

Abstract
The tumor immune microenvironment is influenced by the epigenetic landscape of the tumor. Here, we have identified the SETDB1–TRIM28 complex as a critical suppressor of antitumor immunity. An epigenetic CRISPR-Cas9 screen of 1,218 chromatin regulators identified TRIM28 as a suppressor of PD-L1 expression. We then revealed that expression of the SETDB1–TRIM28 complex negatively correlated with infiltration of effector CD8+ T cells. Inhibition of SETDB1–TRIM28 simultaneously upregulated PD-L1 and activated the cGAS–STING innate immune response pathway to increase infiltration of CD8+ T cells. Mechanistically, SETDB1–TRIM28 inhibition led to micronuclei formation in the cytoplasm, which is known to activate the cGAS–STING pathway. Thus, SETDB1–TRIM28 inhibition bridges innate and adaptive immunity. Indeed, SETDB1 knockout enhanced the antitumor effects of immune checkpoint blockade with anti–PD-L1 in a mouse model of ovarian cancer in a cGAS-dependent manner. Our findings establish the SETDB1–TRIM28 complex as a regulator of antitumor immunity and demonstrate that its loss activates cGAS–STING innate immunity to boost the antitumor effects of immune checkpoint blockade.
Keywords: Epigenetics, Ovarian cancer, Immune checkpoint blockade, SETDB1, cGAS
Introduction
Immunotherapies such as immune checkpoint blockade (ICB) using monoclonal antibodies targeting the PD-1/PD-L1 axis show striking clinical benefit in several cancer types. Despite this advance, the majority of cancers show low response rates to ICB (1). Thus, new therapeutic strategies are urgently needed to expand the utility of ICB in the treatment of cancer. Combining ICB with other therapeutic approaches is an effective strategy to overcome this challenge (2). For example, genetic alterations that increase PD-L1 expression often predict a better response to anti–PD-1/PD-L1 therapy (3). Likewise, therapeutics that enhance PD-L1 expression typically synergize with anti–PD-1/PD-L1 therapy (4). Chromatin regulators are of emerging interest in this regard because they have a key role in shaping antitumor immunity through cell-intrinsic mechanisms (5,6).
Pathways that regulated host innate immunity, such as the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway, play an important role in antitumor immunity and represent an attractive target to boost therapeutic responses to immunotherapies, including ICB (7). cGAS is a cytosolic nucleotidyltransferase that binds double-stranded DNA in a sequence-nonspecific manner (8,9). For example, cGAS senses cytosolic DNA generated by unrepaired DNA lesions and/or mitosis defects during the G2/M phase of the cell cycle; such DNA is often in the form of micronuclei (10,11). Micronuclei are typically positive for both cGAS and DNA damage markers such as γH2AX (10,11). cGAS activation generates the cyclic dinucleotide cyclic GMP–AMP, which in turn triggers a type I IFN response via the adaptor protein STING (12). Activation of cGAS–STING signaling by micronuclei leads to upregulation of unique type I IFN response genes, including CCL5 and CXCL10 (10).
SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) is a protein lysine methyltransferase that methylates histone H3 at lysine 9 to transcriptionally silence expression of its target gene (13). SETDB1 is a component of the human silencing hub (HUSH) complex that includes tripartite motif–containing 28 (TRIM28; also known as KAP1) (14). TRIM28 plays a key role in the recruitment of SETDB1 to specific genomic loci, and both SETDB1 and TRIM28 are implicated as oncogenes in several cancer types (15,16). For example, SETDB1 overexpression correlates with a poor prognosis in non-small cell lung cancer, colon cancer, acute myeloid leukemia and melanoma (17–22). Likewise, TRIM28 overexpression predicts poor prognosis in gastric cancer, ovarian cancer, glioma and hepatocellular carcinoma (23–28). The SETDB1–TRIM28 complex plays a pivotal role in silencing of transposable elements (TEs). Indeed, a recent study shows that SETDB1 loss derepresses latent TE-derived regulatory elements, immunostimulatory genes, and TE-encoded retroviral antigens in these regions, and triggers TE-specific cytotoxic T–cell responses (29). However, the role of SETDB1 in regulating responses to ICB and its underlying mechanism has never been investigated. Here, we show that loss of the SETDB1–TRIM28 complex synergizes with ICB through a two-pronged mechanism: simultaneously upregulating PD-L1 expression and enhancing infiltration of effector CD8+ T cells by activating the cGAS–STING innate immune pathway.
