch. Biochem. Biophys. 2020;685:108354. doi: 10.1016/j.abb.2020.108354. [DOI] [PubMed] [Google Scholar]
- 3.Qiu Y., Cheng R., Liang C., Yao Y., Zhang W., Zhang J., Zhang M., Li B., Xu C., Zhang R. MicroRNA-20b promotes cardiac hypertrophy by the inhibition of mitofusin 2-mediated inter-organelle Ca2+ cross-talk. Mol. Ther. Nucl. Acids. 2020;19:1343–1356. doi: 10.1016/j.omtn.2020.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song G., Zhu L., Ruan Z., Wang R., Shen Y. MicroRNA-122 promotes cardiomyocyte hypertrophy via targeting FoxO3. Biochem. Biophys. Res. Commun. 2019;519:682–688. doi: 10.1016/j.bbrc.2019.09.035. [DOI] [PubMed] [Google Scholar]
- 5.Qi H., Ren J., E M., Zhang Q., Cao Y., Ba L., Song C., Shi P., Fu B., Sun H. MiR-103 inhibiting cardiac hypertrophy through inactivation of myocardial cell autophagy via targeting TRPV3 channel in rat hearts. J. Cell. Mol. Med. 2019;23:1926–1939. doi: 10.1111/jcmm.14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He J., Jiang S., Li F., Zhao X., Chu E., Sun M., Chen M., Li H. MicroRNA-30b-5p is involved in the regulation of cardiac hypertrophy by targeting CaMKIIδ. J. Investig. Med. 2013;61:604–612. doi: 10.2310/JIM.0b013e3182819ac6. [DOI] [PubMed] [Google Scholar]
- 7.Wang K., Liu F., Zhou L., Long B., Yuan S., Wang Y., Liu C., Sun T., Zhang X., Li P. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ. Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 8.Fang Q., Liu T., Yu C., Yang X., Shao Y., Shi J., Ye X., Zheng X., Yan J., Xu D., et al. LncRNA TUG1 alleviates cardiac hypertrophy by targeting miR-34a/DKK1/Wnt-β-catenin signaling. J. Cell. Mol. Med. 2020;24:3678–3691. doi: 10.1111/jcmm.15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W.S., Stockwell B.R. Ferroptosis: death by lipid peroxidation. Trends. Cell. Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon S.J., Stockwell B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 11.Pantopoulos K., Porwal S.K., Tartakoff A., Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51:5705–5724. doi: 10.1021/bi300752r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muckenthaler M.U., Galy B., Hentze M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 13.Yasumura S., Naito Y., Okuno K., Sawada H., Asakura M., Masuyama T., Ishihara M. Effects of heterozygous TfR1 (transferrin receptor 1) deletion in pathogenesis of renal fibrosis in mice. Hypertension. 2020;75:413–421. doi: 10.1161/HYPERTENSIONAHA.119.13670. [DOI] [PubMed] [Google Scholar]
- 14.Conrad M., Proneth B. Broken hearts: iron overload, ferroptosis and cardiomyopathy. Cell. Res. 2019;29:263–264. doi: 10.1038/s41422-019-0150-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang X., Wang H., Han D., Xie E., Yang X., Wei J., Gu S., Gao F., Zhu N., Yin X., et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. U S A. 2019;116:2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai T., Liang R., Zhu R., Wang W., Zhou L., Sun Y. MicroRNA-214-3p enhances erastin-induced ferroptosis by targeting ATF4 in hepatoma cells. J. Cell. Physiol. 2020;235:5637–5648. doi: 10.1002/jcp.29496. [DOI] [PubMed] [Google Scholar]
- 17.Mao C., Wang X., Liu Y., Wang M., Yan B., Jiang Y., Shi Y., Shen Y., Liu X., Lai W. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018;78:3484–3496. doi: 10.1158/0008-5472.CAN-17-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yussman M.G., Toyokawa T., Odley A., Lynch R.A., Wu G., Colbert M.C., Aronow B.J., Lorenz J.N., Dorn G.W. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat. Med. 2002;8:725–730. doi: 10.1038/nm719. [DOI] [PubMed] [Google Scholar]
