

Abstract
While mitochondrial function is essential for life in all multicellular organisms, a mild impairment of mitochondrial function can extend longevity in model organisms. By understanding the molecular mechanisms involved, these pathways might be targeted to promote healthy aging. In studying two long‐lived mitochondrial mutants in C. elegans, we found that disrupting subunits of the mitochondrial electron transport chain results in upregulation of genes involved in innate immunity, which is driven by the mitochondrial unfolded protein response (mitoUPR) but also dependent on the canonical p38‐mediated innate immune signaling pathway. Both of these pathways are required for the increased resistance to bacterial pathogens and extended longevity of the long‐lived mitochondrial mutants, as is the FOXO transcription factor DAF‐16. This work demonstrates that both the p38‐mediated innate immune signaling pathway and the mitoUPR act in concert on the same innate immunity genes to promote pathogen resistance and longevity and that input from the mitochondria can extend longevity by signaling through these pathways. This indicates that multiple evolutionarily conserved genetic pathways controlling innate immunity also function to modulate lifespan.
Keywords: aging, C. elegans, innate immunity, mitochondria, mitochondrial unfolded protein response
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Molecular Biology of Disease; Signal Transduction
Mild impairment of mitochondrial function results in enhanced innate immunity and lifespan extension. Both phenotypes are driven by activation of the mitochondrial unfolded protein response but are also dependent on p38‐mediated innate immune signaling.

Introduction
While aging was long believed to be a stochastic process of damage accumulation, research during the past three decades has demonstrated that lifespan can be strongly influenced by genetics. Single‐gene mutations have been shown to extend longevity in model organisms, including yeast, worms, flies, and mice. Importantly, genes and interventions that increase lifespan tend to be conserved across species. For example, decreasing insulin‐IGF1 signaling, which was first shown to increase lifespan in the worm C. elegans (Klass, 1977; Friedman & Johnson, 1988; Kenyon et al, 1993), has subsequently been shown to extend longevity in flies (Clancy et al, 2001) and mice (Holzenberger et al, 2003), and to be associated with longevity in humans (Flachsbart et al, 2017). This suggests that studying the aging process in model organisms can provide insights that are relevant to human aging.
The first single gene mutation that was shown to extend lifespan was identified in C. elegans (Klass, 1977; Friedman & Johnson, 1988), and since then this organism has been extensively used to find additional genetic pathways associated with lifespan extension and to elucidate the underlying mechanisms. Among the earliest genes that were shown to influence longevity were genes involved in mitochondrial function. Mutations in the clk‐1, nuo‐6, and isp‐1 genes affect different components of the mitochondrial electron transport chain, and all lead to increased lifespan (Wong et al, 1995; Lakowski & Hekimi, 1996; Feng et al, 2001; Yang & Hekimi, 2010b). In the case of nuo‐6 and isp‐1, which encode subunits of complex I and complex III, respectively, a complete loss‐of‐function mutation would be lethal, while point mutations that result in a mild impairment of mitochondrial function extend longevity. Similarly, decreasing the expression of genes involved in mitochondrial function with RNA interference also increases lifespan (Dillin et al, 2002; Lee et al, 2003). Importantly, mutations that affect mitochondrial function have also been shown to increase lifespan in other species, including flies (Copeland et al, 2009) and mice (Liu et al, 2005; Dell'agnello et al, 2007).
While initially it was believed that the mechanism by which mild impairment of mitochondrial function increased lifespan was through a decrease in the production of reactive oxygen species (ROS) and the resulting oxidative damage (Feng et al, 2001), more recent studies show that mutations affecting mitochondrial function actually increase the levels of ROS (Yang & Hekimi, 2010a). The increase in ROS is required for the long lifespan of these mutants, as treatment with antioxidants can decrease their lifespan (Yang & Hekimi, 2010a; Van Raamsdonk & Hekimi, 2012). While multiple factors contributing to the long lifespan of these mitochondrial mutants have been identified (Walter et al, 2011; Baruah et al, 2014; Yee et al, 2014; Munkacsy et al, 2016; Senchuk et al, 2018; Wu et al, 2018), the precise mechanisms of lifespan extension remain incompletely understood. In our previous work, we have shown that two stress‐responsive transcription factors, DAF‐16/FOXO3 and ATFS‐1/ATF5, are required for the long lifespan of nuo‐6 and isp‐1 worms (Senchuk et al, 2018; Wu et al, 2018).
DAF‐16/FOXO3 is a FOXO transcription factor that is directly regulated by phosphorylation in response to insulin‐IGF1 signaling, a growth factor signaling pathway that begins with the insulin‐IGF1 receptor DAF‐2 (Kenyon, 2010). While DAF‐16 normally resides in the cytoplasm, when signaling through the insulin‐IGF1 pathway is reduced DAF‐16 accumulates in nuclei. DAF‐16 also translocates to the nucleus in response to various stresses. In the nucleus, DAF‐16 upregulates genes involved in stress response and metabolism (Murphy et al, 2003; Tepper et al, 2013). DAF‐16 has been shown to be required in multiple different contexts of lifespan extension (Kenyon et al, 1993; Apfeld & Kenyon, 1999; Berman & Kenyon, 2006; Syntichaki et al, 2007; Senchuk et al, 2018).
Activating transcription factor associated with stress 1 (ATFS‐1/ATF5) is the transcription factor that mediates the mitochondrial unfolded protein response (mitoUPR) (Jovaisaite et al, 2014), a stress response pathway that responds to mitochondrial stress in order to restore mitochondrial function. ATFS‐1 contains a nuclear localization signal (NLS) and a mitochondrial targeting sequence (MTS). Under normal conditions, ATFS‐1 is targeted to the mitochondria where it is imported and degraded. Under conditions of mitochondrial stress, mitochondrial import of ATFS‐1 is prevented, resulting in accumulation of ATFS‐1 in the cytoplasm, where the NLS translocates ATFS‐1 into the nucleus in order to restore mitochondrial homeostasis through alterations in expression of genes involved in protein folding and metabolism (Nargund et al, 2012).
While DAF‐16 and ATFS‐1 can both contribute to defense against bacterial pathogens (Pellegrino et al, 2014; Dues et al, 2019), the primary innate immune signaling pathway that responds to bacterial pathogen stress is a mitogen‐activated protein kinase (MAPK) signaling pathway, which has been found to be conserved from invertebrates to mammals (Hoffmann et al, 1999; Kimbrell & Beutler, 2001; Irazoqui et al, 2010). In this pathway, NSY‐1/ASK1 (MAPK kinase kinase) signals to SEK‐1/MKK3/MKK6 (MAPK kinase), which signals to PMK‐1/p38 (MAPK) (Kim et al, 2002; Kim & Ewbank, 2018) (Appendix Fig S1). Downstream of this pathway, the transcription factor ATF‐7/ATF2/ATF‐7/CREB5 acts to modulate the expression of genes involved in innate immunity (Shivers et al, 2010; Fletcher et al, 2019a). While ATF‐7 normally acts as a repressor of gene function, when it is phosphorylated by PMK‐1, ATF‐7 functions as an activator of p38/ATF‐7‐regulated immunity gene expression (Appendix Fig S1).
In this work, we show that the long‐lived mitochondrial mutants, nuo‐6 and isp‐1, exhibit an upregulation of genes involved in innate immunity that is driven by the activation of the mitoUPR, but also dependent on the p38‐mediated innate immune signaling pathway, leading to an increased resistance to bacterial pathogens. We find that the p38‐mediated innate immune signaling pathway is required for the long lifespan of nuo‐6 and isp‐1 mutants. Finally, we demonstrate that the activation of the mitoUPR is sufficient to upregulate innate immunity genes and is also required for their upregulation in nuo‐6 mutants. Overall, this work demonstrates the importance of the mitoUPR in upregulating innate immunity in response to signals from the mitochondria and delineates a clear role of innate immune signaling pathways in determining lifespan.
Results
Long‐lived mitochondrial mutants exhibit broad upregulation of genes involved in innate immunity that is dependent on p38‐mediated innate immune signaling
While mild impairment of mitochondrial function has been shown to extend longevity, the underlying mechanisms are yet to be fully elucidated. When mitochondrial function is impaired, mitochondria are able to communicate with the nucleus to alter nuclear gene expression. To obtain a comprehensive, unbiased view of the transcriptional changes that result from impairment of mitochondrial function, we used RNA sequencing (RNA‐seq) to examine gene expression in two long‐lived mitochondrial mutants, nuo‐6 and isp‐1. After determining which genes were differentially expressed compared with wild‐type worms, we identified groups of genes that showed enrichment. Among the genes that showed enrichment were genes involved in innate immunity. These genes encode proteins that function to inhibit the growth and survival of pathogenic bacteria and to repair or remove damage to the worm (Troemel et al, 2006; Chikka et al, 2016; Fletcher et al, 2019a). Accordingly, we decided to investigate the role of innate immunity in the long lifespan of these long‐lived mitochondrial mutants.
To determine the extent to which genes involved in innate immunity are upregulated in the long‐lived mitochondrial mutants, nuo‐6 and isp‐1, we first examined eight genes, which others have used to monitor innate immune activity (Shivers et al, 2010; Pellegrino et al, 2014; Block et al, 2015; Chikka et al, 2016; Jeong et al, 2017; Wu et al, 2019). These genes included T24B8.5/sysm‐1, K08D8.5, F55G11.8, clec‐65, clec‐67, dod‐22, Y9C9A.8, and C32H11.4. All of these genes are upregulated in response to exposure to the bacterial pathogen Pseudomonas aeruginosa strain PA14, and five of eight have been shown to be direct targets of the p38‐mediated innate immune signaling pathway in a ChIP‐seq analysis of ATF‐7 (Fletcher et al, 2019a). In examining the expression of these genes in our RNA‐seq data, we found that all eight genes were significantly upregulated in both nuo‐6 and isp‐1 worms (Fig 1A).
Figure 1. Innate immunity genes are upregulated in long‐lived mitochondrial mutants.
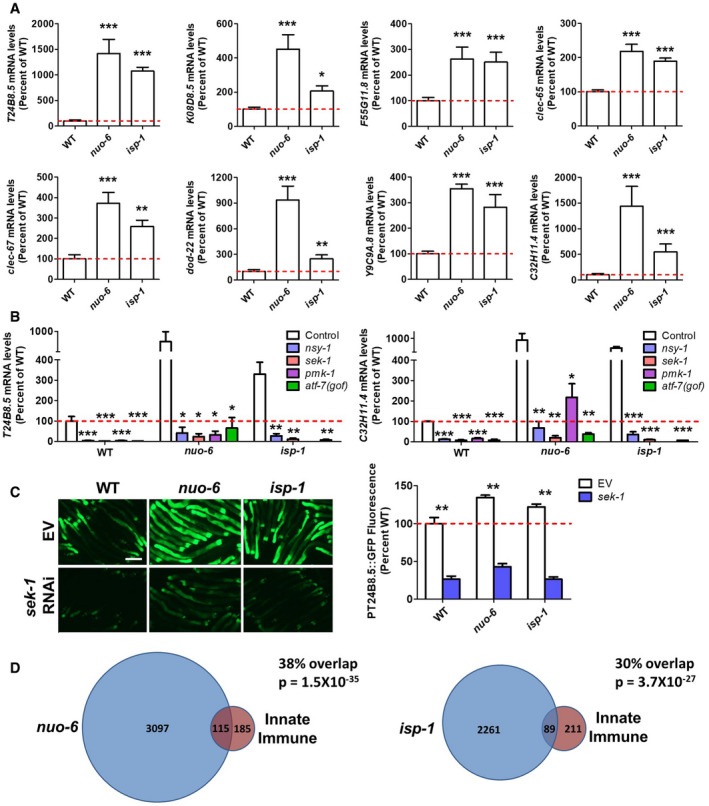
- Genes involved in innate immunity are significantly upregulated in nuo‐6 and isp‐1 worms. Gene expression changes in the mitochondrial mutants were determined by RNA sequencing and compared to wild‐type N2 worms. Results represent counts per million (CPM) expressed as a percentage of wild type of six biological replicates per strain.
- Upregulation of innate immunity genes is dependent on the p38‐mediated innate immune signaling pathway including NSY‐1, SEK‐1, PMK‐1, and ATF‐7. Gene expression changes were examined by quantitative RT–PCR of three biological replicates per strain.
- Using a fluorescent reporter strain for the innate immunity gene T24B8.5 confirms that innate immunity genes are upregulated in the long‐lived mitochondrial mutants and that components of the p38‐mediated innate immune signaling pathway are required for their upregulation. Scale bar indicates 250 µM. Three biological replicates per strain were quantified.
- Differentially expressed genes in long‐lived mitochondrial mutants show significant overlap with genetic targets of the p38‐mediated innate immune signaling pathway. Significantly modulated genes in long‐lived mitochondrial mutants nuo‐6 and isp‐1 (six biological replicates per strain) were compared to genes that are modulated in response to exposure to the bacterial pathogen P. aeruginosa strain PA14 in a PMK‐1‐ and ATF‐7‐dependent manner as identified by Fletcher et al (2019a). There is a highly significant degree of overlap between genes upregulated by activation of the p38‐mediated innate immune pathway and genes upregulated in nuo‐6 and isp‐1 mutants. Similarly, there is a highly significantly degree of overlap between genes downregulated by activation of the p38‐mediated innate immune pathway and genes downregulated in nuo‐6 and isp‐1 mutants. The P‐values indicate the significance of the difference between the observed number of overlapping genes between the two gene sets and the expected number of overlapping genes if the genes were picked at random.
Data information: Error bars indicate SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Statistical significance was determined using a one‐way ANOVA with Dunnett’s multiple comparison test in panels A and B. Statistical significance was determined using a two‐way ANOVA with Bonferroni post‐test in panel C.