Materials and Methods
Cell Culture, transfection and reagents
ID8 (RRID:CVCL_VA22), UPK10 (gift from Dr. J. Conejo-Garcia, Moffitt Cancer Center) and HEK293FT (RRID:CVCL_0045) cells were cultured in DMEM (CORNING, #10-013-CM) supplemented with 10% fetal bovine serum (FBS, R&D Systems, #S11510) and 1% penicillin/streptomycin (CORNING, #30-002-CI) at 37°C with 5% CO2. The human high-grade serous ovarian cancer (HGSOC) cell lines A1847 (RRID:CVCL_9724), PEO4 (RRID:CVCL_2690), OVSAHO (RRID:CVCL_3114), OVCAR10 (RRID:CVCL_4377), OVCAR5 (RRID:CVCL_1628), OVCAR4 (RRID:CVCL_1627), OVCAR3 (RRID:CVCL_0465), CAOV3 (RRID:CVCL_0201), COV362 (RRID:CVCL_2420), COV318 (RRID:CVCL_2419), EF027 (obtained from Dr. Gordon Mills’ laboratory, MD Anderson Cancer Center), Kuramochi (RRID:CVCL_1345) were cultured in RPMI1640 (CORNING, #10-040-CM) supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO2. The human fallopian tube epithelial cells FT246 (RRID:CVCL_UH61) and FT237 (RRID:CVCL_UH59) (30) were gifts from Dr. Ronny Drapkin at the University of Pennsylvania, and they were grown in DMEM/F12 (CORNING, #10-092-CM) with 10% FBS. All the cell lines were authenticated at The Wistar Institute Genomics Facility using short tandem repeat DNA profiling. Mycoplasma testing was performed using LookOut Mycoplasma PCR detection (Millipore Sigma, #MP0035-1KT) every month. Transfection was performed using Lipofectamine 2000 (Invitrogen, #11668027) following the manufacturer’s instructions (see Lentivirus infection below). ID8 cells were treated with 2.5 μg/ml RU.521 (Invitrogen, #inh-ru521) for 48 h to inhibit cGAS activity. To activate the cGAS–STING pathway, ID8 cells were transfected with 10 μg IFN stimulatory DNA (ISD) (InvivoGen, #tlrl-isdn) in a 6 cm-dish using lipofectamine 2000 at a ratio of 1 μl lipofectamine to 1 μg DNA following the manufacturer’s instructions.
Epigenetic CRISPR/Cas9 screen
A previously published library of single guide RNAs (sgRNA) targeting 1,218 genes that encode chromatin regulators was obtained from Dr. Kristian Helin (31). ID8 cells were first transduced by adding filtered DMEM medium (CORNING, #10-013-CM) containing Cas9-expressing lentivirus using a Lenti-EF1a-Cas9-2A-Blast construct (Addgene, #52962, RRID:Addgene_52962). Polybrene (10 μg/mL) (Santa Cruz, #sc-134220) was added to enhance lentiviral injection. A single stable Cas9-expressing ID8 clone was selected by seeding single cells into 96-well to grow into clones. 48 × 106 Cas9-expressing ID8 cells were transduced by adding a pre-determined amount of filtered DMEM medium containing virus expressing the mouse epigenetic gDNA library at a MOI of 0.3 to achieve a library representation of 1000 × (see Lentivirus infection below). The established cell library was then treated with 30 ng/ml IFNγ (STEMCELL, #78021) for 24 h and stained with fluorochrome-conjugated anti–PD-L1 (Biolegend, #124312, RRID:AB_10612741) and sorted with flow cytometry (see Flow cytometry analysis below). The top 15% of PD-L1–expressing cells were sorted using flow cytometry. Unsorted bulk cells were used as internal controls.