- 19.Shirakabe A., Zhai P., Ikeda Y., Saito T., Maejima Y., Hsu C., Nomura M., Egashira K., Levine B., Sadoshima J. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vu T.H., Shipley J.M., Bergers G., Berger J.E., Helms J.A., Hanahan D., Shapiro S.D., Senior R.M., Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strzyz P. Iron expulsion by exosomes drives ferroptosis resistance. Nat. Rev. Mol. Cell. Biol. 2020;21:4–5. doi: 10.1038/s41580-019-0195-2. [DOI] [PubMed] [Google Scholar]
- 22.Li N., Wang W., Zhou H., Wu Q., Duan M., Liu C., Wu H., Deng W., Shen D., Tang Q. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 2020;160:303–318. doi: 10.1016/j.freeradbiomed.2020.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Ma W., Wei S., Zhang B., Li W. Molecular mechanisms of cardiomyocyte death in drug-induced cardiotoxicity. Front. Cell. Dev. Biol. 2020;8:434. doi: 10.3389/fcell.2020.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang W.S., SriRamaratnam R., Welsch M.E., Shimada K., Skota R., Viswanathan V.S., et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H., Liu C., Zhao Y., Gao G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020;99:151058. doi: 10.1016/j.ejcb.2019.151058. [DOI] [PubMed] [Google Scholar]
- 26.Caillon A., Mian M.O.R., Fraulob-Aquino J.C., Huo K.G., Barhoumi T., Ouerd S., Sinnaeve P.R., Paradis P., Schiffrin E.L. γδ T cells mediate angiotensin II-induced hypertension and vascular injury. Circulation. 2017;135:2155–2162. doi: 10.1161/CIRCULATIONAHA.116.027058. [DOI] [PubMed] [Google Scholar]
- 27.Zhou H., Wang S., Zhu P., Hu S., Chen Y., Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–346. doi: 10.1016/j.redox.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ardi V.C., Kupriyanova T.A., Deryugina E.I., Quigley J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. U S A. 2007;104:20262–20267. doi: 10.1073/pnas.0706438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lelongt B., Trugnan G., Murphy G., Lelongt P.M.R.B., Trugnan G., Murphy G., Ronco P.M. Matrix metalloproteinases MMP2 and MMP9 are produced in early stages of kidney morphogenesis but only MMP9 is required for renal organogenesis in vitro. J. Cell. Biol. 1997;136:1363–1373. doi: 10.1083/jcb.136.6.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chua C.C., Hamdy R.C., Chua B.H. Angiotensin II induces TIMP-1 production in rat heart endothelial cells. Biochim. Biophys. Acta. 1996;1311:175–180. doi: 10.1016/0167-4889(95)00205-7. [DOI] [PubMed] [Google Scholar]
- 31.Giraudo E., Inoue M., Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest. 2004;114:623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doll S., Freitas F.P., Shah R., Aldrovandi M., Silva M.C., Ingold I., Grocin A.G., Silva T.N.X., Panzilius E., Scheel C.H., et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 33.Badgley M.A., Kremer D.M., Maurer H.C., Giorno K.E.D., Lee H.J., Purohit V., Sagalovskiy I.R., Ma A., Kapilian J., Firl C.E.M., et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–89. doi: 10.1126/science.aaw9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q., Wu H., Zou M., Li L., Li Q., Sun C., Xia W., Cao Y., Wu L. Folic acid improves abnormal behavior via mitigation of oxidative stress, inflammation, and ferroptosis in the BTBR T+ tf/J mouse model of autism. J. Nutr. Biochem. 2019;71:98–109. doi: 10.1016/j.jnutbio.2019.05.002. [DOI] [PubMed] [Google Scholar]