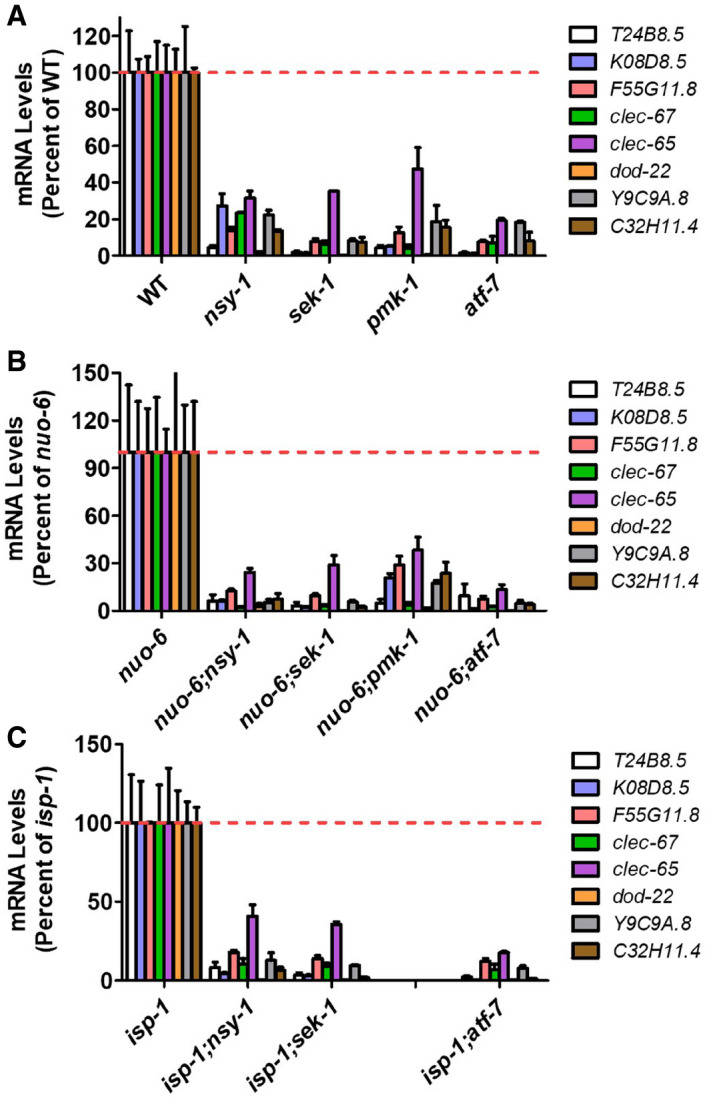
To determine whether the upregulation of innate immunity genes in the long‐lived mitochondrial mutants requires the p38‐mediated innate immune signaling pathway (NSY‐1 → SEK‐1 → PMK‐1 → ATF‐7), we generated double mutants of nuo‐6 and isp‐1 with all of these genes before measuring gene expression with quantitative RT–PCR (qPCR). We used loss‐of‐function mutants for nsy‐1, sek‐1, and pmk‐1. Since ATF‐7 normally acts as a repressor, we used the qd22 gain‐of‐function mutation for this gene. This mutation prevents phosphorylation of ATF‐7 by PMK‐1, thereby making the mutant ATF‐7 a constitutive repressor (Shivers et al, 2010) (Appendix Fig S1). As in the RNA‐seq data, the results from the qPCR experiments showed that innate immune genes are upregulated in nuo‐6 and isp‐1 worms. Importantly, we found that in both nuo‐6 and isp‐1 worms that disruption of genes involved in the p38‐mediated innate immune signaling pathway (nsy‐1, sek‐1, pmk‐1, atf‐7(gof)) prevented the upregulation of innate immunity genes (Figs 1B and EV1).
Figure EV1. Upregulation of innate immunity genes in long‐lived mitochondrial mutants requires the p38‐mediated innate immune signaling pathway.
-
A–CMutation of genes involved in the p38‐mediated innate immune signaling pathway (nsy‐1, sek‐1, pmk‐1, atf‐7(gof)) decreases the expression of genes involved in innate immunity in wild‐type (A), nuo‐6 (B), and isp‐1 (C) worms. Gene expression was determined by quantitative real‐time RT–PCR on three biological replicates of pre‐fertile young adult worms.
Data information: Error bars indicate SEM. All differences from control are significant P < 0.05. Statistical significance was assessed for each gene independently using a one‐way ANOVA with Dunnett’s multiple comparisons test.
To confirm these results using an alternative approach, we crossed a fluorescent reporter strain for one of the innate immunity genes (T24B8.5/sysm‐1) (Shivers et al, 2010) to nuo‐6 and isp‐1 mutants and examined the effect of knocking down sek‐1 through RNA interference (RNAi). Again, we found that T24B8.5/sysm‐1 is upregulated in nuo‐6 and isp‐1 worms and that this upregulation is dependent on SEK‐1 (Fig 1C). Combined, these results demonstrate that innate immunity genes are upregulated in the long‐lived mitochondrial mutants, nuo‐6 and isp‐1, and that this upregulation is dependent on the p38‐mediated innate immune signaling pathway.
To further examine the expression of innate immunity genes in nuo‐6 and isp‐1 mutants, we compared the differentially expressed genes in these mutants to a more comprehensive and unbiased list of genes involved in innate immunity. A recent study defined the changes in gene expression that result from exposure to the bacterial pathogen Pseudomonas aeruginosa strain PA14 and determined which of these changes in gene expression are dependent on PMK‐1 and ATF‐7 (Fletcher et al, 2019a). In total, they reported 300 genes that were upregulated by exposure to PA14 in a PMK‐1‐ and ATF‐7‐dependent manner, and 230 genes that were downregulated by exposure to PA14 in a PMK‐1‐ and ATF‐7‐dependent manner.
We compared these lists of PA14‐modulated, PMK‐1‐dependent, ATF‐7‐dependent genes to genes that we found to be significantly upregulated or downregulated in nuo‐6 and isp‐1 mutants. We found that of the genes that are upregulated by PA14 exposure in a PMK‐1‐ and ATF‐7‐dependent manner, 38% (P = 1.5 × 10−35) and 30% (P = 3.7 × 10−27) are also upregulated in nuo‐6 and isp‐1 mutants, respectively (Fig 1D; P‐values indicate the significance of the difference between the observed number of overlapping genes between the two gene sets and the expected number of overlapping genes if the genes were picked at random). There is a high degree of overlap between genes upregulated in nuo‐6 and isp‐1 mutants (71%), and this includes the genes involved in innate immunity (Dataset EV1; 71 overlapping genes of 89/115 innate immunity genes upregulated in nuo‐6 or isp‐1, respectively). In contrast, there was no significant overlap of the same genes upregulated by PA14 exposure with genes downregulated in nuo‐6 and isp‐1 mutants. In examining the list of genes that are downregulated by exposure to PA14 in a PMK‐1‐ and ATF‐7‐dependent manner, we found that a significant number of these genes are also downregulated in nuo‐6 mutants (20%; P = 8.4 × 10−7) and isp‐1 mutants (20%; P = 7.5 × 10−5).
To more comprehensively compare the PA14‐modulated, PMK‐1‐dependent, ATF‐7‐dependent gene expression changes to gene expression changes in nuo‐6 and isp‐1 mutants, we generated heat maps comparing the expression of PA14‐modulated, PMK‐1‐dependent, ATF‐7‐dependent genes between wild‐type worms and nuo‐6 or isp‐1 mutants. Among the genes that are upregulated by PA14 exposure in a PMK‐1‐ and ATF‐7‐dependent manner, many of these genes are upregulated in nuo‐6 and isp‐1 mutants compared with wild‐type worms, while a small subset show decreased expression (Appendix Figs S2 and S3). Among the genes that are downregulated by PA14 exposure in a PMK‐1‐ and ATF‐7‐dependent manner, many of these genes are downregulated in nuo‐6 and isp‐1 mutants compared with wild‐type worms, while a number of these genes also show increased expression (Appendix Figs S4 and S5).
Overall, these results indicate that a mild impairment of mitochondrial function caused by mutations in nuo‐6 or isp‐1 leads to a broad upregulation of genes involved in innate immunity that is dependent on the p38‐mediated innate immune signaling pathway. At the same time, a smaller proportion of these genes show decreased expression suggesting that the correct balance of innate immune gene expression may be required to optimize stress resistance and lifespan.
Long‐lived mitochondrial mutants show increased resistance to bacterial pathogens, which requires p38‐mediated innate immune signaling
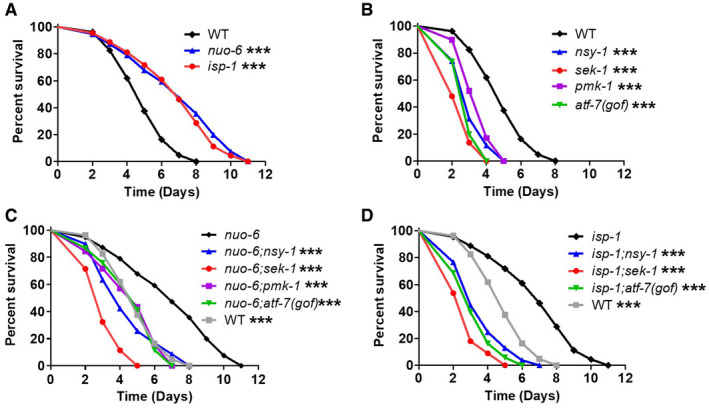
Based on our observation that the long‐lived mitochondrial mutants, nuo‐6 and isp‐1, have increased expression of many genes involved in innate immunity, we next sought to determine whether this increase in their expression resulted in enhanced resistance to bacterial pathogens. To test pathogen resistance, we exposed worms to PA14 in a slow kill assay where worms die from the ingestion and internal proliferation of the PA14 bacteria (Tan et al, 1999a, 1999b; Kirienko et al, 2014). We found that both nuo‐6 and isp‐1 worms exhibited significantly increased survival on PA14 bacteria compared with wild‐type worms (Fig 2A). The increase in survival of nuo‐6 and isp‐1 worms did not result from reduced exposure to the pathogenic bacteria as their tendency to avoid PA14 was equivalent to wild‐type worms (Appendix Fig S6).
Figure 2. Long‐lived mitochondrial mutants exhibit increased resistance to bacterial pathogens that is dependent on the presence of p38‐mediated innate immune signaling pathway.
-
A–DResistance to bacterial pathogens was tested by exposing worms to Pseudomonas aeruginosa strain PA14 in a slow kill assay. Both nuo‐6 and isp‐1 long‐lived mitochondrial mutants show increased resistance compared with wild‐type worms (A). Disruption of components of the p38‐mediated innate immune signaling pathway (nsy‐1, sek‐1, pmk‐1, atf‐7) markedly decreases resistance to PA14 in wild‐type (B), nuo‐6 (C), and isp‐1 (D) worms. All strains were tested in a single parallel experiment. Two biological replicates per strain were performed with a total of at least 175 animals per strain. A bar graph of all of these results can be found in Appendix Fig S7.
Data information: ***P < 0.001. Statistical significance for survival plots was determined with the log‐rank test. Significance is indicated between the strain listed on top and all other strains. Data from panel A are repeated in panels B, C, and D for direct comparison.
Source data are available online for this figure.
To determine the extent to which their enhanced resistance to bacterial pathogens is dependent on the p38‐mediated innate immune signaling pathway, we next examined PA14 resistance in nuo‐6 and isp‐1 worms in which genes in this pathway were disrupted. In wild‐type worms, mutations in nsy‐1, sek‐1, pmk‐1, or atf‐7(gof) significantly decrease the survival of worms exposed to PA14 (Fig 2B). Similarly, we found that disruptions of these innate immune signaling genes in nuo‐6 (Fig 2C) and isp‐1 (Fig 2D) worms also result in a significant decrease in survival on PA14 bacteria. In nuo‐6 worms, survival was decreased back to wild type by mutations in nsy‐1, pmk‐1, or atf‐7(gof), while a larger decrease in survival was observed with the sek‐1 mutation. In isp‐1 worms, mutations in nsy‐1, sek‐1, and atf‐7(gof) all decreased survival to a greater extent than in nuo‐6 mutants, and in each case, the survival of the double mutant was less than in wild‐type worms (Appendix Fig S7). This suggests that isp‐1 worms may be more reliant on the p38‐mediated innate immune signaling pathway for their enhanced pathogen resistance than nuo‐6 worms. For both nuo‐6 and isp‐1 worms, we found that disruption of sek‐1 has a greater impact on pathogen resistance than nsy‐1 and pmk‐1, which is consistent with the fact that SEK‐1 is absolutely required for innate immune signaling, while some partial redundancy exists for NSY‐1 and PMK‐1 (Kim et al, 2002).
Combined, these results show that nuo‐6 and isp‐1 worms have increased resistance to bacterial pathogens, which are dependent on the p38‐mediated innate immune signaling pathway.