Genomic DNA was extracted from the PD-L1high cells and unsorted control cells using salt precipitation. In brief, cell pellets (1–5 × 107 cells) were suspended in 6 ml gDNA lysis buffer (50 mM EDTA, 1% SDS, 50 mM Tris pH 8.0) and 30 μl 20 mg/ml proteinase K (Fisher Scientific, #25-530-049) was added before the mixture was incubated at 55 °C overnight. Then, 30 μl 10 mg/ml RNase A (Thermo Fisher, #EN0531) was added and the mixture incubated at 37 °C for 30 min, before cooling on ice. Protein was precipitated by adding 2 ml 7.5 M stock solution of Ammonium Acetate and centrifuging at > 4000 g for 10 min. The supernatant containing genomic DNA was collected and precipitated by adding 6 ml 100% isopropanol and centrifuging at > 4000 g for 10 min. The DNA pellet was washed with 70% ethanol, air dried and dissolved in H2O for PCR amplification.
Library was constructed by a two-step PCR amplification. The first PCR was performed in 8 PCR reactions each containing 5 μg gDNA, 1.5 μl 10 μM forward and reverse primers (see below for the sequences), 50 μl NEBNext Q5 HotStart HiFi PCR Mastermix (NEB, #M0543L) for 15 cycles of 98 °C, 20 s; 60 °C, 30 s; 65 °C, 45°C using a Bio-RAD T100 thermal cycler PCR machine (#1861096). The first PCR product from the 8 reactions for each sample was pooled together and purified by AMPure XP beads (Beckman Coulter, #A63880) following the manufacturer’s instructions and suspended in 800 μl H2O. To barcode the samples, for each sample, three second PCR reactions containing 10 μl first PCR product, 1μl 10 μM forward and reverse Illumina primers, 50 μl NEBNext Q5 HotStart HiFi PCR Mastermix for 20 cycles of 98 °C, 20 s; 60 °C, 30 s; 65 °C, 45°C. The final PCR product was purified by AMPure XP beads (Beckman Coulter) following the manufacturer’s instructions and sequenced in a 75-base pair single-end run on the Next Seq 500 (Illumina) at the Wistar Genomic facility. The following primers were used: 1st PCR forward: 5’-AATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCG-3’; 1st PCR reverse: 5’-TCTACTATTCTTTCCCCTGCACTGTTGTGGGCGATGTGCGCTCTG-3’; 2nd PCR Universal P7 primer: 5’-CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTCCTTGGTTCTACTATTCTTTCCCCTGCACTGT-3’; 2nd PCR P5 primer for total unsorted sample: 5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTtAAGTAGAGTCTTGTGGAAAGGACGAAACACCG-3’; 2nd PCR P5 primer for PD-L1 high sample: 5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTATACACGATCTCTTGTGGAAAGGACGAAACACCG-3’; 2nd PCR P5 primer for PD-L1 low sample: 5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATCGCGCGGTTCTTGTGGAAAGGACGAAACACCG-3’.
CRISPR-mediated knockouts
pLentiCRISPR v2 (Addgene #52961, RRID:Addgene_52961) and pLentiCRISPR v2-blast (Addgene #83480, RRID:Addgene_83480) were digested with BsmBI (NEB, #R0739) at 55°C for 1h and run on a 1% agarose gel. The digested plasmid was cut out and purified using QIAquick gel extraction kit (QIAGEN, #166047244). Each pair of oligos were phosphorylated using T4 PNK (M0201S) in T4 ligation buffer (New England Biolabs, #B0202S) and annealed in a thermocycler at 37°C for 30 min, 95°C for 5 min, ramped down to 25°C at 5°C/min. Annealed oligonucleotides were diluted 1:200 in RNase/DNase-free water. Ligation of the annealed oligonucleotide and digested pLentiCRISPR v2 plasmid was performed using Quick Ligase (New England Biolabs, #M2200). The following oligonucleotides were used for cloning: mouse cGas gRNA (5’- CGAGGCGCGGAAAGTCGTAA-3’), mouse Setdb1 gRNA#1 (5’-ACTATTGCAACTCAACCACG-3’), mouse Setdb1 gRNA#2 (5’- ACTCTGGCGCCCGACCGCAA-3’), mouse Trim28 gRNA#1 (5’- ATCCCCCGAATTATTCGCTG-3’), mouse Trim28 gRNA#2 (5’- CGCCGCAGCGAATAATTCGG-3’).