- 35.Bao W., Pang P., Zhou X., Hu F., Xiong W., Chen K., Wang J., Wang F., Xie D., Hu Y.Z., et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer's disease. Cell. Death Differ. 2021;28:1548–1562. doi: 10.1038/s41418-020-00685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jens M., Rajewsky N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat. Rev. Genet. 2015;16:113–126. doi: 10.1038/nrg3853. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M., Jiang Y., Guo X., Zhang B., Wu J., Sun J., Liang H., Shan H., Zhang Y., Liu J., et al. Long non-coding RNA cardiac hypertrophy-associated regulator governs cardiac hypertrophy via regulating miR-20b and the downstream PTEN/AKT pathway. J. Cell. Mol. Med. 2019;23:7685–7698. doi: 10.1111/jcmm.14641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomson D.W., Dinger M.E. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 2016;17:272–283. doi: 10.1038/nrg.2016.20. [DOI] [PubMed] [Google Scholar]
- 39.Yao C., Wu W., Tang H., Jia X., Tang J., Ruan X., Li F., Leong D.T., Luo D., Yang D. Self-assembly of stem cell membrane-camouflaged nanocomplex for microRNA-mediated repair of myocardial infarction injury. Biomaterials. 2020;257:120256. doi: 10.1016/j.biomaterials.2020.120256. [DOI] [PubMed] [Google Scholar]
- 40.Cheng W., Nie J., Gao N., Liu G., Tao W., Xiao X., Jiang L., Liu Z., Zeng X., Mei L. A multifunctional nanoplatform against multidrug resistant cancer: merging the best of targeted chemo/gene/photothermal therapy. Adv. Funct. Mater. 2017;27:1704135. [Google Scholar]
- 41.Gilleron J., Querbes W., Zeigerer A., Borodovsky A., Marsico G., Schubert U., Manygoats K., Seifert S., Andree C., Stöter M., et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013;31:638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 42.Cibi D.M., Bi-Lin K.W., Shekeran S.G., Sandireddy R., Tee N., Singh A., Wu Y.J., Srinivasan D.K., Kovalik J.P., Ghosh S., et al. Prdm16 deficiency leads to age-dependent cardiac hypertrophy, adverse remodeling, mitochondrial dysfunction, and heart failure. Cell. Rep. 2020;33:108288. doi: 10.1016/j.celrep.2020.108288. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y., Chen H., Shi J. In vivo bio-safety evaluations and diagnostic/therapeutic applications of chemically designed mesoporous silica nanoparticles. Adv. Mater. 2013;25:3144–3176. doi: 10.1002/adma.201205292. [DOI] [PubMed] [Google Scholar]
- 44.Chen W., Glackin C.A., Horwitz M.A., Zink J.I. Nanomachines and other caps on mesoporous silica nanoparticles for drug delivery. Acc. Chem. Res. 2019;52:1531–1542. doi: 10.1021/acs.accounts.9b00116. [DOI] [PubMed] [Google Scholar]
- 45.Feng L., Dou C., Xia Y., Li B., Zhao M., Yu P., Zheng Y., El-Toni A.M., Atta A., Galal Y., et al. Neutrophil-like cell-membrane-coated nanozyme therapy for ischemic brain damage and long-term neurological functional recovery. ACS. Nano. 2021;15:2263–2280. doi: 10.1021/acsnano.0c07973. [DOI] [PubMed] [Google Scholar]
- 46.Jickling G.C., Liu D., Ander B.P., Stamova B., Zhan X., Sharp F.R. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J. Cereb. Blood Flow. Metab. 2015;35:888–901. doi: 10.1038/jcbfm.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tripathi D., Biswas B., Manhas A., Singh A., Goyal D., Gaestel M., Jagavelu K. Proinflammatory effect of endothelial microparticles is mitochondria mediated and modulated through MAPKAPK2 (MAPK-activated protein kinase 2) leading to attenuation of cardiac hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2019;39:1100–1112. doi: 10.1161/ATVBAHA.119.312533. [DOI] [PubMed] [Google Scholar]
- 48.Seok H., Lee H., Lee S., Ahn S.H., Lee H.S., Kim G.D., Peak J., Park J., Cho Y.K., Jeong Y.D., et al. Position-specific oxidation of miR-1 encodes cardiac hypertrophy. Nature. 2020;584:279–285. doi: 10.1038/s41586-020-2586-0. [DOI] [PubMed] [Google Scholar]
