

Abstract
Background
Thromboembolic events are frequently reported in patients infected with the SARS‐CoV‐2. Recently, we observed that platelets from patients with severe COVID‐19 infection express procoagulant phenotype. The molecular mechanisms that induce the generation of procoagulant platelets in COVID‐19 patients are not completely understood.
Objectives
In this study, we investigated the role of AKT (also known as Protein Kinase B), which is the major downstream effector of PI3K (phosphoinositid‐3‐kinase) (PI3K/AKT) signaling pathway in platelets from patients with COVID‐19.
Patients and Methods
Platelets, Sera and IgG from COVID‐19 patients who were admitted to the intensive care unit (ICU) were analyzed by flow cytometry as well as western blot and adhesion assays.
Results
Platelets from COVID‐19 patients showed significantly higher levels of phosphorylated AKT, which was correlated with CD62p expression and phosphatidylserine (PS) externalization. In addition, healthy platelets incubated with sera or IgGs from ICU COVID‐19 patients induced phosphorylation of PI3K and AKT and were dependent on Fc‐gamma‐RIIA (FcγRIIA). In contrast, ICU COVID‐19 sera mediated generation of procoagulant platelets was not dependent on GPIIb/IIIa. Interestingly, the inhibition of phosphorylation of both proteins AKT and PI3K prevented the generation of procoagulant platelets.
Conclusions
Our study shows that pAKT/AKT signaling pathway is associated with the formation of procoagulant platelets in severe COVID‐19 patients without integrin GPIIb/IIIa engagement. The inhibition of PI3K/AKT phosphorylation might represent a promising strategy to reduce the risk for thrombosis in patients with severe COVID‐19.
Keywords: AKT‐signaling, COVID‐19 platelet adhesion, procoagulant platelet formation, thrombosis
Essentials
-
•
The molecular mechanisms that induce the generation of procoagulant platelets in COVID‐19 patients are still elusive.
-
•
We analyzed platelets, sera and IgG from COVID‐19 patients from the intensive care unit.
-
•
Procoagulant platelet formation in COVID‐19 is dependent on PI3K/AKT pathway.
-
•
Inhibition of PI3K/AKT pathway might be a potential therapeutic target to mitigate increased thrombosis in COVID‐19 patients.
Alt-text: Unlabelled Box
1. INTRODUCTION
The emergence of novel coronavirus disease 2019 (COVID‐19) caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has developed to a pandemic level, profoundly threatening the health of millions of people worldwide.1., 2. Accumulating evidence indicates an association between COVID‐19 and hypercoagulation as well as thrombosis.3., 4., 5.
Thrombotic complications were observed in patients with severe COVID‐19 admitted to the intensive care unit (ICU).5 Patients with thrombosis show higher rates of procoagulant platelets.6., 7. However, the molecular mechanisms that regulate the formation of procoagulant platelets in COVID‐19 patients remain poorly understood. Recently, we demonstrated that immunoglobulin G (IgG) fractions from severe COVID‐19 patients induce procoagulant phenotype in platelets.8., 9. Furthermore, we found that increased thrombus formation in severe COVID‐19 patients is Fc‐gamma RIIA‐ (FcγRIIA) dependent, and enhanced level of cyclic‐adenosine‐monophosphate (cAMP) prevents antibody‐induced procoagulant platelet generation and clot formation.10
Considering the central role of PI3K/AKT signaling pathway in platelet function, it makes this pathway a potential therapeutic target to mitigate increased thrombosis in COVID‐19 patients.11
In this study, we investigated the role of PI3K/AKT signaling pathway in platelets from COVID‐19 patients and evaluated the effect of its inhibition on antibody‐mediated procoagulant platelet formation.
2. MATERIALS AND METHODS
2.1. Patients
Patients were diagnosed for SARS‐CoV‐2 infection by real time PCR on material collected by nasal swabs. Functional as well as signaling assays were performed using residual citrated blood samples collected from 24 COVID‐19 patients including patients admitted to ICU (n = 13; ICU COVID‐19) as well as normal ward (n = 11; non‐ICU COVID‐19). For control groups, blood samples were collected from severe ill ICU patients that presented an high sequential organ failure assessment (SOFA)‐score (n = 9, ICU control, Table S1 and Figure S1), which predicts the risk of morbidity and mortality due to sepsis and COVID‐19 as previously described,12 as well as from healthy volunteers at the blood donation center in Tuebingen, after written consent was obtained.
2.2. Evaluation of the clinical data
Clinical data related to treatments, demography and outcome were obtained from electronic medical records of the university hospital of Tuebingen. Data were independently reviewed by two physicians (K.A. and S.H.). Status of critical illness was assessed using the SOFA score. In brief, SOFA score was calculated by the assessment of functional data of the respiratory‐system, nervous‐system cardio‐vascular system, kidney function, liver function as well as coagulation system.
2.3. Platelet assays
Platelets were isolated from citrated whole blood of ICU COVID‐19 patients or controls and tested within 3 h after blood withdrawal. Briefly, whole blood sample was incubated with buffer or thrombin receptor activating peptide (TRAP‐6, 10 µM; Hart Biologicals) for 15 min at room temperature (RT). Next, cells were stained with a monoclonal antibody (moAb) against CD62p (anti‐CD62p 1:20 BD) and annexin‐V (1:20; Immunotools) that detects externalized phosphatidylserine (PS) in the presence of 1 mM CaCl2, (Sigma) for 15 min at RT in the dark. Finally, the expression of CD62p and PS on the platelet surface was determined by flow cytometry (FC, Navios, Beckman Coulter).