Lentivirus infection
HEK293FT cells were transfected with target plasmids (see below), packaging plasmids psPAX2 (Addgene #12260, RRID:Addgene_12260) and pCMV-VSV (Addgene #8454, RRID:Addgene_8454) using Lipofectamine 2000 at a ratio of 1 μl lipofectamine to 1 μg DNA for 6 h a, after which the medium was replaced. Lentivirus was harvested and filtered through a 0.45 μm filter 72h post transfection. The filtered medium was then used to infect cells for 48 h by culturing cells using virus-containing medium and fresh medium at a 1:1 ratio. After infection, the cells were selected in medium contains 1 μg/ml puromycin for one week. The following target short-hairpin RNA (shRNA)-expressing plasmids (available from Sigma-Aldrich) were obtained from the Molecular Screening Facility of Wistar Institute were used: pLKO.1-shTrim28 (mouse, TRCN0000071363 and TRCN0000071364), pLKO.1-shTRIM28 (human, TRCN0000017998 and TRCN0000018001), pLKO.1-shMavs (mouse, TRCN0000124771), pLKO.1-shSETDB1 (human, TRCN0000148112 and TRCN0000276169).
Immunoblots
Cells were trypsinized and washed two times with PBS. Protein was extracted by incubation with RIPA lysis buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS and 1 mM phenylmethylsulfonyl fluoride (PMSF)] on ice for 30 min. Protein concentration was measured by the BCA assay (Pierce, #23225). Samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membrane (Millipore). Membranes were blocked with 4% BSA/TBS-T and then incubated with primary antibodies and then secondary antibodies (Cell Signaling, #7074S). The following primary antibodies were used: mouse anti-β-actin (Sigma, #A5316), rabbit anti-TRIM28 (Abcam, #ab10484, RRID:AB_297223), mouse anti-SETDB1(Santa Cruz, #sc-271553, RRID:AB_10649961), rabbit anti-tubulin (CST, #2125S, RRID:AB_2619646), rabbit anti-γ-H2AX (CST, #9718S, AB_2118009), rabbit anti–PD-L1 (CST, #13684S, RRID:AB_2687655), rabbit anti-cGAS (CST, # 31659S, RRID:AB_2799008), anti-STING (CST, #13647, RRID:AB_2732796), and anti-phospho IRF3 (CST, #4947S, RRID:AB_823547).
Quantitative reverse-transcriptase PCR (qRT-PCR)
Total RNA from three replicates was extracted using RNeasy Kit (Qiagen, #74104) following the manufacturer’s instructions. 1 μg of purified RNA was used for reverse-transcriptase PCR (RT-PCR) with High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher, #4374967). Quantitative PCR (qPCR) was performed using iTaq Universal SYBR Green Supermix (Bio-Rad, #1725121) and run on QuantStudio 3 Real-Time PCR System. Gene expression fold change was calculated using the 2^-ΔΔCT. The sequences of the primers used for RT-qPCR are as follows: mCd274 forward: 5’-GCATTATATTCACAGCCTGC-3’ and reverse: 5’-CCCTTCAAAAGCTGGTCCTT-3’; mCxcl10 forward: 5’-TCAGCACCATGAACCCAAG-3’ and reverse: 5’-CTATGGCCCTCATTCTCACTG-3’; mCcl5 forward: 5’-CCACTTCTTCTCTGGGTTGG-3’ and reverse: 5’-GTGCCCACGTCAAGGAGTAT-3’; mMavs forward: 5’-GCTCCTTGGTCTCAGAACCC-3’ and reverse: 5’-CTGGGGCTTTCGTCTACCTG-3’; mTrim28 forward: 5’-CGCTCACAAGGACCATCAGT-3’ and reverse: 5’-AGCTTCGAACCTCCTTGGTG-3’; hTRIM28 forward: 5’-GGAAGGCTATGGCTTTGGGT-3’ and reverse: 5’-GGGAAGACCTTGAAGACGGG-3’; hCXCL10 forward: 5’-TGCCATTCTGATTTGCTGCC-3’ and reverse: 5’-TGCAGGTACAGCGTACAGTT-3’; hCCL5 forward: 5’-CGTGCCCACATCAAGGAGTA-3’ and reverse: 5’-TCGGGTGACAAAGACGACTG-3’; hGAPDH forward: 5’-GTCTCCTCTGACTTCAACAGCG-3’ and reverse: 5’-ACCACCCTGTTGCTGTAGCCAA-3’; mActb forward: 5’-CTCCTATGTGGGTGACGAGG-3’ and reverse: 5’-ACGGTTGGCCTTAGGGTTC-3’. Human GADPH and mouse Actb were used as an internal control to normalize the expression in human and mouse cells, respectively.