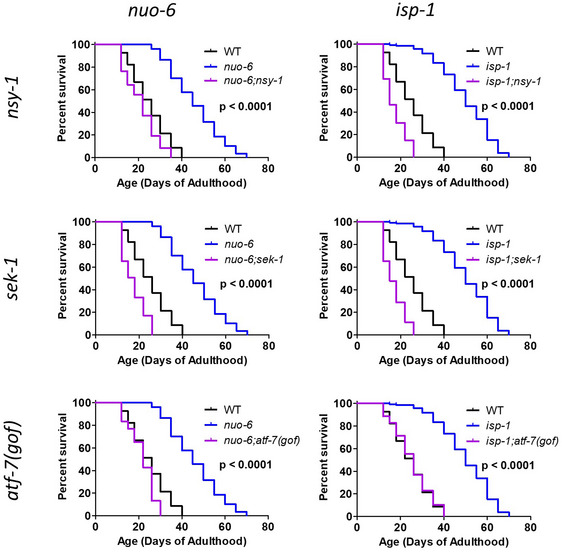
p38‐mediated innate immune signaling is required for extended longevity in long‐lived mitochondrial mutants
The p38‐mediated innate immune signaling pathway is largely required for the lifespan extension resulting from decreased insulin‐IGF1 signaling (daf‐2 mutants) and is essential in dietary restriction (Troemel et al, 2006; Wu et al, 2019). Interestingly, in both daf‐2 mutants and dietary restricted worms, the expression of p38‐regulated innate immunity genes is mostly decreased, and the activation of this pathway to higher levels is deleterious for lifespan extension, suggesting that a lower level of immune activation is optimal under these conditions (Wu et al, 2019). We decided to examine the role of the p38‐mediated innate immune signaling pathway in the long lifespan of nuo‐6 and isp‐1 mutants, in which we found these immunity genes were largely upregulated. To do this, we genetically disrupted components of the p38‐mediated innate immune signaling pathway in nuo‐6 and isp‐1 mutants and quantified the resulting effect on lifespan.
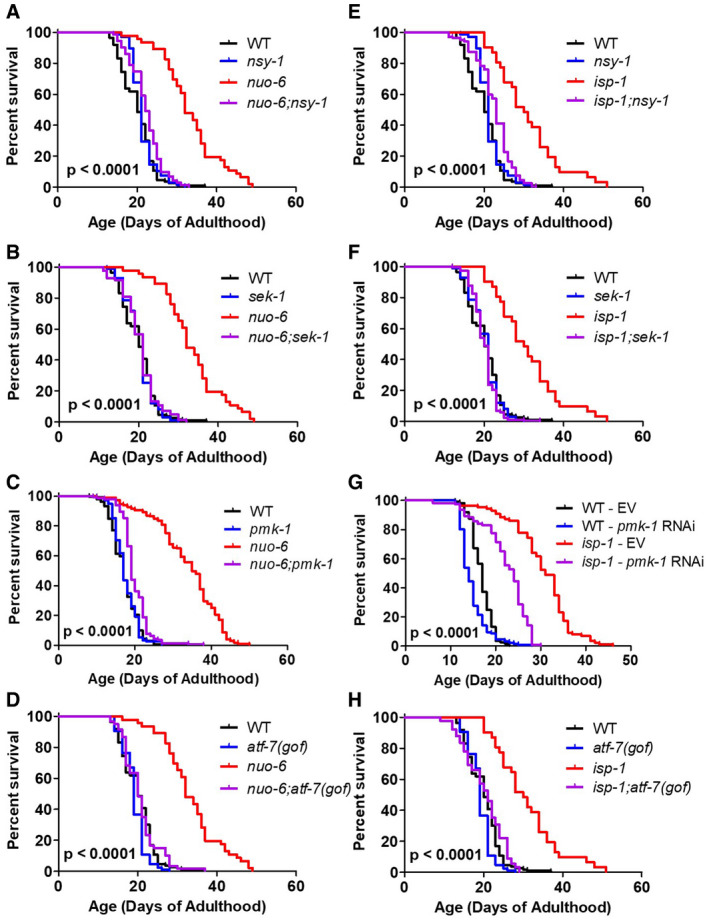
In wild‐type worms, we found that mutations in nsy‐1, sek‐1, pmk‐1, or atf‐7(gof) resulted in little or no effect on lifespan (Fig 3; nsy‐1: +7%, NS; sek‐1: +1%, NS; pmk‐1: +2%, NS; atf‐7(gof): −5%, P = 0.008). The fact that genes involved in the p38‐mediated innate immune signaling pathway are not required to achieve a normal lifespan on OP50 bacteria suggests that OP50 bacteria may not be pathogenic enough to require innate immune activation for normal longevity.
Figure 3. Disruption of the p38‐mediated innate immune signaling pathway markedly ablates extended longevity of long‐lived mitochondrial mutants.
-
A–HTo examine the role of the p38‐mediated innate immune signaling pathway in the long lifespan of the nuo‐6 and isp‐1 mitochondrial mutants, we crossed the long‐lived mitochondrial mutants to worms with mutations in nsy‐1, sek‐1, pmk‐1, and atf‐7(gof). In each case, mutation of genes involved in p38‐mediated innate immunity signaling markedly reduces the lifespan of the long‐lived mitochondrial mutant but has little or no impact on wild‐type lifespan. In panel G, we utilized pmk‐1 RNAi instead of the pmk‐1 mutation because of the close proximity of isp‐1 and pmk‐1 on the same chromosome. EV indicates empty vector. The lifespan results for panels A, B, D, E, F, and H were performed in a single parallel experiment. Results are from a minimum of three biological replicates per strain.
Data information: Statistical significance for survival plots was determined with the log‐rank test. P‐values indicate significance between red and purple lines. Control strains are shown in multiple panels for direct comparison.
Source data are available online for this figure.
In nuo‐6 mutants, we found that mutation of nsy‐1, sek‐1, pmk‐1, or atf‐7(gof) reduced the long lifespan of nuo‐6 worms to near wild‐type lifespan, almost completely preventing any increase in lifespan resulting from the nuo‐6 mutation (Fig 3A–D). Similarly, we found that mutations in the p38‐mediated innate immune signaling pathway also markedly reduced the lifespan of isp‐1 worms (Fig 3E–H). Mutations in nsy‐1, sek‐1, or atf‐7(gof) decreased isp‐1 lifespan to near wild‐type lifespan, while pmk‐1 RNAi reduced the lifespan extension in isp‐1 worms by half (we were unable to generate isp‐1 pmk‐1 double mutants because of the proximity of the two genes on the same chromosome).
Although OP50 bacteria, which are typically used for C. elegans maintenance and lifespan assays, are considered to be non‐pathogenic, it can have detrimental effects on lifespan through proliferation in the pharynx and intestine (Gems & Riddle, 2000; Garigan et al, 2002). To determine whether the extended lifespan of nuo‐6 and isp‐1 worms results from enhanced resistance to OP50 bacteria and to determine whether the effect of disrupting the p38‐mediated innate immune signaling pathway on the lifespan of nuo‐6 and isp‐1 mutants results from losing protection against OP50 bacteria, we measured the lifespan of nuo‐6 and isp‐1 mutants on non‐proliferating OP50 bacteria, as well as the dependency of their long lifespan on the p38‐mediated innate immunity pathway under these conditions. We found that even when no proliferating bacteria are present, nuo‐6 and isp‐1 worms still exhibit a robust extension of lifespan (Fig EV2). Similarly, disruption of the p38‐mediated innate immune signaling pathway still markedly decreases isp‐1 and nuo‐6 lifespan when worms are fed non‐proliferating bacteria (Fig EV2). This suggests that the p38‐mediated innate immune signaling pathway is required for their long lifespan independently of its ability to protect against bacterial infection. Similarly, we have previously shown that lifespan extension resulting from dietary restriction is dependent on the p38‐mediated innate immune signaling pathway even when non‐proliferating bacteria are used (Wu et al, 2019).
Figure EV2. Disruption of genes involved in the p38‐mediated innate immune signaling pathway abolishes the extended longevity of long‐lived mitochondrial mutants independently of bacterial proliferation.
Quantification of nuo‐6 and isp‐1 lifespan on non‐proliferating bacteria revealed that their long lifespan is independent of bacterial proliferation. Similarly, lifespan extension in nuo‐6 and isp‐1 mutants is completely dependent on having a function p38‐mediated innate immune signaling pathways as deletion of nsy‐1, sek‐1, or atf‐7(gof) completely prevented the increase in lifespan in long‐lived nuo‐6 and isp‐1 mutants. Lifespans were performed in liquid culture with worms fed ad libitum. Bacteria proliferation was prevented through treatment with cold and antibiotics. All strains were tested in a single parallel experiment. Two biological replicates per strain were measured with a total of at least 274 animals per strain.
Data information: Statistical significance for survival plots was determined with the log‐rank test. P‐value indicates significance of difference between blue and purple lines. Control strains are shown in multiple panels for direct comparison.
Source data are available online for this figure.
Combined, these results clearly indicate that the p38‐mediated innate immune signaling pathway is required for the extended longevity in the long‐lived mitochondrial mutants, nuo‐6 and isp‐1. However, the fact that disruption of the p38‐innate immune signaling pathway on nuo‐6 and isp‐1 lifespan is independent of bacterial proliferation suggests that the lifespan extension resulting from mild impairment of mitochondrial function is not necessarily a direct consequence of their enhanced resistance to bacterial pathogens, but more likely a result of other functions of the p38‐mediated innate immune signaling pathway.
p38‐mediated innate immune signaling pathway does not show enhanced activation in long‐lived mitochondrial mutants
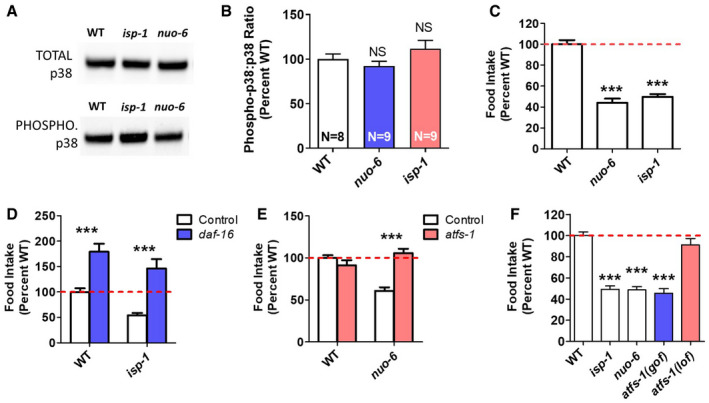
Since target genes of the p38‐mediated innate immune signaling pathway are upregulated in nuo‐6 and isp‐1 mutants, we sought to determine the extent to which this pathway might be activated in these mutants by quantifying the ratio of phosphorylated (active) PMK‐1/p38 to total PMK‐1/p38 by Western blotting (Inoue et al, 2005). To validate this approach, we showed that decreasing innate immune signaling with sek‐1 RNAi and increasing innate immune signaling with vhp‐1 RNAi (VHP‐1 is a phosphatase that inhibits PMK‐1/p38 signaling) alters the ratio of phospho‐p38 to p38 as expected (Appendix Fig S8).
Having shown that known modulators of PMK‐1/p38 activation alter PMK‐1/p38 phosphorylation in a predictable manner, we examined PMK‐1/p38 activation in nuo‐6 and isp‐1 mutants. In both cases, we found that neither the levels of PMK‐1/p38 protein nor the proportion of PMK‐1/p38 protein that is phosphorylated are increased compared with wild type (Fig 4A and B). Thus, even though nuo‐6 and isp‐1 mutants exhibit increased expression of ATF‐7 target genes, there is no detectable increase in PMK‐1/p38 activation. Since the p38‐mediated innate immune signaling pathway is required for the increased expression of ATF‐7 target genes even though p38 signaling is not increased, this suggests that this pathway is playing a permissive role and that other mechanisms are driving the enhanced innate immune activation in the long‐lived mitochondrial mutants.
Figure 4. p38‐mediated innate immune signaling pathway is not activated in long‐lived mitochondrial mutants.
-
A, BTo determine the extent to which the p38‐mediated innate immunity pathway is activated in nuo‐6 and isp‐1 mutants, we measured the ratio of phosphorylated PMK‐1/p38 to total PMK‐1/p38 by Western blotting. Representative blots are shown (A). There is no difference in the ratio of phosphorylated to total PMK‐1/p38 in the long‐lived mitochondrial mutants compared with wild type (B). Eight biological replicates of wild‐type worms and nine of mitochondrial mutants were quantified.
-
CBoth long‐lived mitochondrial mutants, nuo‐6 and isp‐1, exhibit markedly decreased food consumption compared with wild‐type worms. Six biological replicates per strain were measured, each including at least three technical replicates.
-
DDeletion of daf‐16 significantly increases food consumption in both wild‐type and isp‐1 mutants. Three biological replicates per strain were measured, each including at least three technical replicates.
-
EWhile disruption of atfs‐1 does not affect food intake in wild‐type worms, it increases food consumption in nuo‐6 worms back to wild type. atfs‐1 loss‐of‐function allele was gk3094. Three biological replicates per strain were measured, each including at least three technical replicates.
-
FConstitutive activation of atfs‐1 is sufficient to decrease food consumption to the same level as in long‐lived mitochondrial mutants. Three biological replicates per strain were measured, each including at least three technical replicates. atfs‐1(lof) allele is gk3094, and atfs‐1(gof) allele is et17.
Data information: Error bars indicate SEM. ***P < 0.001. NS = not significant. Statistical significance was assessed using a one‐way ANOVA with Dunnett’s multiple comparison test in panels B, C, and F. Statistical significance was assessed using a two‐way ANOVA with Bonferroni post‐test in panels D and E.
Source data are available online for this figure.
Since the p38‐mediated innate immune signaling pathway can be activated by nutrient consumption independently of pathogen exposure (Wu et al, 2019), we wondered whether altered feeding affects the level of activation of this pathway in the long‐lived mitochondrial mutants. Accordingly, we quantified food consumption in nuo‐6 and isp‐1 worms directly by monitoring the decrease in OP50 bacteria from day 3 to day 12 of adulthood (Wu et al, 2019). We found that both long‐lived mutants show a dramatic decrease in food consumption compared with wild‐type worms (Fig 4C). This decrease in food consumption does not result from a difference in occupancy of the bacteria as nuo‐6 and isp‐1 worms spend the same amount of time on the bacteria as wild‐type worms (Appendix Fig S6). The decrease in food consumption in nuo‐6 and isp‐1 worms would be predicted to decrease p38‐mediated innate immune signaling. The fact that the activation of the p38‐mediated innate immune signaling pathway is not decreased in the long‐lived mitochondrial mutants, and that target genes of this pathway are upregulated, again suggests that other factors are acting in parallel to p38 signaling to upregulate the expression of innate immunity genes.