2.4. Platelet signaling pathway assay
For the detection of AKT phosphorylation on Ser473 in platelets, whole blood samples were fixed using the commercially available PerFix‐nc kit (Beckmann Coulter) and subsequently stained with CD41‐PC5 (BD), pAKT‐PE and AKT‐APC (both Invitrogen) at RT for 30 min. Additionally, to induce maximum phosphorylation levels of AKT, blood samples were incubated for 30 min at RT with TRAP‐6 (10 µM; Hart Biologicals). Test results were normalized to the MFI of healthy controls. Subsequently, samples were analyzed to determine the intracellular level of pAKT and the expression of CD62p as well as PS on the platelet surface (for details see Figure S2).
2.5. Procoagulant platelet formation
Platelets were isolated from citrated whole blood of healthy controls and incubated with sera or IgG from ICU COVID‐19 patients or controls. As positive controls, platelets were incubated with TRAP‐6 (10 µM; Hart Biologicals) or ionomycin (16 μM; Sigma‐Aldrich) for 15 min at RT. Next, cells were stained with moAb against CD62p (anti‐CD62p, 1:20 BD) and annexin‐V (1:20, Immunotools). When indicated, activated GPIIb/IIIa was assessed using the mAb PAC‐1 (1:20 BD) in the presence of 1 mM CaCl2 (Sigma) for 15 min at RT in the dark. Finally, the expression of CD62p, PS and activated PAC‐1 on the platelet surface was determined by flow cytometry (FC, Navios, Beckman Coulter) (for details see Figure S2).
2.6. Sera preparation and incubation with washed platelets
All sera were heat‐inactivated at 56°C for 30 min, followed by a sharp centrifugation step at 5000g to exclude any unspecific effects like the activation of platelets via complement and immune complexes. The supernatant was collected and incubated with washed platelets. Washed platelets were prepared as previously described.13 In brief, whole blood from healthy controls was centrifuged at 120g for 20 min without break at RT. The supernatant platelet rich plasma (PRP) was gently collected and immediately supplemented with apyrase (5 µl/ml PRP, [Sigma‐Aldrich]) and pre‐warmed ACD‐A (111 µl/ml PRP). Subsequently, platelets were separated from PRP via centrifugation (650g, 7 min, RT, without brake), resuspended in 5 ml of wash‐solution (modified Tyrode buffer: 5 ml bicarbonate buffer, 20% bovine serum albumin, 10% glucose solution, 2.5 U/ml apyrase, 1 U/ml hirudin [Pentapharm, Basel, Swiss], pH 6.3) and allowed to rest for 15 min at 37°C. Following a final centrifugation step (650g, 7 min, RT, without brake), platelets were resuspended in 2 ml of resuspension‐buffer (50 ml of modified Tyrode buffer, 0.5 ml of 1 mM MgCl2, 1 ml of 2 mM CaCl2, pH 7.2).
2.7. IgG preparation and assessment of antibody‐mediated signaling
A commercially available IgG‐purification‐kit (MelonTM‐Gel IgG Spin Purification Kit; Thermo Fisher Scientific) was used for IgG purification as described in the manufacturer's recommendations (for details, see Appendix).
To assess the role of FcγRIIA, platelets were preincubated with the FcγRIIA blocking mAb anti‐CD32 (mAb IV.3; Stemcell™ Technologies) for 45 min at 37°C, prior to incubation with COVID‐19 sera.
To analyze the impact of pAKT/PI3K pathway on the Ab‐mediated procoagulant platelet formation two inhibitors were used, BYL719 and BAY1125976. BYL719 (Alpelisib, Piqray® medication sold by Novartis) is an alpha‐specific PI3K inhibitor. It was previously shown to target p‐AKT(S473) (8)/p‐AKT(T308) (9); p100β/p110α/p85 p‐BAD (23,24). BAY1125976 (BAY) blocks PI3K/AKT signaling pathway14 and it was previously shown to target AKT1‐S473, T308 and to inhibit the 4EBP1‐T70.14 Platelets were incubated with BAY1125976 (50 µM, Cayman Chemical Company) or BYL719 (100 µM; Adooq Bioscience) for 10 min at 37°C. Afterwards, samples were washed once (7 min, 650 g, RT, without brake) and gently resuspended in 75 µl of phosphate‐buffered saline (PBS, Biochrom). Platelets were then incubated with patients sera or IgGs as described above.
2.8. Western blot analysis
Protein levels of pAKT, AKT and PI3K in platelets from COVID‐19 patients and healthy controls were determined by western blot (WB). In brief, following platelet isolation, cells were centrifuged (5 min, 700 g at 4°C) and the platelet pellet resuspended in ice‐cold RIPA lysis buffer (ThermoFisher Scientific) containing Halt™ protease and phosphatase inhibitor cocktail (100×; ThermoFisher Scientific) and EDTA (0.5 M, 100× ThermoFisher Scientific). The proteins were separated by electrophoresis using 12% SDS‐PAGE gels in glycine‐tris buffer and subsequently transferred to polyvinylidene difluoride (PVDF) membranes (0.45 µm; Merck). Finally, the membranes were incubated with rabbit phospho‐AKT antibody (1:1000; Invitrogen), mouse AKT antibody (1:1000; Abcam, Signaling), rabbit pPI3K (p85,N‐SH, 1:1000; Merck) and mouse α‐Tubulin (Cell Signaling) which was followed by the incubation with appropriate secondary anti‐rabbit IRDye®800 and anti‐mouse antibody IRDye®680 (1:3000, LI.COR®). Protein bands were detected with Odyssey infrared imaging system (LI.COR®). Relative intensity of western blots was quantified using ImageJ software (NIH).