RNA sequencing (RNA-seq)
Total RNA of control ID8 and two Setdb1 knockout ID8 cell lines was extracted using RNeasy mini Kit (Qiagen, #74106) according to the manufacturer’s instructions and digested with DNase I (Qiagen, #79254). RNA-seq libraries were constructed using ScriptSeq complete Gold kit (Epicentre, #SCL24EP) and subjected to a 75 bp paired-end sequencing run on NextSeq 500, using Illumina’s NextSeq 500 high output sequencing kit (#20024906) following the manufacturer’s instructions.
Cell proliferation assay
For cell proliferation analysis, 20,000 control or Setdb1-knockout ID8 cells were seeded into a 6 cm-dish. Cell number was counted every two days by hemocytometer. Three repeats were performed, and P values were calculated using a two-way ANOVA.
Cell cycle analysis
For cell-cycle analysis, control and Setdb1-knockout ID8 cells were suspended in 500 μl PBS and fixed by gently adding 5 ml cold 70% ethanol dropwise. Cells were then washed twice with PBS and stained by adding 1 ml propidium iodide (PI) solution [3.8mM sodium citrate, 50 μg/ml PI (Invitrogen, #P3566) in PBS] and 50 μl RNase A stock solution (Fisher scientific, # FEREN0531) at room temperature for 30 min. PI staining was detected using a Becton-Dickinson LSR18 machine, and analyzed with FlowJo version 7 software (Tree Star, Inc.)
Flow cytometry analysis
For immune infiltration analysis, surgically removed tumors were minced into small (1 to 2 mm) pieces and digested with 1 mg/mL collagenase IV (Sigma-Aldrich, #C5138), 0.1 mg/mL Hyaluronidase (Sigma-Aldrich, #H6254) and 0.01 mg/mL deoxyribonuclease I (Sigma-Aldrich, #D5025). The cells were sequentially filtered through 40 μm cell strainers. Analysis of tumor-infiltrating lymphocytes (TILs) involved viability staining (Thermo Fisher, #L34957), Fc blocking (BD, #553142, RRID:AB_394657) and then surface staining in FACS buffer (3% FBS in PBS) with fluorochrome-conjugated antibodies specific for mouse CD45 (Biolegend, #103147, RRID:AB_2564383), mouse CD3 (BD, #552774, RRID:AB_394460), mouse CD8 (Biolegend, #100708, RRID:AB_312747), and mouse PD-L1 (Biolegend, #124321, RRID:AB_2563635). For intracellular staining, the TILs were cultured in RMPI1640 plus 10% FBS with cell activation cocktail (Biolegend, #423303) overnight, and then were fixed and permeabilized with Cyto-Fast™ Fix/Perm Buffer Set (Biolegend, #426803) for mouse Granzyme B staining (Biolegend, # 515403, RRID:AB_2114575). For PD-L1 expression in cancer cells obtained from the syngeneic model, CD45− cells were surface stained in FACS buffer with fluorochrome-conjugated antibody specific for mouse PD-L1 (Biolegend, #124312, RRID:AB_10612741). All FACS analyses were performed on a BD LSR II or a Canto II Flow Cytometer, and data were analyzed with FlowJo software (Tree Star, Inc., version 7).