Decreased feeding in long‐lived mitochondrial mutants is mediated by the activation of the mitoUPR
We next wanted to define the mechanism by which feeding is decreased in the long‐lived mitochondrial mutants. Since DAF‐16 is activated in nuo‐6 and isp‐1 mutants (Senchuk et al, 2018) and disruption of daf‐16 has been shown to increase food intake in wild‐type and daf‐2 worms (Wu et al, 2019), we sought to determine whether daf‐16 is required for the decreased food intake observed in the long‐lived mitochondrial mutants. We found that deletion of daf‐16 increased food consumption in both wild‐type and isp‐1 worms (Fig 4D). While this result indicates that DAF‐16 is required for the decreased food consumption in isp‐1 mutants, it is unclear whether DAF‐16 is driving this change as loss of DAF‐16 similarly increased food consumption in wild‐type worms.
Since activation of ATFS‐1 is sufficient to upregulate DAF‐16 target genes (Wu et al, 2018) and ATFS‐1 is activated in the long‐lived mitochondrial mutants (Wu et al, 2018), we wondered whether ATFS‐1 contributes to the decreased food intake in the long‐lived mitochondrial mutants. While disruption of atfs‐1 (using the gk3094 deletion mutation) had no effect on feeding in a wild‐type background, loss of atfs‐1 reverted food consumption in nuo‐6 worms to wild type (Fig 4E). This suggests that the activation of ATFS‐1 is driving the decreased food intake in the long‐lived mitochondrial mutants.
Having shown that activation of ATFS‐1 contributes to the decreased food intake in nuo‐6 mutants, we wondered whether the activation of ATFS‐1 alone is sufficient to decrease food consumption. To test this idea, we measured food consumption in a constitutively activated atfs‐1 mutant (et17). In this mutant, the mitochondrial targeting sequence of ATFS‐1 is disrupted resulting in nuclear localization and constitutive activation of ATFS‐1 target genes (Rauthan et al, 2013). We found that the constitutively activated atfs‐1 mutant et17 showed a marked decrease in food consumption, equivalent to that observed in nuo‐6 and isp‐1 mutants (Fig 4F). Combined, this indicates that the activation of ATFS‐1 is sufficient to decrease food consumption and is required for the diminished food intake in nuo‐6 worms.
Since nuo‐6 and isp‐1 worms have decreased food consumption, and dietary restriction is sufficient to increase resistance to bacterial pathogens (Wu et al, 2019), we wondered whether dietary restriction could further enhance bacterial pathogen resistance in the long‐lived mitochondrial mutants. We found that while dietary restriction increased bacterial pathogen resistance in wild‐type worms, it did not further increase the enhanced pathogen resistance in nuo‐6 or isp‐1 worms (Appendix Fig S9). Thus, the enhanced pathogen survival rates resulting from dietary restriction and mild impairment of mitochondrial function are not additive.
Although both dietary restriction and mild mitochondrial impairment increase resistance to bacterial pathogens, they have opposite effects on the expression of innate immunity genes.
We previously reported that dietary restriction primarily downregulates innate immunity genes (Wu et al, 2019). Here, we show that mutations in nuo‐6 or isp‐1 primarily result in upregulation of innate immunity genes (Fig 1). To more globally examine gene expression changes between dietary restriction and mild impairment of mitochondrial function, we compared differentially expressed genes. We found that there was very little overlap of either upregulated or downregulated genes between dietary restriction and nuo‐6 or isp‐1 mutants (Appendix Fig S10).
Since the levels of ROS are increased in nuo‐6 and isp‐1 mutants (Yang & Hekimi, 2010a) and ROS have been shown to activate p38 signaling (Inoue et al, 2005; Hourihan et al, 2016), we sought to determine the extent to which elevated ROS contributes to bacterial pathogen resistance, PMK‐1/p38 activation, and food intake in the long‐lived mitochondrial mutants. To do this, we treated wild‐type, nuo‐6, and isp‐1 worms with the antioxidant vitamin C, at a concentration that we have previously shown is effective at decreasing isp‐1 and clk‐1 lifespan, without affecting the lifespan of wild‐type worms (10 mM; Schaar et al, 2015; Van Raamsdonk & Hekimi, 2012). We found that treatment with vitamin C had no effect on bacterial pathogen resistance (Appendix Fig S11), PMK‐1/p38 activation (Appendix Fig S12), or food intake (Appendix Fig S13) in nuo‐6 or isp‐1 worms. This suggests that the elevated levels of ROS in the long‐lived mitochondrial mutants are not driving these phenotypes, though it is also possible that the dose or timing of antioxidant treatment required to affect these phenotypes is different than the dose required to affect longevity. Interestingly, treatment with vitamin C increased bacterial pathogen resistance and decreased food intake in wild‐type worms (Appendix Figs S11 and S13). This suggests the possibility that the upregulation of antioxidant genes in nuo‐6 and isp‐1 worms (Dues et al, 2017; Wu et al, 2018) may be contributing to these two phenotypes.
The FOXO transcription factor DAF‐16 is required for bacterial pathogen resistance in long‐lived mitochondrial mutants but does not account for activation of innate immunity genes
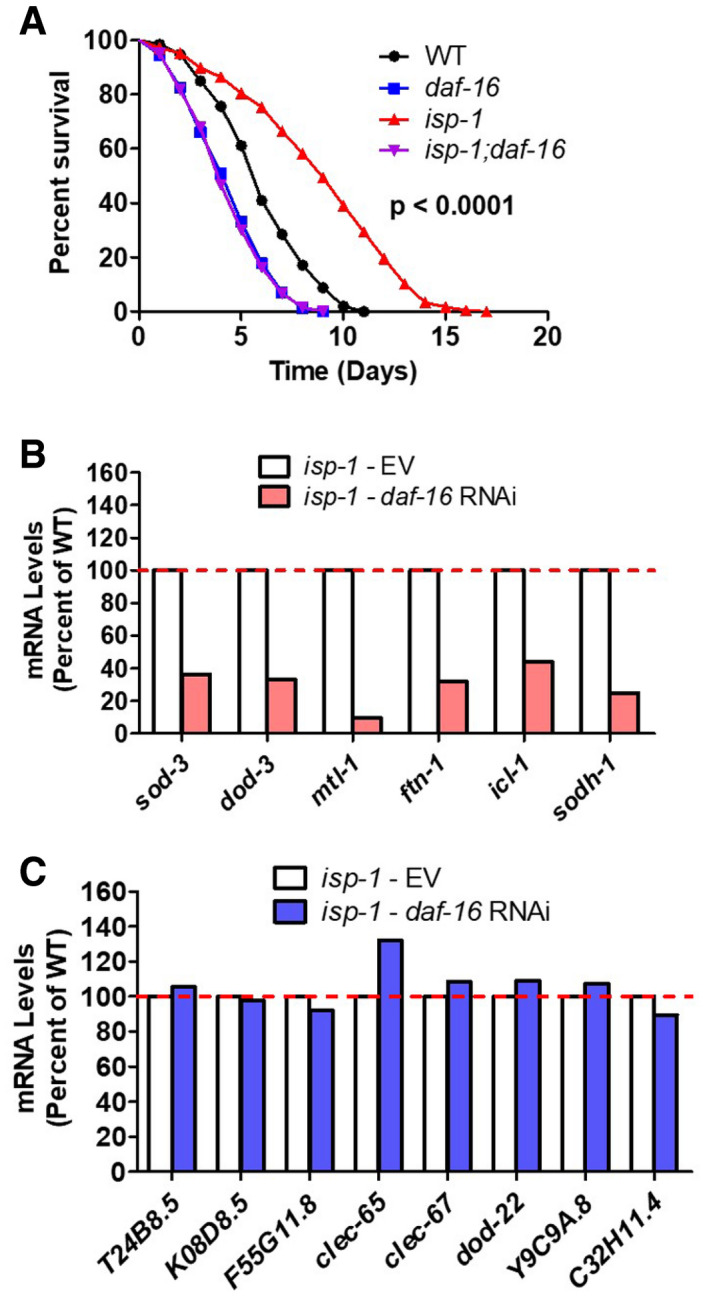
As we have previously shown that target genes of the FOXO transcription factor DAF‐16 are upregulated in the long‐lived mitochondrial mutants (Senchuk et al, 2018), and others have shown that DAF‐16 is required for the enhanced bacterial pathogen resistance of long‐lived daf‐2 mutants (Troemel et al, 2006), we next examined the contribution of DAF‐16 to bacterial pathogen resistance in long‐lived mitochondrial mutants. We found that disruption of daf‐16 completely abolished the increased resistance to bacterial pathogens in isp‐1 worms (Fig 5A), thereby indicating that DAF‐16 is required for the enhanced bacterial pathogen resistance in isp‐1 worms (note that we only used isp‐1 mutants to examine the effect of daf‐16 because we were unable to generate nuo‐6 daf‐16 double mutants due to close proximity of the two genes on the same chromosome).
Figure 5. DAF‐16/FOXO is required for increased resistance to bacterial pathogens in long‐lived mitochondrial mutants.
-
AA deletion of daf‐16 completely abolishes the increased resistance to bacterial pathogens in isp‐1 mutants. P‐value indicates the significance of difference between red and purple lines. Three biological replicates per strain were performed.
-
B, CWhile daf‐16 RNAi effectively decreases the expression of DAF‐16 target genes (B, sod‐3, dod‐3, mtl‐1, ftn‐1, icl‐1, sodh‐1), it does not affect the expression of any of the innate immunity genes (C, T24B8.5, K08D8.5, F55G11.8, clec‐65, clec‐67, dod‐22, Y9C9A.8, and C32H11.4). This indicates that DAF‐16 is not required for expression of innate immune signaling pathway target genes in isp‐1 mutants. daf‐16 expression was knocked down using RNAi beginning at the L4 stage of the parental generation. RNA was isolated from six biological replicates at the young adult stage of the experimental generation. RNA from the six biological replicates was pooled for RNA sequencing.
Data information: Statistical significance in panel A was assessed using log‐rank test.
Source data are available online for this figure.
To determine the extent to which the effects of DAF‐16 on bacterial pathogen resistance might be mediated through the same genes that are modulated by the p38‐mediated innate immune signaling pathway, we examined the effect of daf‐16 RNAi on the expression of innate immune genes in wild‐type, nuo‐6, and isp‐1 worms. While daf‐16 RNAi was effective in decreasing the expression of DAF‐16 target genes (sod‐3, dod‐3, mtl‐1, ftn‐1, icl‐1, sodh‐1) in all three strains, there was little or no effect on genes involved in innate immunity (T24B8.5/sysm‐1, K08D8.5, F55G11.8, clec‐65, clec‐67, dod‐22, Y9C9A.8, and C32H11.4) (Figs 5B and C, and EV3). This finding is consistent with a previous study that compared microarray data of genes modulated by DAF‐16 and PMK‐1 and observed no overlap, despite both pathways being required for pathogen resistance in daf‐2 mutants (Troemel et al, 2006). Thus, although DAF‐16 activation contributes to the enhanced pathogen resistance in the long‐lived mitochondrial mutants, it does not contribute to the upregulation of p38/ATF‐7‐regulated innate immunity genes in these worms.
Figure EV3. DAF‐16 is not required for expression of innate immune signaling pathway target genes.
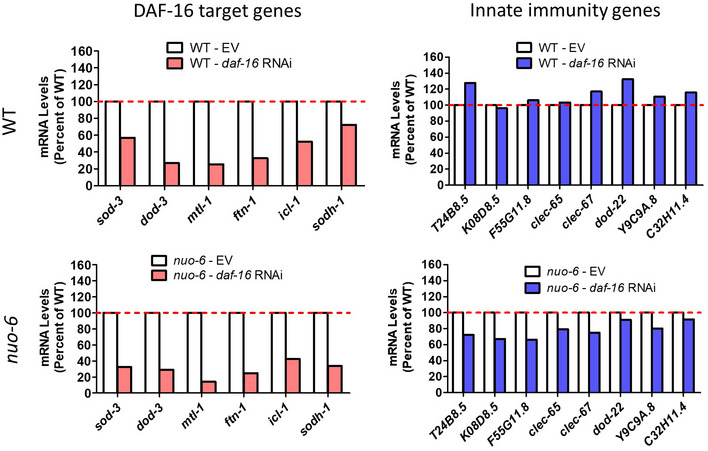
daf‐16 expression was knocked down using RNAi beginning at the L4 stage of the parental generation. While daf‐16 RNAi effectively decreases the expression of DAF‐16 target genes (Left; red bars; sod‐3, dod‐3, mtl‐1, ftn‐1, icl‐1, sodh‐1) in both wild‐type and nuo‐6 mutants, it does not markedly affect the expression of any of the innate immunity genes (Right; blue bars; T24B8.5, K08D8.5, F55G11.8, clec‐65, clec‐67, dod‐22, Y9C9A.8, and C32H11.4). This suggests that DAF‐16 is not required for expression of innate immune signaling pathway target genes in wild‐type worms and nuo‐6 mutants. RNA was isolated from six biological replicates at the young adult stage of the experimental generation. RNA from the six biological replicates was pooled for RNA sequencing.
DAF‐16 may instead be enhancing resistance to pathogens through activation of different immunity genes or through a general increase in resistance to stress and or survival.