2.9. Microscopy and platelet adhesion test
For the assessment of the adhesion ability of platelets, an adhesion assay was performed. In brief, after incubation of platelets with COVID‐19 ICU or control serum, platelets (3 × 106 platelets/well) were seeded in 8 well chamber slides (Corning) that were pre‐coated with 100 µg/ml of fibrinogen (Sigma Aldrich) overnight. Platelets were allowed to rest for 1 h at RT before staining. TRAP‐6 (10 µM; Hart Biologicals) was used as a positive control. Where indicated, platelets were preincubated with inhibitors BAY and BYL as previously described. In further experiments, GPIIb/IIIa was blocked by 10 min of pre‐incubation at 37°C, with eptifibatide (2.4 µM, accord; Middlesex). Afterwards, slides were fixed with 2% paraformaldehyde (PFA; Morphisto) for 20 min at RT in the dark. For adhesion quantification, images were captured from five randomly chosen microscopic fields and analyzed with the CellSens Standard software (Olympus) (×60; Olympus IX73).
2.10. Statistical analyses
Flow cytometry measurements were normalized to two healthy controls tested in parallel. Statistical analysis was performed using GraphPad Prism, Version 8.1 (GraphPad). Non‐parametric tests were used when data failed to follow a normal distribution as assessed by the D’Agostino and Pearson omnibus normality test. Group comparison was performed using the Wilcoxon rank‐sum test. All analyses were two‐tailed and a p‐value of <.05 was defined to indicate a statistically significant difference.
2.11. Ethics statement
The study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all volunteers prior to any study‐related procedure. The study protocol was approved by the Institutional Review Board of the University of Tuebingen.
3. RESULTS
3.1. Clinical and laboratory characteristics of patient cohort
A total of 24 patients were enrolled in this study. Thirteen out of 24 (54.2%) patients developed thrombotic complications after having been infected with SARS‐CoV2 (10 in ICU and three non‐ICU COVID‐19, Table S1) and IgGs as well as IgAs antibodies from COVID‐19 sera were detected by bead‐based multiplex assay (see Figure S1). Clinical data of 10 out of 24 patients has been reported in a previous study.9 The mean age of ICU COVID‐19 patients was 62 years (range 32–84 years). In total 18 (75%) patients had known risk factors for a severe COVID‐19 infection as described previously, including hypertension, obesity, coronary artery disease or diabetes mellitus. Thrombotic complications were more frequent (p < .01) in the ICU COVID‐19 patients as compared to control ICU group.
3.2. AKT pathway is activated in platelets from COVID‐19 patients
The ratio of pAKT/AKT in platelets from ICU COVID‐19 was significantly higher than that in platelets from healthy controls, ICU controls or non‐ICU COVID‐19 patients (Ratio pAKT/AKT; ICU COVID‐19 1.78 ± 0.14 vs. healthy controls 1.03 ± 0.03; p < .0001; vs. ICU controls: 0.91 ± 0.07, p < .0001; and vs. non‐ICU COVID‐19 patients 1.17 ± 0.06, p < .001 respectively, Figure 1A ).
FIGURE 1.

Platelet signaling through phosphorylation of AKT in ICU COVID‐19 patients. The ratio of pAKT/AKT (A) was measured through intracellular staining via flow cytometry. The results were compared to non‐ICU COVID‐19 patients, ICU control patients as well as healthy controls, respectively. Platelet activation, determined as CD62p expression, was correlated with AKT phosphorylation in COVID‐19 patients (B). Generation of procoagulant platelets, determined as phosphatidylserine's (PS) expression was correlated with AKT phosphorylation in COVID‐19 patients (C). Platelet count was correlated with the AKT phosphorylation in COVID‐19 patients (D). Data are presented as mean ± SEM of the measured fold increase compared to control; ns, not significant, *p < .05, **p < .01, ***p < .001 and ****p < .0001
Furthermore, CD62p levels positively correlated with AKT phosphorylation in ICU and non‐ICU COVID‐19 patients (r = .55, p < .01, Figure 1B). Similarly, PS externalization showed a positive correlation with AKT phosphorylation in ICU and non‐ICU COVID‐19 patients (r = .42, p < .05, Figure 1C). Interestingly, platelet count was negatively correlated with AKT phosphorylation in ICU and non‐ICU COVID‐19 patients (r = −.65, p < .001, Figure 1D).
3.3. Sera from severe COVID‐19 patients stimulate AKT signaling in platelets
To analyze whether a serum‐derived factor might have contributed to increased phosphorylation of AKT in COVID‐19 patients, sera and IgGs of ICU COVID‐19 patients were incubated with healthy platelets. We focused on the ICU cohort since these patients showed the highest rates of clinical overt thrombosis and the highest pAKT/AKT levels when compared to controls.