Immunofluorescence staining
Cells on cover slips were fixed with 3% paraformaldehyde in PBS, permeabilized in PBS containing 0.5% Triton X-100, blocked with 3% bovine serum albumin (BSA) in PBS and incubated with rabbit anti-cGAS (CST, # 31659S, RRID:AB_2799008), and anti-γ-H2AX (CST, #9718S, AB_2118009) at 4°C overnight. Cells were then washed twice with PBS and incubated with secondary antibodies conjugated with AlexaFluor dyes (Molecular Probes, #A11008, RRID:AB_143165; Molecular Probes, #A11004, RRID:AB_2534072) at room temperature for 1 h. Slips were then washed with PBS for 3 times, and incubated with diluted DAPI DNA staining dye in PBS for 15 min at room temperature. Stained slides were analyzed using a Leica TCS SP5 II scanning confocal microscope.
Immunohistochemical (IHC) staining
HGSOC tumor tissue microarrays (TMA) were kindly provided by Dr. Benjamin G Bitler from The University of Colorado (COMIRB# 17-7788). Detailed information on the tumors contained on the TMA has been published previously (32,33). IHC was performed using the Dako EnVision+ system following the manufacturer’s instructions. Briefly, the sections were dewaxed, rehydrated and immersed in 3% hydrogen peroxide in methanol to quench endogenous peroxidase activity. Antigen retrieval was performed in sodium citrate buffer (Thermo Fisher, #005000) and boiled for 45 min. The sections were incubated with blocking buffer for 1 h, primary antibody against TRIM28 (Abcam, #ab10484, RRID:AB_297223), SETDB1 (Biological Novus, #NBP2-20322) or PD-L1 (CST, #13684S, RRID:AB_2687655) at 4°C overnight and secondary antibody for 1h (DAKO, #K4011). Counterstaining was performed using Mayer’s Hematoxylin (Dako, #3309S). Expression of the stained markers was scored using a histologic score (H score). Based on the H scores, Kaplan-Meier survival curves were generated for SETDB1 and TRIM28 high and low patient groups using GraphPad Prism 7 (GraphPad) software. P value was calculated by log-rank test.
Syngeneic orthotopic ovarian cancer mouse model
Animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of The Wistar Institute. C57BL/6 mice were purchased from Charles River Laboratories. 1 × 106 ID8 cells were unilaterally injected into the ovarian bursa sac of 6–8-week old female C57BL/6 mice (n = 5 mice per group). 4 weeks after injection, mice were treated with IgG or anti-Pd-l1 (BioXcell, B7-H1, RRID:AB_10949073) (10 mg per kg, twice per week) for 2 weeks. After treatment, tumors from 5 mice per group were surgically dissected and tumor weight was measured as a surrogate for tumor burden.
Data mining
For expression correlation analysis, TCGA HGSOC RNA-seq data (PanCancer Atlas, n = 300) (34) was downloaded from cBioPortal (https://www.cbioportal.org/) and Cancer Cell Line Encyclopedia (CCLE, n = 912) RNA-seq data was downloaded from (https://sites.broadinstitute.org/ccle/datasets). Pearson’s correlation was used for calculating P and R values in Microsoft Excel. Pearson’s correlations of SETDB1 or TRIM28 with CD8A or GZMB were analyzed in 24 out of 33 types of TCGA PanCancer Atlas datasets with more than 100 patients. The analysis was performed on cBioPortal website.
A melanoma proteomics dataset with clinical response to anti-PD1 ICB was downloaded from the supplemental information section of the original paper (35), the protein levels of TRIM28 in responders (CR: complete responder, n = 10 and PR: partial responder, n = 30) and non-responders (progressive disease, n = 27) was compared.