The mitoUPR transcription factor ATFS‐1 mediates upregulation of innate immune genes in long‐lived mitochondrial mutants
In our previous studies, we have shown that the mitoUPR is activated in nuo‐6 and isp‐1 worms and that this activation is required for their increased lifespan (Wu et al, 2018). As activation of the mitoUPR by RNAi against spg‐7 (the worm homolog of SPG7/paraplegin) has been shown to increase resistance to PA14 and upregulate innate immunity genes (Pellegrino et al, 2014), we hypothesized that the ATFS‐1 activation that occurs in nuo‐6 and isp‐1 mutants (Wu et al, 2018) might contribute to their enhanced resistance to PA14.
To test this, we examined the effect of inhibiting the mitoUPR on the survival of nuo‐6 worms under bacterial pathogen stress (we were unable to test the effects of mitoUPR inhibition in isp‐1 worms as isp‐1; atfs‐1 double mutants arrest during development (Wu et al, 2018)). We found that disruption of atfs‐1 completely ablates the bacterial pathogen resistance of nuo‐6 worms such that nuo‐6; atfs‐1 worms have decreased survival compared with wild‐type or atfs‐1 single mutants (Fig 6A). This indicates that the activation of the mitoUPR is required for the enhanced pathogen resistance in nuo‐6 mutants. The fact that nuo‐6; atfs‐1 mutants have decreased survival compared with wild‐type worms and atfs‐1 mutants suggests the possibility that the nuo‐6 mutation makes worms more sensitive to bacterial pathogens, perhaps due to decreased energy production, and that activation of ATFS‐1 is required not only to enhance their resistance to pathogens but also to increase their resistance back to wild‐type levels.
Figure 6. ATFS‐1 is required for increased bacterial pathogen resistance and upregulation of innate immunity genes in long‐lived nuo‐6 mitochondrial mutants.
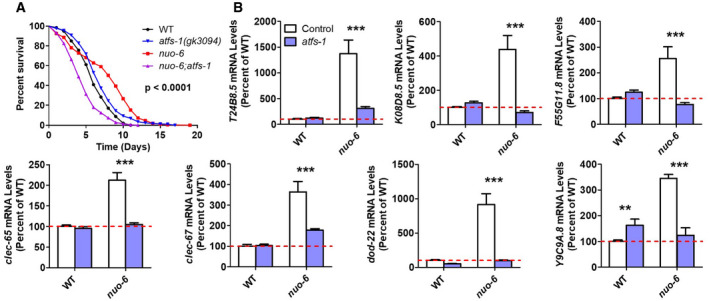
- To examine the role of the ATFS‐1 transcription factor and the mitoUPR response in the enhanced bacterial pathogen resistance in nuo‐6 mutants, we examined the effect of disrupting atfs‐1 in nuo‐6 mutants. Loss of atfs‐1 completely abolishes the increased resistance to bacterial pathogens in nuo‐6 worms. P‐value indicates significance of difference between red and purple lines. nuo‐6; atfs‐1 contains the gk3094 deletion allele. Three biological replicates per strain were measured.
- To examine the role of ATFS‐1 in the upregulation of innate immunity genes in nuo‐6 mutants, we compared gene expression between nuo‐6 and nuo‐6; atfs‐1 mutants with wild‐type worms and atfs‐1 mutants as controls. Deletion of atfs‐1 significantly decreases the expression of genes involved in innate immunity in nuo‐6 worms but does not decrease the expression of these genes in wild‐type worms. Gene expression changes were determined by RNA sequencing of six biological replicates of each genotype. Results represent counts per million (CPM) expressed as a percentage of wild type. Worms in a wild‐type background (control) are shown with white bars, while worms with atfs‐1 deletion are shown with blue bars.
Data information: Error bars indicate SEM. **P < 0.01, ***P < 0.001. Statistical significance in panel A was assessed using log‐rank test. Statistical significance in panel B was assessed using a two‐way ANOVA with Bonferroni post‐test.
Source data are available online for this figure.
To determine whether ATFS‐1 is also responsible for the upregulation of innate immunity genes in nuo‐6 mutants, we examined the effect of atfs‐1 deletion. Deletion of atfs‐1 did not decrease the expression of any of the innate immunity genes examined in wild‐type worms (Fig 6B), but did result in a small increase in the expression of Y9C9A.8, possibly as a result of a compensatory upregulation of specific stress response genes, as we previously observed for hsp‐16.11 and hsp‐16.12 (Wu et al, 2018). In contrast, loss of atfs‐1 markedly decreased the expression of all of the innate immunity genes examined in nuo‐6 worms, in most cases reverting expression levels to wild type (Fig 6B). Thus, ATFS‐1 is required for the upregulation of innate immunity genes in nuo‐6 mutants but is dispensable for the normal nutrient‐driven regulation of innate immune gene expression that is seen in wild‐type worms (Wu et al, 2019).
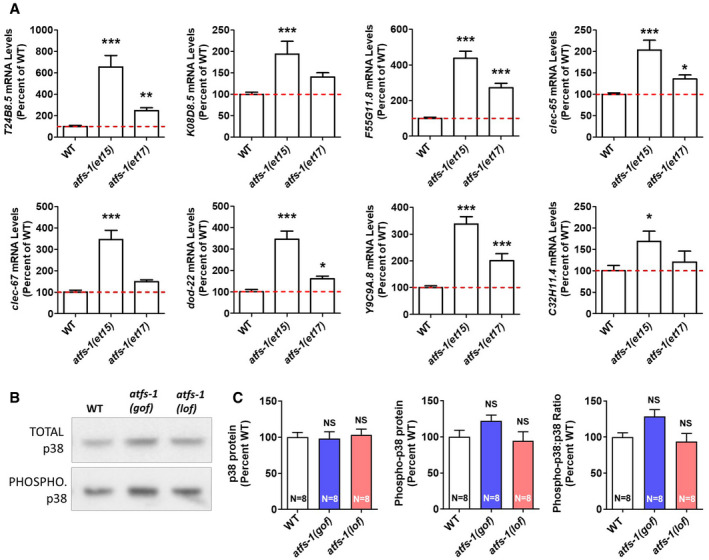
Activation of ATFS‐1 is sufficient to upregulate innate immune signaling genes
Since loss of atfs‐1 prevented the upregulation of innate immunity genes in nuo‐6 mutants, we next sought to determine whether the activation of ATFS‐1 is sufficient to cause upregulation of innate immunity genes. To do this, we examined gene expression in two constitutively active atfs‐1 mutants, et15 and et17 (Rauthan et al, 2013). We found constitutive activation of ATFS‐1 resulted in a significant upregulation of innate immunity genes (Fig 7A). As a positive control, we found that the ATFS‐1 target gene hsp‐6 is significantly upregulated in both constitutively activated atfs‐1 mutants (Appendix Fig S14).
Figure 7. Constitutive activation of mitochondrial unfolded protein response results in upregulation of innate immunity genes.
-
AExamination of mRNA levels of genes involved in innate immunity in two constitutively active atfs‐1 mutants (et15 and et17) revealed that innate immunity genes are upregulated when ATFS‐1 is activated. This indicates that the activation of the mitoUPR can cause activation of genes that are regulated by the p38‐mediated innate immune pathway. Gene expression changes were determined by RNA sequencing of six biological replicates of each genotype. Results represent counts per million (CPM) expressed as a percentage of wild type.
-
B, CConstitutive activation of ATFS‐1 does not increase activation of PMK‐1/p38 as measured by the ratio of phosphorylated PMK‐1/p38 to total PMK‐1/p38 using Western blotting. Representative blots are shown in panel (B). There is also no significant difference in the levels of PMK‐1/p38 or phosphorylated PMK‐1/p38 in atfs‐1(gof) and atfs‐1(lof) mutants compared with wild type (C). atfs‐1(gof) is the et17 allele. atfs‐1(lof) is the gk3094 allele. Eight biological replicates per strain were quantified.
Data information: Error bars indicate SEM. *P < 0.05 **P < 0.01, ***P < 0.001. NS = not significant. Statistical significance in panels A and C was assessed using a one‐way ANOVA with Dunnett’s multiple comparison test.
Source data are available online for this figure.
To determine whether the activation of ATFS‐1 causes upregulation of innate immunity genes through enhanced activation of the p38‐mediated innate immune signaling pathway, we examined PMK‐1/p38 phosphorylation in a constitutively activated atfs‐1 mutant (et17) and an atfs‐1 loss‐of‐function mutant (gk3094). While there is a trend toward a modest increase in phosphorylated p38 in the constitutively active atfs‐1 mutant compared with wild type, quantification of eight biological replicates indicated that the levels of PMK‐1/p38 protein, phosphorylated PMK‐1/p38 protein, and the ratio of phosphorylated to total PMK‐1/p38 are equivalent to wild type in both atfs‐1 mutants (Fig 7B and C). This result is consistent with the fact that the levels of phosphorylated p38 are not significantly increased in nuo‐6 or isp‐1 mutants, both of which show activation of ATSF‐1 (Fig 4). This suggests that the activation of ATFS‐1 can upregulate innate immunity genes without increasing activation of the p38‐mediated innate immune signaling pathway.
To determine whether ATFS‐1 might be able to directly bind to the same innate immunity genes as ATF‐7, we compared data from two previous CHiP‐seq studies (Nargund et al, 2015a; Fletcher et al, 2019a). For this comparison, we defined innate immunity genes as genes that are upregulated in response to a 4‐h exposure to PA14 (Fletcher et al, 2019a). After exposure to PA14, ATF‐7 was found to be bound to 345 of these innate immunity genes (Fletcher et al, 2019a; Data ref: Fletcher et al, 2019b). Following exposure to mitochondrial stress induced by spg‐7 RNAi, ATFS‐1 was found to be bound to 50 of these innate immunity genes (Nargund et al, 2015a; Data ref: Nargund et al, 2015b). Of the 50 innate immunity genes that can be bound by ATFS‐1, 48% (24 genes) can also be bound by ATF‐7 (Fig EV4; Dataset EV2).
Figure EV4. ATFS‐1 and ATF‐7 bind to the same genes involved in innate immunity.
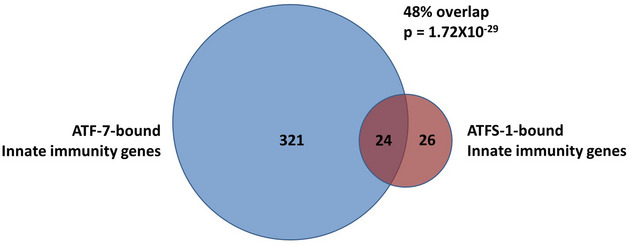
Innate immunity genes are defined as genes that are upregulated by a 4‐h exposure to PA14. This list was obtained from Fletcher et al (2019b). ATF‐7‐bound genes are genes bound by ATF‐7 after exposure to PA14 from a CHIP‐seq experiment conducted by Fletcher et al (2019b). ATFS‐1‐bound genes are genes bound by ATFS‐1 after mitochondrial stress resulting from RNAi against spg‐7 as determined by a CHIP‐seq experiment conducted by Nargund et al (2015b). ATF‐7 was found to be bound to 345 innate immunity genes after exposure to PA14. ATFS‐1 was found to be bound to 50 innate immunity genes after exposure to mitochondrial stress. Of these 50 genes, 24 were found to be in common with ATF‐7‐bound innate immunity genes. This highly significant overlap (P = 1.72 × 10−29) clearly demonstrates that ATF‐7 and ATFS‐1 can bind to and regulate the same innate immunity genes. See Dataset EV2 for the complete gene lists.
Data information: The P‐value indicates the significance of the difference between the observed number of overlapping genes between the two gene sets and the expected number of overlapping genes if the genes were picked at random.
To gain further insight into how ATFS‐1 and ATF‐7 might be modulating the expression of the same innate immunity genes, we mapped out the regions identified by the CHiP‐seq studies and the location of the consensus binding sites within those regions. Of the 24 genes that were in common between the two ChIP‐seq studies, there were 11 genes in which ATFS‐1 and ATF‐7 bind to the same gene but in non‐overlapping regions. For the other 13 genes, the binding peaks for ATFS‐1 and ATF‐7 are in close proximity, and for 10 of the 13 genes, the binding peaks occur in the promoter region (Fig EV5; Appendix Fig S15). For genes in which the binding peaks exhibited overlap, there did not appear to be overlap in the consensus binding site. This suggests that the binding of ATFS‐1 and ATF‐7 most likely occurs at different sites within the same gene. This conclusion is supported by the observation that the predicted binding site motifs of ATFS‐1 and ATF‐7 bear little similarity (Nargund et al, 2015a; Fletcher et al, 2019a). Nonetheless, the fact that ATFS‐1 and ATF‐7 are able to bind to the same genes involved in innate immunity indicates that the mitoUPR and p38‐mediated innate immune signaling pathway act in parallel to modulate the expression of innate immunity genes, which are critical for lifespan extension.
Figure EV5. ATF‐7 and ATFS‐1 can bind to the promoter region of the same innate immunity genes.