Increased ratio of pAKT/AKT was exclusively observed in washed platelets that were incubated with serum from ICU COVID‐19 patients (ratio of pAKT/AKT: 1.61 ± 0.08 vs. 1.00 ± 0.00, p < .0001; and vs. the ICU control group 0.99 ± 0.03, p < .0001, respectively, Figure 2A ). The alterations were nearly absent in platelets treated with sera from healthy controls and ICU control patients. Interestingly, IgGs from ICU COVID‐19 patients induced a significant increase in the ratio of pAKT/AKT when compared to IgGs from the healthy control group (ratio of pAKT/AKT: 1.62 ± 0.15 vs. 1.00 ± 0.00, p < .001) or to the ICU control groups (ratio of pAKT/AKT: 0.92 ± 0.04, p < .001, Figure 2B). To further confirm our findings, we conducted WB analysis for pAKT and AKT in washed platelets that were incubated with ICU COVID‐19 patient sera. In accordance with our flow cytometric analysis, the ratio of pAKT/AKT was significantly higher in washed platelets that were incubated with sera from ICU COVID‐19 patients compared to healthy control (Relative intensity pAKT/AKT from healthy control:18.6 ± 8.15 vs. 1 ± 0.0, p < .001, Figure 2C,D). Moreover, an involvement of PI3K was observed, as we detected increased phosphorylated (p85) PI3K protein abundance in platelets that were incubated with sera from ICU COVID‐19 patients (relative intensity pPI3K/tubulin from healthy control: 12.23 ± 5.78 vs. 1 ± 0.0, p < .05, Figure 2E,F).
FIGURE 2.

ICU COVID‐19 sera and IgG mediated effect of AKT‐ and PI3K‐phosphorylation on platelets. The ratio of pAKT/AKT (A,B) was measured through intracellular staining via flow cytometry. The results were compared to sera (A) or IgGs (B) from ICU COVID‐19 patients and healthy controls or ICU controls, respectively. Representative WB assays that were performed to confirm the FACS measurements of AKT/pAKT (C) and of PI3K (E) and the quantification of their protein levels, respectively (D,F). Data are presented as mean ± SEM of the measured fold increase compared to control; ns, not significant, *p < .05, **p < .01, ***p < .001 and ****p < .0001
3.4. Phosphorylation of PI3K and AKT proteins is mediated via FcγRIIA
Next, we sought to investigate whether ICU COVID‐19 patient sera and the corresponding IgG fractions activate the PI3K/AKT signaling pathway via the crosslinking FcγRIIA. We blocked FcγRIIA using the mAb IV.3 prior to co‐incubation with patient sera which was followed by measurements for PI3K/AKT pathway. Quantifying pAKT and AKT protein levels showed that pre‐treatment of platelets with mAb IV.3 prevented AKT phosphorylation upon incubation with sera from ICU COVID‐19 (relative intensity pAKT/AKT from healthy control: 3.93 ± 0.78 vs. 0.56 ± 0.07, p < .05, Figure 3A,B ). Furthermore, we showed that phosphorylated (p85) PI3K abundance was also reduced after pretreatment with mAb IV.3 (relative intensity pPI3K/Tubulin: 3.85 ± 0.80 vs. 0.79 ± 0.17, p < .05, Figure 3C,D).
FIGURE 3.

FcγRIIA blocking decreased PI3K/AKT phosphorylation in ICU COVID‐19 patients. Effects of FcγRIIA‐receptor blocking on the expression of PI3K/AKT signaling on platelets incubated with ICU COVID‐19 sera. Prior to incubation with patients’ sera, washed platelets from healthy donors were pretreated with a monoclonal antibody IV.3 (mAb IV.3) against the FcγRIIA‐receptor. WB assays were performed to quantify AKT/pAKT (A,B) and PI3K protein levels (C,D). Data are presented as mean ± SEM of the measured fold increase compared to control with and without pretreatment; ns, not significant, *p < .05, **p < .01, ***p < .001 and ****p < .0001
3.5. Sera from severe COVID‐19 patients induce the generation of procoagulant platelets without PAC‐1 activation
Next we analyzed the impact of COVID‐19 sera on GPIIb/IIIa activation in procoagulant platelets. The CD32‐crosslinking mAb AT10 and sera of ICU COVID‐19 patients were incubated with healthy platelets and stained using PAC1, annexinV and anti‐CD62p antibodies. CD32‐crosslinking by AT10 as well as by ICU COVID‐19 sera did not induce conformational changes of GPIIb/IIIa in procoagulant platelets compared to isotype control mAb and sera from healthy donors, respectively (% of PAC‐1 expression on procoagulant platelet gate: 5.47 ± 0.93 vs. 1.67 ± 0.54, p > .05; and 5.11 ± 0.95 vs. 1.91 ± 1.13, p > .05, respectively, Figure 4A ). In addition, the percentage of platelets double positive for CD62p and PS after CD32‐crosslinking AT10 was not affected by the GPIIb/IIIa inhibitor eptifibatide (% of/PS double positive platelets: 22.66 ± 4.12 vs. 18.87 ± 1.80, respectively, p > .05, Figure 4B). Furthermore, the generation of procoagulant platelets after incubation with ICU COVID‐19 patients, sera was not affected by eptifibatide treatment (% of CD62p/PS double positive platelets: 17.38 ± 5.21 vs. 14.58 ± 5.00, respectively, p > .05, Figure 4C). Taken together, these results demonstrate that the formation of procoagulant phenotype in platelets by COVID‐19 sera is independent of integrin engagement.
FIGURE 4.