HGSOC tumor tissue microarrays (TMA) were kindly provided by Dr. Benjamin G Bitler from The University of Colorado (COMIRB# 17-7788). The TMAs contain 131 patient tumor samples. In addition, data was available for the quantification of infiltrated immune cell populations by flow cytometry such as CD8+ T cell, CD8+/Granzyme B+ T cells and regulatory T cells for 120 out of the 131 patients. The percentages of indicated infiltrated immune cell populations in SETDB1 or TRIM28 high (H score ≥ 200) and low groups (H score ≤ 200) were compared.
Bioinformatics and Statistical Analysis
For analysis of the CRISPR/Cas9 screen, the raw. fastq file was parsed to obtain sgRNA abundance values using regular expression (stagger+BARCODE)(primer)(sgRNA), where stagger+BARCODE sequences were (atAAGTAGAG|gatAAGTAGAG) corresponding to PD-L1 and control samples correspondingly, primer sequence TCTTGTGGAAAGGACGAAACACCG and sgRNA sequence of 20bp length from the Epigenetic sgRNA library (31). Only perfect matches were considered for calculating sgRNA counts. Significance of difference between PD-L1 vs control samples on gene level was estimated using Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) software (36) using “mageck test -k counts.txt -t PD-L1 -c ctr -n GeneRankOutput” command. Genes that passed FDR<5% cutoff were considered significant.
For RNA-seq, data was aligned using bowtie2 (37) against mm10 version of the mouse genome and RSEM v1.2.12 software (38) was used to estimate raw read counts and RPKM values using Ensemble transcriptome. Abundance of TEs in samples was estimated using TEtranscripts software with default parameters (39). DESeq2 (40) was used to estimate significance of differential expression between groups pairs and calculate normalized counts. Overall gene expression changes were considered significant if passed FDR<5% thresholds unless stated otherwise. TE expression changes were considered significant if P value < 0.05 and FDR < 5%. The significantly changed TEs were shown as a heatmap and volcano plot using ggplot2 package (v3.3.3; Wickham, 2016). Enrichment analysis was performed using gene-set enrichment analysis (GSEA) (41) using MSigDB Hallmark gene sets (42) and results passing FDR<5% were reported.
For statistical analysis, experiments were repeated at least three times unless otherwise stated. Statistical analysis was performed using the GraphPad Prism 7 (GraphPad) software. Quantitative data are expressed as mean ± SEM unless otherwise stated. For all statistical analysis, the cutoff for significance was set at 0.05. For correlation studies, Pearson’s correlation was used for calculating P and R values in Microsoft Excel. Animal experiments were randomized. Combination index (CI) (43) for PD-L1 treatment and Setdb1 knockout was calculated as follows: CI = [(A+B) – A*B]/AB. A or B is the effect of the single condition, AB is the effect of the combination of A and B. Effect was calculated by the decreased percentage of the tumor weight: (1- treated tumor / control tumor) * 100%.
Data access
Next-generation sequencing (NGS) data for the CRISPR screen and RNA-seq have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE182198 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE182198)
Results
TRIM28 loss promotes PD-L1 expression
To systematically explore chromatin regulators of PD-L1 expression, we performed a CRISPR-Cas9 screen in the mouse ovarian cancer cell line ID8 using an sgRNA library targeting 1,218 genes that encode chromatin regulators, with typically 10 sgRNAs per gene (Fig. 1A). IFNγ was used to stimulate PD-L1 expression. To limit experimental variations, we sorted the 15% of ID8 cells with highest PD-L1 expression and compared the sgRNA profile in these cells with that in the bulk, unsorted cell population to ensure identical background and conditions for comparison. MAGeCK analysis revealed a list of sgRNAs that were either downregulated or upregulated in the PD-L1high cell population (Fig. 1B and Supplementary Table S1). Genes that were known regulators of PD-L1 were successfully identified as top ranked hits in our screen. For example, both Jak2 and Trp53 were identified as hits whose targeting sgRNAs were downregulated in the PD-L1high cell population (Fig. 1B). This is consistent with previous reports that both Jak2 and Trp53 are required for IFNγ-induced PD-L1 expression (44,45).