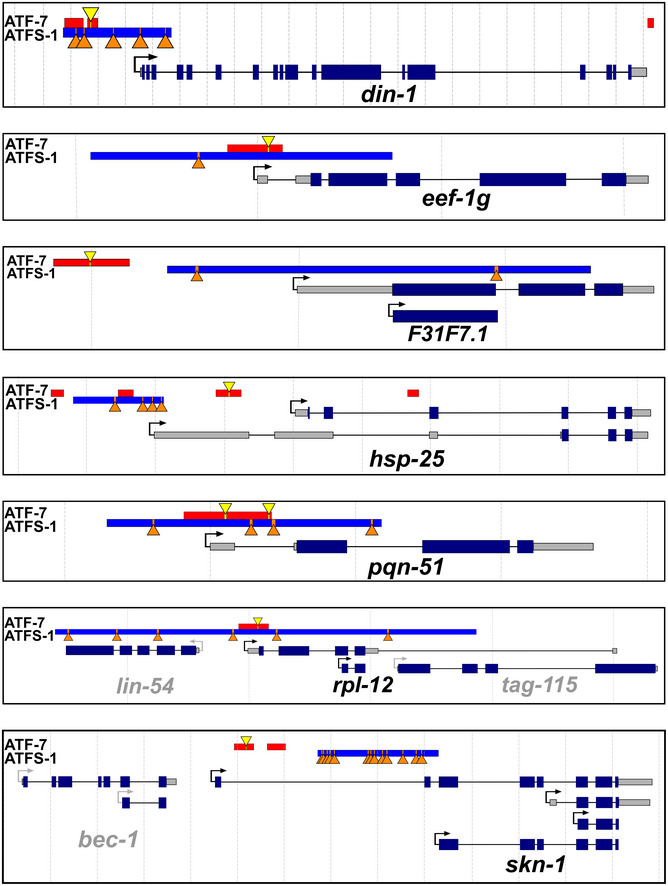
To better understand how ATF‐7 and ATFS‐1 act to modulate the same innate immunity genes, we mapped out the regions identified by ChIP‐seq studies as well as the location of the consensus binding sites. Ten of the 24 genes that show binding of both ATF‐7 and ATFS‐1 demonstrated binding of ATF‐7 and ATFS‐1 in close proximity in the promoter region of innate immunity target genes. Diagrams of seven of those genes are shown here, while diagrams of the other three genes can be found in Appendix Fig S15. The binding regions identified for ATF‐7 and ATFS‐1 by ChIP‐seq experiments are shown by red and blue bars, respectively. The consensus binding sites for ATF‐7 and ATFS‐1 are indicated by yellow triangles and orange triangles, respectively. The distance between dotted lines is 1 kb. Exons are indicated by dark blue bars. Untranslated regions are indicated by gray bars, and introns are indicated by black lines joining together two exons. Note that not all transcripts for each gene are illustrated, only those with differing promoter regions and or markedly different structures.
Discussion
Aging may be defined as a progressive decline of physiologic functions that increases the probability of illness and death. The major factors that contribute to aging have been categorized into hallmarks or pillars of aging (Kennedy et al, 2014). Interestingly, at least two of the seven pillars are directly related to the immune system, but in opposite directions. First, the ability of the immune system to fight off external stresses such as exposure to bacterial pathogens declines with age, which can be accounted for by an age‐dependent decrease in basal and activated PMK‐1/p38 levels (Youngman et al, 2011; Dues et al, 2016). Second, there is an increase in the basal activity of the immune system that occurs with advancing age, eventually leading to chronic inflammation (Franceschi et al, 2007). Thus, the relationship between aging and the immune response can be seen as a fine balance between maintaining the ability to respond to stresses, while limiting non‐specific immune activation.
Our ability to precisely define the relationship between immune activity and aging has been greatly enhanced by the discovery of genes that increase lifespan in model organisms, which allow for investigation of the molecular mechanisms. In this work, we demonstrate that mild impairment of mitochondrial function through mutations affecting either complex I (nuo‐6) or complex III (isp‐1) of the electron transport chain results in increased expression of a broad panel of genes involved in innate immunity. Treatment with the complex I inhibitor rotenone was previously shown to cause neurodegeneration and the upregulation of a small panel of genes involved in innate immunity, but the mechanism by which these genes were upregulated was not determined (Chikka et al, 2016). While it is well established that the degeneration of neurons can activate an innate immune response (Heneka et al, 2014; Labzin et al, 2018), our work demonstrates that the impairment of mitochondrial function can also lead to a broad upregulation of innate immunity genes. Further, our results indicate that the upregulation of innate immunity genes in response to mild mitochondrial impairment is driven by activation of the mitoUPR transcription factor ATFS‐1, and dependent upon all of the components of the p38‐mediated innate immune signaling pathway (NSY‐1, SEK‐1, PMK‐1, ATF‐7).
While both the long‐lived mitochondrial mutants and constitutively active atfs‐1 mutants exhibit increased expression of ATF‐7 target genes overall, it is intriguing that these mutants show no increase in p38‐mediated innate immune signaling (Fig 8). This suggests that the p38‐mediated innate immunity pathway is playing a permissive role. The p38‐mediated innate immunity pathway appears to be required for baseline expression of innate immunity genes, while activation of ATFS‐1 is driving the enhanced expression of innate immunity genes that we observe in the long‐lived mitochondrial mutants and constitutively active atfs‐1 mutants. Our data showing that disruption of nsy‐1, sek‐1, pmk‐1, or atf‐7 decreases the expression of innate immunity genes in wild‐type worms suggest that ATF‐7 binding and relief from its repressor activity is required for any transcription of p38‐mediated innate immunity target genes to occur. Furthermore, our data showing that disruption of atfs‐1 does not affect the expression of innate immunity genes in wild‐type worms but prevents nuo‐6 mutants from having increased expression suggest that the role of ATFS‐1 is for enhanced expression of innate immunity genes such that when ATFS‐1 is bound to the promoter region, or perhaps enhancer elements, the baseline expression of innate immunity genes that results from the binding of ATF‐7 is increased. This model is supported by the fact that ATFS‐1 can bind to the same innate immunity genes as ATF‐7 (Figs EV4 and EV5).
Figure 8. Mild impairment of mitochondrial function increases bacterial pathogen resistance and lifespan.
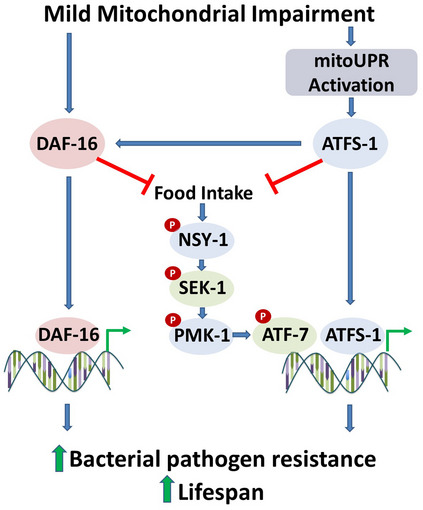
Impairment of mitochondrial function through mutation of nuo‐6 or isp‐1, which affects the mitochondrial electron transport chain, leads to the activation of the mitoUPR and nuclear localization of DAF‐16. The mitoUPR transcription factor ATFS‐1 can bind directly to innate immunity genes to increase their expression. The activation of ATFS‐1 is also sufficient to decrease food consumption. As decreasing food consumption decreases activation of the p38‐mediated innate immune signaling pathway, the ATFS‐1‐mediated decrease in food consumption should result in decreased signaling through the p38‐mediated innate immune signaling pathway. This signaling pathway terminates with the ATF‐7 transcription factor that can bind to the same genes as ATFS‐1. ATFS‐1 also acts to facilitate the nuclear localization of DAF‐16, which modulates the expression of a separate set of genes that promotes survival of bacterial pathogens and longevity. DAF‐16 acts to decrease food intake, thereby decreasing signaling through the p38‐mediated innate immune signaling pathway. DAF‐16, ATFS‐1, and all of the components of the p38‐mediated innate immune signaling pathway (NSY‐1, SEK‐1, PMK‐1, and ATF‐7) are required for the increased resistance to bacterial pathogens and increased lifespan in nuo‐6 and isp‐1 mutants.
Previous work examining the relationship between the mitoUPR and p38‐mediated innate immune signaling pathway has generated conflicting results. In an earlier study, activation of the mitoUPR by spg‐7 RNAi was found to increase the survival of pmk‐1 and sek‐1 mutants on PA14, leading the authors to conclude that the mitoUPR was increasing pathogen resistance independently of the p38‐mediated innate immune signaling pathway (Pellegrino et al, 2014). Subsequently, it was shown that overexpression of the mitoUPR target gene hsp‐60 is sufficient to activate the p38‐mediated innate immune signaling pathway leading to increased expression of genes involved in innate immunity and increased PA14 resistance, all of which was dependent on p38‐mediated innate immune signaling pathway (nsy‐1, sek‐1 and pmk‐1) (Jeong et al, 2017). Our current results resolve this discrepancy by demonstrating that the mitoUPR can affect the expression of innate immunity genes and pathogen resistance both directly through binding of ATFS‐1 to innate immunity genes and indirectly through the p38‐mediated innate immune signaling and ATF‐7 (Fig 8). A role for ATFS‐1 in modulating the expression of innate immunity genes is also supported by our observation that ATFS‐1 is required for upregulation of innate immunity genes in response to bacterial pathogen stress (Soo et al, 2021).
The increased expression of innate immunity genes in nuo‐6 and isp‐1 worms results in enhanced resistance to bacterial pathogens, which is dependent on both ATFS‐1 and the p38‐mediated innate immune signaling pathway. In addition to its ability to protect against bacterial pathogen stress, activation of ATFS‐1 can also protect against oxidative stress, endoplasmic reticulum stress, osmotic stress, and anoxia through upregulation of target genes from multiple stress response pathways (Soo et al, 2021). The increased resistance to bacterial pathogens in nuo‐6 and isp‐1 mutants is also dependent on DAF‐16, which has increased nuclear localization in long‐lived mitochondrial mutants (Senchuk et al, 2018). However, the mechanism by which DAF‐16 protects against bacterial pathogens appears to be distinct from the mechanism of ATFS‐1, as loss of DAF‐16 does not affect the expression of the innate immunity genes that are upregulated by ATFS‐1 and ATF‐7 (Figs 5 and EV3) (Troemel et al, 2006). The effect of DAF‐16 on bacterial pathogen resistance may instead be due to a general effect of DAF‐16 on resistance to stress or modulation of an independent set of innate immunity genes. Previous work has shown that disruption of HIF‐1/HIF1 or AAK‐2/AMPK decreases the survival of isp‐1 worms grown on pathogenic bacteria (Hwang et al, 2014). In future work, it will be interesting to examine whether HIF‐1 and AAK‐2 act in concert with ATFS‐1 and the p38‐mediated innate immune signaling pathway to mediate pathogen resistance in isp‐1 worms, or whether they are acting independently of these pathways similar to DAF‐16.
In contrast to the large ATFS‐1‐dependent increase in bacterial pathogen resistance in nuo‐6 and isp‐1 mutants, constitutive activation of ATFS‐1 has only mild and variable effects on bacterial pathogen resistance in a wild‐type background which are dependent on assay conditions and specific allele tested (Soo et al, 2021). This suggests that the ability of ATFS‐1 to protect against bacterial pathogen stress may be enhanced under conditions of mitochondrial impairment (e.g., nuo‐6, isp‐1 mutants). Alternatively, there may be differences in the level or nature of the activation of ATFS‐1 between the long‐lived mitochondrial mutants and the constitutively active ATFS‐1 mutants that account for the differences in protection against bacterial pathogens.
We have previously shown that both ATFS‐1 and DAF‐16 are required for the long lifespan of nuo‐6 and isp‐1 mutants (Senchuk et al, 2018; Wu et al, 2018). Here, we show that their lifespan is also completely dependent on the p38‐mediated innate immune signaling pathway (NSY‐1, SEK‐1, PMK‐1, ATF‐7). As all of these factors are also required for the enhanced bacterial pathogen resistance in nuo‐6 and isp‐1 worms, this indicates that there are multiple conserved genetic pathways acting in parallel to upregulate innate immunity and promote longevity in response to impaired mitochondrial function.
It appears that there are multiple ways to optimize innate immune signaling with respect to longevity. As in the long‐lived mitochondrial mutants that we studied here, the p38‐mediated innate immune signaling pathway is also required for the longevity of other genes and interventions that increase lifespan. daf‐2 mutants have increased lifespan (Kenyon et al, 1993) and increased resistance to bacterial pathogens (Garsin et al, 2003). Unlike the nuo‐6 and isp‐1 mutants, daf‐2 mutants show a downregulation of many, but not all, SEK‐1‐dependent genes involved in innate immunity through decreased feeding and decreased phosphorylation of PMK‐1/p38 (Wu et al, 2019). Nonetheless, genes involved in the p38‐mediated innate immune signaling pathway (SEK‐1, PMK‐1) are required for the long lifespan, and increased resistance to bacterial pathogens in daf‐2 worms, as is DAF‐16 (Troemel et al, 2006; Zarse et al, 2012; Dues et al, 2019).
Similar to daf‐2 mutants, the increase in lifespan caused by dietary restriction resulting from decreased nutrient availability is dependent on the p38‐mediated innate immune signaling pathway (NSY‐1, SEK‐1, PMK‐1, ATFS‐7), despite the fact that dietary restriction was found to decrease PMK‐1/p38 activation and downregulate more innate immune genes (162 genes) than were upregulated (46 genes) (Wu et al, 2019). Based on this and other supporting data, it was concluded that lifespan extension resulting from dietary restriction was not mediated by pathogen resistance per se, but by a metabolic/nutrient‐related function of the p38‐mediated innate immune signaling pathway.
Combined, this clearly indicates that it is not a simple linear relationship between innate immune activation and maximum longevity. Increased lifespan can be achieved with upregulation of innate immunity genes, as in the long‐lived mitochondrial mutants, or downregulation of innate immunity genes that are nevertheless essential, as in daf‐2 mutants and dietary restriction. While a functional p38‐mediated innate immune signaling pathway may be universally required for long life, different levels of its activity are required to achieve maximum lifespan extension under different circumstances or genetic backgrounds. While many previous studies have only examined a few genes involved in innate immunity, the fact that different innate immune genes can be both upregulated and downregulated in the same mutants demonstrates the importance of looking more broadly across all genes involved in innate immunity to gain a more in‐depth understanding of how these changes are affecting longevity. Moreover, it will be important to understand the specific roles of each of these genes in pathogen defense and longevity. It will also be interesting to explore whether specific tissues are important in sensing mitochondrial impairment in order to upregulate genes involved in innate immunity, and the extent to which changes in the expression of innate immunity genes occur ubiquitously or in specific tissues.