Sera from ICU COVID‐19 patients induce the generation of procoagulant platelets without PAC‐1 activation. The expression of PAC1 was determined on the surface of double positive CD62p/PS platelets upon incubation with sera from healthy donors or ICU COVID‐19 patients, in the presence and in the absence of eptifibatide. Quantification of the percentage of PAC‐1 expression from the procoagulant cell population (A) and CD62p/PS double positive platelets by CD32‐crosslinking by AT10 (B) as well as by ICU COVID‐19 sera (C). The results were compared to sera from ICU COVID‐19 patients and healthy controls, respectively. Data are presented as mean ± SEM of the measured fold increase compared to control; ns, not significant and *p < .05
3.6. AKT and PI3K inhibitors prevent COVID‐19‐induced procoagulant platelet formation
To examine the impact of pAKT and PI3K inhibition on the generation of platelets with procoagulant properties, platelets were pretreated with BAY and BYL prior to incubation with ICU COVID‐19 sera. The CD62p/PS double positive procoagulant platelet population was reduced in the presence of the AKT inhibitor BAY as well as the PI3K inhibitor BYL (% of CD62p/PS platelets: ICU COVID‐19 sera: 100 ± 0 vs. BAY: 4.19 ± 0.61, p < .05; and BYL: 2.96 ± 0.62, p < .01, respectively, Figure 5A ).
FIGURE 5.

Inhibition of PI3K and AKT reduced antibody‐mediated procoagulant ability in platelets from severe ICU COVID‐19 patients. Upon pre‐treatment of platelets with inhibitors BAY or BYL and sera or IgG incubation from ICU COVID‐19 patients or healthy controls, the double positive CD62p/PS cells after sera (A) or IgG incubation (B) were determined. The ratio of pAKT/AKT with sera (C) and IgG (D) were measured through intracellular staining via flow cytometry. The data are presented as mean ± standard error of mean. WB assays were performed to determine the protein levels of phosphorylated AKT (pAKT) and AKT (E) as well as pPI3K and α‐Tubulin (G). The quantification of pAKT (F) and pPI3K (H) abundance between ICU COVID‐19 patients sera, that were pretreated with BAY and BYL compared to ICU COVID‐19 patient sera. Data are presented as mean ± SEM of the measured fold increase compared to control with and without pretreatment of BAY and BYL; ns, not significant, **p < .01
Furthermore, we analyzed the IgG fractions from ICU COVID‐19 patients. There was a significantly higher expression of CD62p/PS on platelets incubated with ICU COVID‐19 IgGs compared to healthy control group (% of CD62p/PS platelets: COVID‐19 ICU IgG: 9.02 ± 1.56, vs. healthy control 1.0 ± 0.00, p < .001, Figure 5B). After pretreatment with the AKT inhibitor BAY as well as the PI3K inhibitor BYL a significant decrease was observed compared to buffer (% of CD62p/PS platelets: COVID‐19 ICU IgG: 9.02 ± 1.56 vs. BAY: 5.99 ± 0.66, p < .01 and vs. BYL: 3.36 ± 0.36, p < .001, Figure 5B).
Next, we tested the inhibitory effect in % of BAY and BYL on sera and IgG mediated AKT phosphorylation. BAY and BYL significantly inhibited ICU COVID‐19 sera and IgGs induced AKT phosphorylation (COVID‐19 ICU sera vs. BAY: 100 ± 0 vs. 79.8 ± 1.91, p < .01 and COVID‐19 ICU sera vs. BYL: 100 ± 0 vs. 70.8 ± 1.39, p < .01 Figure 5C; COVID‐19 IgG vs. BAY: 100 ± 0 vs. 42.71 ± 10.54, p < .01 and COVID‐19 ICU sera vs. BYL: 100 ± 0 vs. 49.38 ± 12.78, p < .01, respectively, Figure 5D).
In order to confirm that the enhanced surface expression levels of CD62p and PS induce an intracellular signal, which triggers an increase of the downstream effectors, pAKT and PI3K expression was quantified by western blots. Pretreatment with BAY and BYL reduced significantly the phosphorylation level of AKT (COVID‐19 ICU sera vs. BAY: 100 ± 0 vs. 42.49 ± 10.40, p < .01 and BYL: 100 ± 0 vs. 31.77 ± 14.23, p < .01, Figure 5E,F) as well as PI3K phosphorylation (COVID‐19 ICU sera vs. BAY: 100 ± 0 vs. 89.93 ± 19.29, p > .05 and COVID‐19 ICU sera vs. BYL: 100 ± 0 vs. 37.83 ± 5.89, p < .01 Figure 5G,H). Together, these findings show for the first time that the pronounced antibody‐mediated formation of procoagulant platelets from ICU COVID‐19 patient sera is initiated by the PI3K/AKT signaling pathway and can be prevented by PI3K and AKT inhibitors.
3.7. Induction of PI3K/AKT signaling pathway leads to increased platelet adhesion
To verify the biological relevance of our findings, platelet adhesion to fibrinogen was examined. Platelets from healthy subjects were analyzed after incubation with sera from ICU COVID‐19 patients or ICU controls (Figure 6 ). Platelet adhesion was significantly increased after incubation with sera from ICU COVID‐19 patients (% of adherent activated platelets: 70% ± 4%) compared to healthy controls (% of adherent activated platelets: 42% ± 7.4%; p < .05) and ICU controls (% of adherent activated platelets: 36% ± 4%; p < .05). While the COVID‐19 sera‐mediated adhesion of platelet was significantly reduced in the presence of BAY (% of adherent activated platelets: 48% ± 2.95%, p < .05) as well as BYL (% of adherent activated platelets: 38.81% ± 10.76%; p < .05). Pretreatment of platelets with eptifibatide showed no effect on both COVID‐19 sera (% of adherent activated platelets:76% ± 1.8% vs. 75% ± 1.1%, respectively, p > .05, Figure S3) and CD32‐crosslinking AT10 induced platelet adhesion to fibrinogen (% of adherent activated platelets: 62% ± 4.6% vs. 77% ± 3.2%, respectively, p > .05, Figure S3). These results may indicate that the increased platelet adhesion in the presence of COVID‐19 sera is independent of integrin engagement.