- 49.Zhang M., Cheng K., Chen H., Tu J., Shen Y., Pang L., Wu W. MicroRNA-27 attenuates pressure overload-Induced cardiac hypertrophy and dysfunction by targeting galectin-3. Arch. Biochem. Biophys. 2020;689:108405. doi: 10.1016/j.abb.2020.108405. [DOI] [PubMed] [Google Scholar]
- 50.Tian C., Hu G., Gao L., Hackfort B.T., Zucker I.H. Extracellular vesicular MicroRNA-27a∗ contributes to cardiac hypertrophy in chronic heart failure. J. Mol. Cell. Cardiol. 2020;143:120–131. doi: 10.1016/j.yjmcc.2020.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ponting C.P., Oliver P.L., Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 52.Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 53.Clemson C.M., Hutchinson J.N., Sara S.A., Ensminger A.W., Fox A.H., Chess A., Lawrence J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell. 2009;33:717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gong C.G., Maquat L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanduri C. Kcnq1ot1: a chromatin regulatory RNA. Semin. Cell. Dev. Biol. 2011;22:343–350. doi: 10.1016/j.semcdb.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 56.Khaitan D., Dinger M.E., Mazar J., Crawford J., Smith M.A., Mattick J.S., Perera R.J. The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res. 2011;71:3852–3862. doi: 10.1158/0008-5472.CAN-10-4460. [DOI] [PubMed] [Google Scholar]
- 57.Song C., Qi H., Liu Y., Chen Y., Shi P., Zhang S., Ren J., Wang L., Cao Y., Sun H. Inhibition of lncRNA Gm15834 attenuates autophagy-mediated myocardial hypertrophy via miR-30b-3p/ULK1 axis in mice. Mol. Ther. 2021;29:1120–1137. doi: 10.1016/j.ymthe.2020.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun X., Lv J., Dou L., Chen D., Zhu Y., Hu X. LncRNA NEAT1 promotes cardiac hypertrophy through microRNA-19a-3p/SMYD2 axis. Eur. Rev. Med. Pharmacol. Sci. 2020;24:1367–1377. doi: 10.26355/eurrev_202002_20194. [DOI] [PubMed] [Google Scholar]
- 59.Whitehead K.A., Langer R., Anderson D.G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanasty R., Dorkin J.R., Vegas A., Anderson D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 61.Shi J., Kantoff P.W., Wooster R., Farokhzad O.C. Cancer nanomedicine: progress, challenges and opportunities. Nat. Rev. Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Q., Qi H., Cao Y., Shi P., Song C., Ba L., Chen Y., Gao J., Li S., Li B., et al. Activation of transient receptor potential vanilloid 3 channel (TRPV3) aggravated pathological cardiac hypertrophy via calcineurin/NFATc3 pathway in rats. J. Cell. Mol. Med. 2018;22:6055–6067. doi: 10.1111/jcmm.13880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou H., Li D., Zhu P., Ma Q., Toan S., Wang J., Hu S., Chen Y., Zhang Y. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J. Pineal. Res. 2018;65:e12503. doi: 10.1111/jpi.12503. [DOI] [PubMed] [Google Scholar]
- 64.Zhou H., Hu S., Jin Q., Shi C., Zhang Y., Zhu P., Ma Q., Tian F., Chen Y. Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J. Am. Heart Assoc. 2017;6:e005328. doi: 10.1161/JAHA.116.005328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo J., Yan D., Li S., Liu S., Zeng F., Cheung C.W., Liu H., Irwin M.G., Huang H., Xia Z. Allopurinol reduces oxidative stress and activates Nrf2/p62 to attenuate diabetic cardiomyopathy in rats. J. Cell. Mol. Med. 2020;24:1760–1773. doi: 10.1111/jcmm.14870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cerezo A.B., Hornedo-Ortega R., Álvarez-Fernández M.A., Troncoso A.M., García-Parrilla M.C. Inhibition of VEGF-induced VEGFR-2 activation and HUVEC migration by melatonin and other bioactive indolic compounds. Nutrients. 2017;9:249. doi: 10.3390/nu9030249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.