Conclusions
Overall, this work demonstrates the importance of the p38‐mediated innate immune signaling pathway and the mitoUPR for both pathogen resistance and lifespan. It shows that the activation of innate immunity is a key function of the mitoUPR that works in concert with the p38‐mediated innate immune signaling pathway as a key defense mechanism, which is activated by mitochondrial impairment. The relationship between innate immune gene expression, pathogen resistance, and lifespan is complex, and there are multiple ways to optimize the levels of innate immune activation with respect to longevity. As all of the signaling pathways studied here are conserved up to mammals, advancing our understanding of the links between innate immune gene expression, pathogen resistance and longevity in model organisms, may advance our understanding of these processes in humans. Understanding the relationship between mitochondrial function, innate immunity, and longevity will provide important insights into fundamental aspects of how lifespan can be extended.
Materials and Methods
Strains
WT(N2)
MQ1333 nuo‐6(qm200)
JVR171 isp‐1(qm150)
T24B8.5p::GFP
JVR521 nsy‐1(ok593)
JVR520 sek‐1(km4)
JVR165 pmk‐1(km25)
ZD442 atf‐7(qd22); agIs219[T24B8.5p::GFP+ttx‐3p::GFP]
JVR455 atfs‐1(gk3094)
JVR479 nuo‐6(qm200);atfs‐1(gk3094)
QC1115 atfs‐1(et15)
QC1117 atfs‐1(et17)
JVR533 nuo‐6(qm200);nsy‐1(ok593)
JVR543 nuo‐6(qm200);sek‐1(km4)
JVR281 nuo‐6(qm200);pmk‐1(km25)
JVR527 nuo‐6(qm200);atf‐7(qd22); agIs219[T24B8.5p::GFP+ttx‐3p::GFP]
JVR532 isp‐1(qm150);nsy‐1(ok593)
JVR534 isp‐1(qm150);sek‐1(km4)
JVR538 isp‐1(qm150);atf‐7(qd22); agIs219[T24B8.5p::GFP+ttx‐3p::GFP]
math‐33(tm6724)
JVR456 isp‐1(qm150); math‐33(tm6724)
JVR457 nuo‐6(qm200); math‐33(tm6724)
daf‐16(mu86)
JVR380 isp‐1(qm150);daf‐16(mu86)
Strains were grown on OP50 bacteria at 20°C. All of the strains were genotyped or sequenced to confirm the presence of the mutation in the gene of interest. Double mutants were generated by crossing WT males to nuo‐6 or isp‐1 mutants and crossing the resulting heterozygous male progeny (nuo‐6/+ or isp‐1/+) to the mutant of interest (nsy‐1, sek‐1, pmk‐1, atf‐7). After selfing, slow‐growing progeny were singled and genotyped or sequenced to identify double mutants. We were unable to generate isp‐1 pmk‐1 and nuo‐6 daf‐16 double mutants due to the close proximity of these two genes on the same chromosome.
Bacteria pathogenesis—slow kill assay
Pathogenesis assays with P. aeruginosa strain PA14 were performed as described previously (Wu et al, 2019). In brief, the overnight PA14 culture was seeded to the center of a 35‐mm NGM agar plate containing 20 mg/l FUdR. Seeded plates were incubated at 37°C overnight and then incubated at room temperature overnight. Approximately 40‐day three adult worms were transferred to these prepared plates, and the assays were conducted at 20°C. Animals that did not respond to gentle prodding from a platinum wire were scored as dead.
Lifespan
All lifespan assays were performed at 20°C. Except where noted lifespan assays were performed on NGM plates seeded with OP50 bacteria. Lifespan assays included FUdR to limit the development of progeny. Lifespan assays on solid plates utilized 25 µM FUdR to minimize potential effects of FUdR on lifespan (Van Raamsdonk & Hekimi, 2011). Animals were excluded from the experiment if they crawled off the plate or died of internal hatching of progeny or expulsion of internal organs.
Lifespan assays with arrested bacteria
Growth‐arrested bacterial food was prepared through treatment with antibiotics and cold as described previously (Wu et al, 2019). We previously showed that there is only a very small decrease in OD600 of antibiotic‐treated, cold‐treated OP50 after 10 days (Moroz et al, 2014). Since dead bacteria are rapidly broken down leading to a decrease in OD600, this result is consistent with the bacteria being alive but not proliferating. All worms were passaged at 20°C for at least two generations before lifespan assays were initiated. Synchronized populations of L1 animals were obtained by hypochlorite treatment and then allowed to develop at 20°C on standard NGM plates seeded with OP50‐1. The young adults were transferred to NGM agar plates containing 50 mg/l ampicillin, 10 mg/l kanamycin, 1 mg/l tetracycline, 50 mg/l nystatin, and 100 mg/l FUdR and seeded with the cold‐ and antibiotic‐treated OP50‐1. Three days later, the animals were washed twice in S basal and transferred to a 24‐well plate with 1 ml S basal in each well containing the arrested OP50‐1 at OD600 3. 20–40 worms were placed in each well.
Western Blotting
Protein levels were measured by Western blotting in whole worm lysates obtained from 8 to 9 independent biological replicates. Briefly, whole worm lysates obtained from approximately 1,000 animals on day 1 of adulthood were subjected to SDS–PAGE in polyacrylamide gels (4%‐12%), immunoblotted with specific antibodies against phospho‐p38 (Cell Signaling) or total PMK‐1/p38 (Inoue et al, 2005), and visualized following standard procedures. Quantification analysis of all blots was performed with the use of ImageJ software. Results were corrected to Ponceau red staining (0.5%, w:v). See Appendix for complete images of Western blots.
Food intake assay
Food intake was quantified in liquid culture by measuring the relative amount of growth‐arrested bacteria that are present in a culture before and after incubation with C. elegans, as previously described (Wu et al, 2019). For the previous study, we compared assays for measuring food intake on solid agar media versus the liquid culture approach used in the current study to determine which method is the most robust. While both assays produced similar results, performing the food intake assay on solid agar plates was much more variable as it is challenging to scrape off all of the uneaten bacteria from solid plates in order to measure it. Since the approach of measuring food intake in liquid media produces more consistent and reliable results, we chose to use this assay for the current study.
To measure food intake, synchronized populations of L1 animals were obtained by hypochlorite treatment and allowed to develop at 20°C on standard NGM agar plates seeded with OP50. The worms were transferred onto NGM plates containing FUdR at the L4 stage of development. On day 3 of adulthood, animals were washed twice in S basal (5.85 g/l NaCl, 1 g/l K2HPO4, 6 g/l KH2PO4) and 30–50 worms were transferred to each well of a 24‐well plate containing 1 ml of arrested OP50 at optical density 3 (600 nm) re‐suspended in S basal complete medium (S basal containing 5 mg/l cholesterol, 50 mg/l ampicillin, 10 mg/l kanamycin, 1 mg/l tetracycline, 50 mg/l nystatin, FUdR). On day 12 of adulthood, bacteria were removed from the wells by washing and diluted 10× with S basal prior to measuring the optical density (600 nm). The relative food intake was determined by the change in OP50 optical density between day 3 and day 12 of adulthood and normalized to the number of worms per well.
Bacterial avoidance assay
A colony of PA14 was placed in 5 ml of Luria Broth (LB) or a colony of OP50 was placed in 5 ml 2YT, and the cultures were incubated, shaking, for 16 h at 37°C. 100 µl of the culture was seeded onto the center of 6 cm NGM plates, which were grown for 24 h at room temperature. Animals were synchronized by picking stage L4 animals the day before the experiment and were placed on OP50 plates at 20°C. The synchronized day 1 animals were washed off of the OP50 plates with M9 buffer and washed three times in M9 buffer, and then, ∼40 animals were deposited on each of the assay plates using a glass pipette. The animals were placed on the agar ∼1 cm from the edge of the bacterial spots. The plates were then placed at 20°C. The number of animals on and off of the bacteria were counted after 24 h.
Quantification of reporter fluorescence
T24B8.5p::GFP animals were randomly selected and imaged with ZEN 2012 software on an Axio Imager M2 microscope with a 10×/0.25 objective (Zeiss, Jena, Germany). Fluorescence brightness was quantified blindly using National Institutes of Health (NIH) ImageJ software.
RNA isolation
mRNA was collected from pre‐fertile young adult worms using TRIzol as previously described (Schaar et al, 2015). For quantitative real‐time RT–PCR, we collected three biological replicates for each strain on separate days. For RNA sequencing experiments, we collected 3‐6 biological replicates.
Quantitative Real‐Time RT–PCR
The mRNA was converted to cDNA using a High‐Capacity cDNA Reverse Transcription Kit (Life Technologies/Invitrogen) according to the manufacturer’s directions. qPCR was performed using a FastStart Universal SYBR Green kit (Roche) in a Bio‐Rad iCycler real‐time PCR detection system (Machiela et al, 2016). Primer sequences for other genes tested include the following:
T24B8.5 (TACACTGCTTCAGAGTCGTG, CGACAACCACTTCTAACATCTG);
K08D8.5 (CAAAATATCCTCCGGGAAGTC, TTCACGGAATCACCATCGTA);
F55G11.8 (GGAAATGGTTGCAAACTTGG, TGCAGAATCGACAGTTTGGA);
clec‐67 (TTTGGCAGTCTACGCTCGTT, CTCCTGGTGTGTCCCATTTT);
clec‐65 (GCAATCAACCTCGTGATGTG, CGCAGAAGCAGTTTGTATCC);
dod‐22 (TCCAGGATACAGAATACGTACAAGA, GCCGTTGATAGTTTCGGTGT);
Y9C9A.8 (CGGGGATATAACTGATAGAATGG, CAAACTCTCCAGCTTCCAACA); and
C32H11.4 (GGCAATACTTGCAAGACTGGAA, CCAGCTACACGATTGGTCCT).
RNA sequencing and bioinformatic analysis
RNA sequencing was performed as previously described (Dues et al, 2017). RNA‐seq data are available on NCBI GEO: GSE93724 (Senchuk et al, 2018) and GSE110984 (Wu et al, 2018) and were analyzed by the Harvard School of Public Health Bioinformatics core for this paper.
Read mapping and expression level estimation
All samples were processed using an RNA‐seq pipeline implemented in the bcbio‐nextgen project (https://bcbio‐nextgen.readthedocs.org/en/latest/). Raw reads were examined for quality issues using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to ensure library generation, and sequencing data were suitable for further analysis. If necessary, adapter sequences, other contaminant sequences such as polyA tails, and low‐quality sequences were trimmed from reads using cutadapt https://cutadapt.readthedocs.org/. Trimmed reads were aligned to the Ensembl build WBcel235 (release 90) of the C. elegans genome using STAR (Dobin et al, 2013). Quality of alignments was assessed by checking for evenness of coverage, rRNA content, genomic context of alignments (e.g., alignments in known transcripts and introns), complexity, and other quality checks. Expression quantification was performed with Salmon (Patro et al, 2017) to identify transcript‐level abundance estimates and then collapsed down to the gene level using the R Bioconductor package tximport (Soneson et al, 2015). Principal components analysis (PCA) and hierarchical clustering methods validated clustering of samples from the same batches and across different mutants.
Differential gene expression and functional enrichment analysis
Differential expression was performed at the gene level using the R Bioconductor package DESeq2 (Love et al, 2014). For each wild‐type mutant, comparison significant genes were identified using an FDR threshold of 0.01. For datasets in which experiments were run across two batches, we included batch as a covariate in the linear model.
Overlap table
Lists of differentially expressed genes were separated by direction of expression change and compared to genes that are modulated in response to exposure to the bacterial pathogen P. aeruginosa strain PA14 in a PMK‐1‐ and ATF‐7‐dependent manner, as identified by Fletcher et al (2019a). Significance of overlap was computed using the hypergeometric test.
Heatmaps
For each dataset (long‐lived mutant comparison to wild type), the raw counts were regularized log (rlog)‐transformed using the DESeq2 package (Love et al, 2014). This transformation moderates the variance across the mean, improving the clustering in an unbiased manner. The rlog matrix was then subset to retain only those genes from the Fletcher et al gene lists which were also significantly differentially expressed between mutant and wild type. Heatmaps were plotted using the pheatmap package in R.
Localization of predicted binding sites for ATFS‐1 and ATF‐7 in innate immunity genes
To identify possible binding regions for ATF‐7 and ATFS‐1, we analyzed the previously reported binding regions for ATFS‐1 and ATF‐7 (Nargund et al, 2015a; Fletcher et al, 2019a). We searched for possible binding sites that matched the position‐weighted matrices (PWMs) of the respective transcription factors using the FIMO software from the MEME Suite (Bailey et al, 2009). We recreated the PWM for ATFS‐1 from the published ATFS‐1 ChIP‐seq binding data as previously described (Nargund et al, 2015a) and used the published PWM for ATF‐7 (MA1438.1) (Fletcher et al, 2019a).
Statistical analysis
Experiments were conducted such that the experimenter was blinded to the genotype of the animals being tested. The worms for each experiment were randomly selected from maintenance plates at stages when all worms appeared healthy. Except where noted, we completed a minimum of three biological replicates for each assay (independent population of worms tested on a different day). Statistically significance of differences between groups of more than two was determined by one‐way ANOVA with Dunnett’s multiple comparison post hoc test or two‐way ANOVA with Bonferroni post‐doc test using GraphPad Prism. For survival analysis, a log‐rank test was used. On all bar graphs, the bar indicates the average (mean) and error bars indicate standard error of the mean (SEM). This study was not pre‐registered. No sample size calculations were performed. This study did not include a pre‐specified primary endpoint.