FIGURE 6.

Analysis of platelets adhesion to fibrinogen. Quantification (upper panel) and representative bright field microscopic images (lower panel) of adherent platelets in the presence of sera from healthy control or ICU control and ICU COVID‐19 patients with and without pretreatment with the AKT (BAY) and the PI3K (BYL) inhibitors (scale 10 µm). Activation with TRAP‐6 served as positive control. The % of adherent activated platelets per field was determined from five different microscopic fields. The data are presented as mean ± standard error of mean *p < .05
4. DISCUSSION
Patients with COVID‐19 are prone to a heightened risk for developing thromboembolic complications.5., 15., 16. However, the mechanisms by which COVID‐19 affects the generation of procoagulant platelets are not yet fully understood. Procoagulant platelets are characterized by high CD62p expression and PS externalization and have higher ability to bind plasma‐derived activated clotting factors.17 In this study, we showed for the first time that increased generation of procoagulant platelets observed in severe COVID‐19 patients is mediated by IgG antibodies through PI3K/AKT signaling pathway in an FcγRIIA‐dependent manner.
It is known that phosphorylation of AKT is involved in the thrombus formation through the PI3K/AKT signaling pathway that activates platelets.11., 18., 19., 20., 21. We investigated the antibody‐mediated signaling downstream of the FcγRIIA by blocking PI3K and AKT activation with the specific inhibitors, BAY1125976 and BYL719, respectively (Figure 6). Interestingly, the inhibition of PI3K and AKT reduced the generation of procoagulant platelets and suppressed AKT phosphorylation on Ser473.
In accordance with our results, Agbani et al.,17 postulated that procoagulant platelets adhere to fibrinogen without an activation of PAC‐1 (GPIIb/IIIa).
It has been shown that on activated platelets, PS acts as a binding site for plasma coagulation factors and propagates thrombus formation.22., 23., 24.
Furthermore, inhibition of the FcγRIIA prevented AKT phosphorylation and the generation of procoagulant platelets, suggesting that antibodies from severe COVID‐19 patients trigger platelet adhesion by crosslinking the FcγRIIA and subsequent AKT phosphorylation.
These data suggest that the PI3K/AKT signaling pathway promotes procoagulant platelets and inhibitors of PI3K or AKT potently abolish the enhanced fibrinogen binding of platelets induced by incubation with ICU COVID‐19 sera. A minor finding of our inhibitory analyses is that BAY and BYL induced enhanced inhibition with the IgGs compared to the sera. This could be due to the presence of other co‐factors in addition to IgGs that are inducing pAKT in the sera or due to a stronger effect of the inhibitors in buffer compared to serum.
More importantly, the effects of PI3K and AKT inhibition not only underline that the antibody‐mediated procoagulant platelets generation in severe ICU COVID‐19 patients involves the AKT/PI3K signaling cascade, but also show that the pro‐coagulant status can be prevented.
However, our study has some limitations. First, patients in our study cohort have comorbidities such as diabetes mellitus, which can possibly affect AKT signaling pathway. This should be considered while interpreting our results. Second, the low samples size reduced the power of our statistical analysis. Further studies should also investigate the downstream targets of PI3K and AKT in COVID‐19. Furthermore, in order to understand the platelet‐mediated pathological mechanisms responsible from the progression of COVID‐19, clinical studies with larger sample sizes are warranted. We hope though that our findings will contribute to shed light on the role of PI3K/AKT in the pathogenesis of COVID‐19 associated coagulopathy. Taking together our findings suggest that PI3K and AKT are promising drug targets to treat and prevent critical thrombosis in ICU COVID‐19 patients. Interestingly, based on our results, the small molecule inhibitors BAY1125976 and BYL719 may be encouraging drug candidates to suppress the antibody mediated α‐granules externalization and PS exposure that contributes to thrombosis in COVID‐19 infection (Figure 7 ).
FIGURE 7.

Schematic illustration of proposed role of PI3K and AKT signaling pathway in procoagulant platelets in severe COVID‐19. Proposed mechanism presenting the role of PI3K and AKT signaling pathways in platelet function via stimulation of FcγRIIA‐receptor in severe COVID‐19 patients. After stimulation by SARS‐CoV‐2 infection, FcγRIIA interacts with immunoreceptor tyrosine‐based activation motif (ITAM), upon which phosphatidylinositol 3‐kinase (PI3K) is activated and subsequently induces downstream signal pathways, activating the serine/threonine protein kinase B (PKB/AKT) on Ser473. This leads to serine and/or threonine phosphorylation of a range of downstream substrates which often themselves are kinase/phosphatases and signaling molecules which mediate the main platelet activation responses by α‐granules externalization and PS exposure, which leads to monocytes adhesion and endothelia dysfunction. AKT, AKT8 virus oncogene cellular homolog, kinase; FC, flow cytometry; mAb, mouse antibody; PI3K, phosphoinositide‐3‐kinase; PS, phosphatidylserine; RT, room temperature
In summary, we provide evidence for the involvement of the PI3K/AKT pathway in antibody‐mediated generation of procoagulant platelets. We propose that the inhibition of PI3K and AKT offers a treatment opportunity to prevent thromboembolic events in severe COVID‐19.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
L.P., K.A. and T.B. designed the study. L.P., A.S., J.F., A.W., J.Z., K.W., and W.A. performed the experiments and collected the data. K.A. and S.H. organized the clinical material. L.P., A.S., I.M., G.U., and T.B. analyzed the data, interpreted the results, and wrote the manuscript. All authors read and approved the manuscript.
ACKNOWLEDGMENTS
We thank Aleyna Karakuyu for assistance in adhesion assays. This work was supported by grants from the German Research Foundation, the Ministerium for Wissenschaft and Kunst in Baden‐Würtemberg, and the German Heart Foundation to TB (BA5158/4). Open access funding enabled and organized by ProjektDEAL.
German Research Foundation
Ministerium for Wissenschaft and Kunst in Baden‐Würtemberg
German Heart FoundationBA5158/4
Footnotes
Manuscript handled by: Wolfgang Bergmeier
Supporting Information
Fig S1
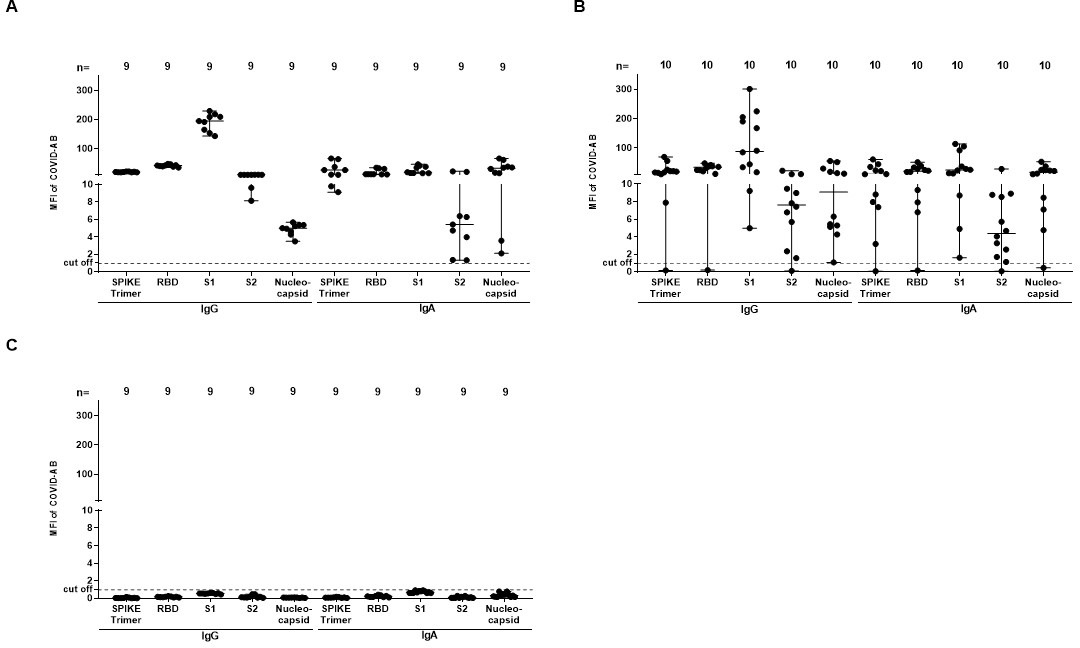{kind=link}
Fig S2
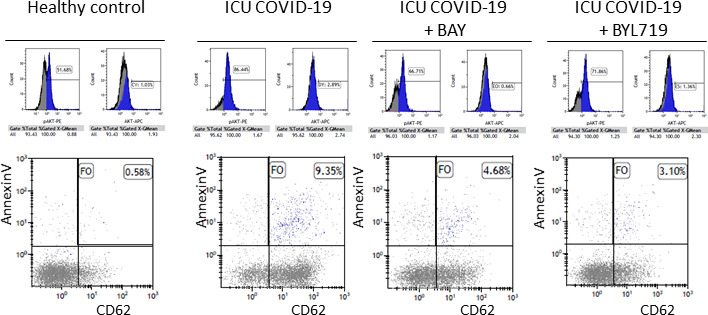{kind=link}
Fig S3
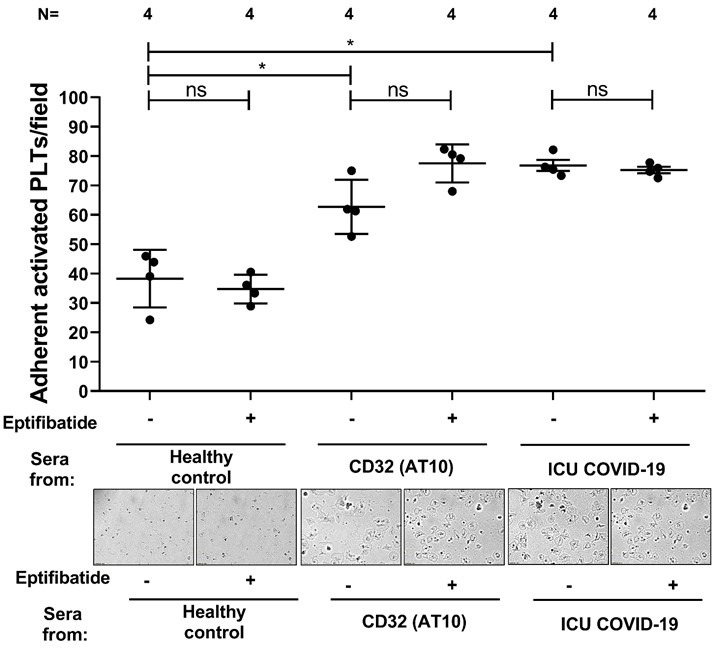{kind=link}
Table S1
Supplementary Material
REFERENCES
- 1.Rostami A., Sepidarkish M., Leeflang M.M.G., et al. SARS‐CoV‐2 seroprevalence worldwide: a systematic review and meta‐analysis. Clin Microbiol Infect. 2021;27(3):331–340. doi: 10.1016/j.cmi.2020.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel U., Malik P., Mehta D., et al. Early epidemiological indicators, outcomes, and interventions of COVID‐19 pandemic: a systematic review. J Glob Health. 2020;10 doi: 10.7189/jogh.10.020506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abou‐Ismail M.Y., Diamond A., Kapoor S., Arafah Y., Nayak L. The hypercoagulable state in COVID‐19: incidence, pathophysiology, and management. Thromb Res. 2020;194:101–115. doi: 10.1016/j.thromres.2020.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berkman S.A., Tapson V.F. COVID‐19 and its implications for thrombosis and anticoagulation. Semin Respir Crit Care Med. 2021;42(02):316–326. doi: 10.1055/s-0041-1722992. [DOI] [PubMed] [Google Scholar]
- 5.Malas M.B., Naazie I.N., Elsayed N., Mathlouthi A., Marmor R., Clary B. Thromboembolism risk of COVID‐19 is high and associated with a higher risk of mortality: a systematic review and meta‐analysis. EClinicalMedicine. 2020;29:100639. doi: 10.1016/j.eclinm.2020.100639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prodan C.I., Vincent A.S., Dale G.L. Coated‐platelet levels are elevated in patients with transient ischemic attack. Transl Res. 2011;158:71–75. doi: 10.1016/j.trsl.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Prodan C.I., Stoner J.A., Cowan L.D., Dale G.L. Higher coated‐platelet levels are associated with stroke recurrence following nonlacunar brain infarction. J Cereb Blood Flow Metab. 2013;33:287–292. doi: 10.1038/jcbfm.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connors J.M., Levy J.H. COVID‐19 and its implications for thrombosis and anticoagulation. Blood. 2020;135:2033–2040. doi: 10.1182/blood.2020006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Althaus K., Marini I., Zlamal J., et al. Antibody‐induced procoagulant platelets in severe COVID‐19 infection. Blood. 2021;137(8):1061–1071. doi: 10.1182/blood.2020008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zlamal J., Althaus K., Jaffal H., et al. Upregulation of cAMP prevents antibody‐mediated thrombus formation in COVID‐19. Blood Adv. 2021 doi: 10.1182/bloodadvances.2021005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guidetti G.F., Canobbio I., Torti M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv Biol Regul. 2015;59:36–52. doi: 10.1016/j.jbior.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Vincent J.L., Moreno R., Takala J., et al. The SOFA (Sepsis‐related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis‐Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22:707–710. doi: 10.1007/BF01709751. [DOI] [PubMed] [Google Scholar]