Author contributions
JVR and TKB conceptualized the study. JCC, ZW, PDR, SKS, MM, TKB, and JVR contributed to methodology. JCC, ZW, PDR, SKS, MM, and JVR investigated the study. JCC, ZW, PDR, SKS, MM, and JVR visualized the study. JVR wrote—original draft. JCC, ZW, PDR, SKS, MM, TKB, and JVR wrote—review and editing. JCBF, TKB, and JVR supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View and Appendix
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 4 and 7
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (CIHR; http://www.cihr‐irsc.gc.ca/; JVR; Application 399148 and 416150), the Natural Sciences and Engineering Research Council of Canada (NSERC; https://www.nserc‐crsng.gc.ca/index_eng.asp; JVR; Application RGPIN‐2019‐04302), and the National Institute of General Medical Sciences (NIGMS; https://www.nigms.nih.gov/; JVR; Grant number R01 GM121756). JVR is the recipient of a Senior Research Scholar career award from the Fonds de Recherche du Québec Santé (FRQS) and Parkinson Quebec. SKS received a scholarship from FRQS. PDR received a fellowship award from FRQS. TKB, JCC, and ZW were supported by research (R35 GM122610, R01 AG054215) and center (P30 DK036836) grants from the NIH. JCC was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2019/18444‐9). ZW was sponsored by Shanghai Pujiang Program (20PJ1405400). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P30 OD010440). We would also like to acknowledge the C. elegans knockout consortium and the National Bioresource Project of Japan for providing strains used in this research.
EMBO reports (2021) 22: e52964.
Footnotes
Correction added on the 6th of December 2021, after first online publication: the grant number has been added.
Contributor Information
T Keith Blackwell, Email: keith.blackwell@joslin.harvard.edu.
Jeremy M Van Raamsdonk, Email: jeremy.vanraamsdonk@mcgill.ca.
Data availability
RNA‐seq data have been deposited on NCBI GEO: GSE93724 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE93724), GSE110984 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110984). All other data and strains generated in the current study are included with the manuscript or available from the corresponding author on request. Source data for stress assays and lifespan are available online.
References
- Apfeld J, Kenyon C (1999) Regulation of lifespan by sensory perception in Caenorhabditis elegans . Nature 402: 804–809 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruah A, Chang H, Hall M, Yuan J, Gordon S, Johnson E, Shtessel LL, Yee C, Hekimi S, Derry WB et al (2014) CEP‐1, the Caenorhabditis elegans p53 homolog, mediates opposing longevity outcomes in mitochondrial electron transport chain mutants. PLoS Genet 10: e1004097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman JR, Kenyon C (2006) Germ‐cell loss extends C. elegans life span through regulation of DAF‐16 by kri‐1 and lipophilic‐hormone signaling. Cell 124: 1055–1068 [DOI] [PubMed] [Google Scholar]
- Block DH, Twumasi‐Boateng K, Kang HS, Carlisle JA, Hanganu A, Lai TY, Shapira M (2015) The Developmental intestinal regulator ELT‐2 controls p38‐dependent immune responses in adult C. elegans . PLoS Genet 11: e1005265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikka MR, Anbalagan C, Dvorak K, Dombeck K, Prahlad V (2016) The mitochondria‐regulated immune pathway activated in the C. elegans intestine is neuroprotective. Cell Rep 16: 2399–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L (2001) Extension of life‐span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292: 104–106 [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW (2009) Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol 19: 1591–1598 [DOI] [PubMed] [Google Scholar]
- Dell'agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M (2007) Increased longevity and refractoriness to Ca(2+)‐dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet 16: 431–444 [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes‐Oliveira N, Lehrer‐Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Rates of behavior and aging specified by mitochondrial function during development. Science 298: 2398–2401 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dues DJ, Andrews EK, Schaar CE, Bergsma AL, Senchuk MM, Van Raamsdonk JM (2016) Aging causes decreased resistance to multiple stresses and a failure to activate specific stress response pathways. Aging 8: 777–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dues DJ, Andrews EK, Senchuk MM, Van Raamsdonk JM (2019) Resistance to stress can be experimentally dissociated from longevity. J Gerontol A Biol Sci Med Sci 74: 1206–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dues DJ, Schaar CE, Johnson BK, Bowman MJ, Winn ME, Senchuk MM, Van Raamsdonk JM (2017) Uncoupling of oxidative stress resistance and lifespan in long‐lived isp‐1 mitochondrial mutants in Caenorhabditis elegans . Free Radic Biol Med 108: 362–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bussiere F, Hekimi S (2001) Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans . Dev Cell 1: 633–644 [DOI] [PubMed] [Google Scholar]
- Flachsbart F, Dose J, Gentschew L, Geismann C, Caliebe A, Knecht C, Nygaard M, Badarinarayan N, ElSharawy A, May S et al (2017) Identification and characterization of two functional variants in the human longevity gene FOXO3. Nat Commun 8: 2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher M, Tillman EJ, Butty VL, Levine SS, Kim DH (2019a) Global transcriptional regulation of innate immunity by ATF‐7 in C. elegans . PLoS Genet 15: e1007830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher M, Tillman EJ, Butty VL, Levine SS, Kim DH (2019b) Gene Expression Omnibus GSE119293 [DATASET] https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE119293
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M et al (2007) Inflammaging and anti‐inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev 128: 92–105 [DOI] [PubMed] [Google Scholar]
- Friedman DB, Johnson TE (1988) A mutation in the age‐1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 118: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat‐shock factor and bacterial proliferation. Genetics 161: 1101–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM (2003) Long‐lived C. elegans daf‐2 mutants are resistant to bacterial pathogens. Science 300: 1921 [DOI] [PubMed] [Google Scholar]
- Gems D, Riddle DL (2000) Genetic, behavioral and environmental determinants of male longevity in Caenorhabditis elegans . Genetics 154: 1597–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14: 463–477 [DOI] [PubMed] [Google Scholar]
- Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA (1999) Phylogenetic perspectives in innate immunity. Science 284: 1313–1318 [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y (2003) IGF‐1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421: 182–187 [DOI] [PubMed] [Google Scholar]
- Hourihan JM, Moronetti Mazzeo LE, Fernandez‐Cardenas LP, Blackwell TK (2016) Cysteine sulfenylation directs IRE‐1 to activate the SKN‐1/Nrf2 antioxidant response. Mol Cell 63: 553–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang AB, Ryu EA, Artan M, Chang HW, Kabir MH, Nam HJ, Lee D, Yang JS, Kim S, Mair WB et al (2014) Feedback regulation via AMPK and HIF‐1 mediates ROS‐dependent longevity in Caenorhabditis elegans . Proc Natl Acad Sci USA 111: E4458–E4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK, Matsumoto K (2005) The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN‐1 in oxidative stress response. Genes Dev 19: 2278–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irazoqui JE, Urbach JM, Ausubel FM (2010) Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol 10: 47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong DE, Lee D, Hwang SY, Lee Y, Lee JE, Seo M, Hwang W, Seo K, Hwang AB, Artan M et al (2017) Mitochondrial chaperone HSP‐60 regulates anti‐bacterial immunity via p38 MAP kinase signaling. EMBO J 36: 1046–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovaisaite V, Mouchiroud L, Auwerx J (2014) The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol 217: 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE et al (2014) Geroscience: linking aging to chronic disease. Cell 159: 709–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ (2010) The genetics of ageing. Nature 464: 504–512 [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366: 461–464 [DOI] [PubMed] [Google Scholar]
- Kim DH, Ewbank JJ (2018) Signaling in the innate immune response. WormBook 2018: 1–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka‐Hino M, Hisamoto N, Matsumoto K, Tan MW et al (2002) A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297: 623–626 [DOI] [PubMed] [Google Scholar]
- Kimbrell DA, Beutler B (2001) The evolution and genetics of innate immunity. Nat Rev Genet 2: 256–267 [DOI] [PubMed] [Google Scholar]
- Kirienko NV, Cezairliyan BO, Ausubel FM, Powell JR (2014) Pseudomonas aeruginosa PA14 pathogenesis in Caenorhabditis elegans . Methods Mol Biol 1149: 653–669 [DOI] [PubMed] [Google Scholar]
- Klass MR (1977) Aging in the nematode Caenorhabditis elegans: major biological and environmental factors influencing life span. Mech Ageing Dev 6: 413–429 [DOI] [PubMed] [Google Scholar]
- Labzin LI, Heneka MT, Latz E (2018) Innate immunity and neurodegeneration. Annu Rev Med 69: 437–449 [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S (1996) Determination of life‐span in Caenorhabditis elegans by four clock genes. Science 272: 1010–1013 [DOI] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G (2003) A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33: 40–48 [DOI] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S (2005) Evolutionary conservation of the clk‐1‐dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev 19: 2424–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela E, Dues DJ, Senchuk MM, Van Raamsdonk JM (2016) Oxidative stress is increased in C. elegans models of Huntington's disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol Dis 96: 1–11 [DOI] [PubMed] [Google Scholar]
- Moroz N, Carmona JJ, Anderson E, Hart AC, Sinclair DA, Blackwell TK (2014) Dietary restriction involves NAD ‐dependent mechanisms and a shift toward oxidative metabolism. Aging Cell 13: 1075–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkacsy E, Khan MH, Lane RK, Borror MB, Park JH, Bokov AF, Fisher AL, Link CD, Rea SL (2016) DLK‐1, SEK‐3 and PMK‐3 are required for the life extension induced by mitochondrial bioenergetic disruption in C. elegans . PLoS Genet 12: e1006133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of DAF‐16 to influence the lifespan of Caenorhabditis elegans . Nature 424: 277–283 [DOI] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM (2015a) Mitochondrial and nuclear accumulation of the transcription factor ATFS‐1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 58: 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM (2015b) Gene Expression Omnibus GSE63803 [DATASET] https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63803
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM (2012) Mitochondrial import efficiency of ATFS‐1 regulates mitochondrial UPR activation. Science 337: 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods 14: 417–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM (2014) Mitochondrial UPR‐regulated innate immunity provides resistance to pathogen infection. Nature 516: 414–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauthan M, Ranji P, Aguilera Pradenas N, Pitot C, Pilon M (2013) The mitochondrial unfolded protein response activator ATFS‐1 protects cells from inhibition of the mevalonate pathway. Proc Natl Acad Sci USA 110: 5981–5986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar CE, Dues DJ, Spielbauer KK, Machiela E, Cooper JF, Senchuk M, Hekimi S, Van Raamsdonk JM (2015) Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet 11: e1004972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senchuk MM, Dues DJ, Schaar CE, Johnson BK, Madaj ZB, Bowman MJ, Winn ME, Van Raamsdonk JM (2018) Activation of DAF‐16/FOXO by reactive oxygen species contributes to longevity in long‐lived mitochondrial mutants in Caenorhabditis elegans . PLoS Genet 14: e1007268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivers RP, Pagano DJ, Kooistra T, Richardson CE, Reddy KC, Whitney JK, Kamanzi O, Matsumoto K, Hisamoto N, Kim DH (2010) Phosphorylation of the conserved transcription factor ATF‐7 by PMK‐1 p38 MAPK regulates innate immunity in Caenorhabditis elegans . PLoS Genet 6: e1000892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, Robinson MD (2015) Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000Res 4: 1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo SK, Traa A, Rudich PD, Mistry M, Van Raamsdonk JM (2021) Mitochondrial unfolded protein response transcription factor ATFS‐1 increases resistance to exogenous stressors through upregulation of multiple stress response pathways. Life Sci Alliance 4: e202101182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntichaki P, Troulinaki K, Tavernarakis N (2007) eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans . Nature 445: 922–926 [DOI] [PubMed] [Google Scholar]
- Tan MW, Mahajan‐Miklos S, Ausubel FM (1999a) Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci USA 96: 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MW, Rahme LG, Sternberg JA, Tompkins RG, Ausubel FM (1999b) Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc Natl Acad Sci USA 96: 2408–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper RG, Ashraf J, Kaletsky R, Kleemann G, Murphy CT, Bussemaker HJ (2013) PQM‐1 complements DAF‐16 as a key transcriptional regulator of DAF‐2‐mediated development and longevity. Cell 154: 676–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH (2006) p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans . PLoS Genet 2: e183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S (2011) FUdR causes a twofold increase in the lifespan of the mitochondrial mutant gas‐1. Mech Ageing Dev 132: 519–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S (2012) Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci USA 109: 5785–5790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Baruah A, Chang HW, Pace HM, Lee SS (2011) The homeobox protein CEH‐23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans . PLoS Biol 9: e1001084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A, Boutis P, Hekimi S (1995) Mutations in the clk‐1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139: 1247–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Isik M, Moroz N, Steinbaugh MJ, Zhang P, Blackwell TK (2019) Dietary restriction extends lifespan through metabolic regulation of innate immunity. Cell Metab 29: 1192–1205.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Senchuk MM, Dues DJ, Johnson BK, Cooper JF, Lew L, Machiela E, Schaar CE, DeJonge H, Blackwell TK et al (2018) Mitochondrial unfolded protein response transcription factor ATFS‐1 promotes longevity in a long‐lived mitochondrial mutant through activation of stress response pathways. BMC Biol 16: 147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi S (2010a) A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans . PLoS Biol 8: e1000556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi S (2010b) Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans . Aging Cell 9: 433–447 [DOI] [PubMed] [Google Scholar]
- Yee C, Yang W, Hekimi S (2014) The intrinsic apoptosis pathway mediates the pro‐longevity response to mitochondrial ROS in C. elegans . Cell 157: 897–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman MJ, Rogers ZN, Kim DH (2011) A decline in p38 MAPK signaling underlies immunosenescence in Caenorhabditis elegans . PLoS Genet 7: e1002082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, Ristow M (2012) Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L‐proline catabolism to induce a transient ROS signal. Cell Metab 15: 451–465 [DOI] [PMC free article] [PubMed] [Google Scholar]