- 13.Greinacher A., Michels I., Kiefel V., Mueller‐Eckhardt C. A rapid and sensitive test for diagnosing heparin‐associated thrombocytopenia. Thromb Haemost. 1991;66:734–736. [PubMed] [Google Scholar]
- 14.Politz O., Siegel F., Barfacker L., et al. BAY 1125976, a selective allosteric AKT1/2 inhibitor, exhibits high efficacy on AKT signaling‐dependent tumor growth in mouse models. Int J Cancer. 2017;140:449–459. doi: 10.1002/ijc.30457. [DOI] [PubMed] [Google Scholar]
- 15.Manne B.K., Denorme F., Middleton E.A., et al. Platelet gene expression and function in patients with COVID‐19. Blood. 2020;136:1317–1329. doi: 10.1182/blood.2020007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loo J., Spittle D.A., Newnham M. COVID‐19, immunothrombosis and venous thromboembolism: biological mechanisms. Thorax. 2021;76(4):412–420. doi: 10.1136/thoraxjnl-2020-216243. [DOI] [PubMed] [Google Scholar]
- 17.Agbani E.O., Poole A.W. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. 2017;130:2171–2179. doi: 10.1182/blood-2017-05-787259. [DOI] [PubMed] [Google Scholar]
- 18.Chen Z., Li T., Kareem K., Tran D., Griffith B.P., Wu Z.J. The role of PI3K/Akt signaling pathway in non‐physiological shear stress‐induced platelet activation. Artif Organs. 2019;43:897–908. doi: 10.1111/aor.13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niu H., Chen X., Gruppo R.A., et al. Integrin alphaIIb‐mediated PI3K/Akt activation in platelets. PLoS One. 2012;7 doi: 10.1371/journal.pone.0047356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pelzl L., Sahu I., Ma K., et al. Beta‐glycerophosphate‐induced ORAI1 expression and store operated Ca(2+) entry in megakaryocytes. Sci Rep. 2020;10:1728. doi: 10.1038/s41598-020-58384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin J., Vinogradova O., Plow E.F. Integrin bidirectional signaling: a molecular view. PLoS Biol. 2004;2:e169. doi: 10.1371/journal.pbio.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monroe D.M., Hoffman M., Roberts H.R. Transmission of a procoagulant signal from tissue factor‐bearing cell to platelets. Blood Coagul Fibrin. 1996;7:459–464. doi: 10.1097/00001721-199606000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Butenas S., Branda R., van ’t Veer C., Cawthern K., Mann K. Platelets and phospholipids in tissue factor‐initiated thrombin generation. Thromb Haemost. 2001;86:660–667. [PubMed] [Google Scholar]
- 24.Solum N.O. Procoagulant expression in platelets and defects leading to clinical disorders. Arterioscler Thromb Vasc Biol. 1999;19:2841–2846. doi: 10.1161/01.atv.19.12.2841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Table S1
Supplementary Material