Figure 1: Identification of Trim28 as a suppressor of PD-L1 expression.
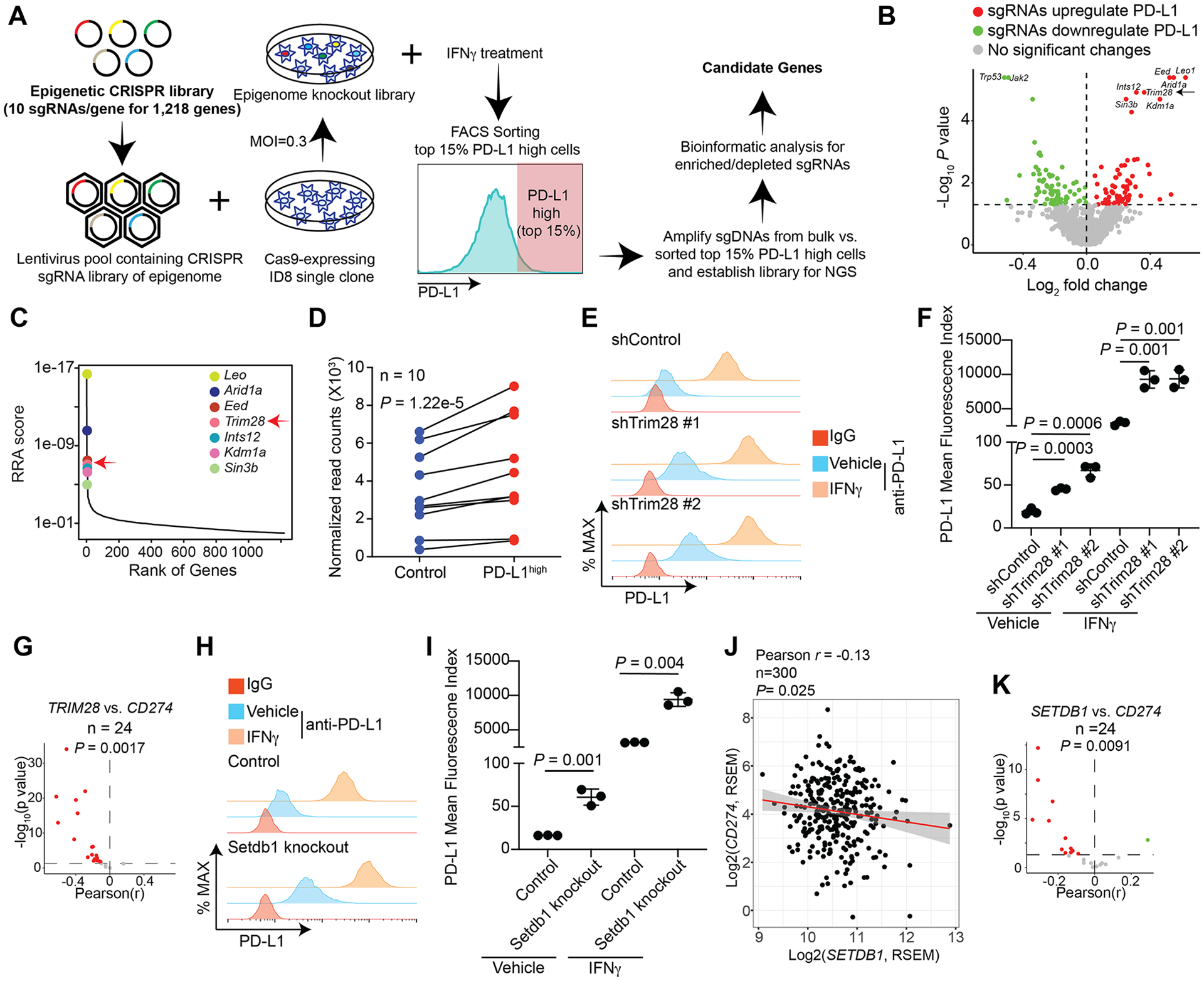
A, Schematic of the experimental design for the epigenome-wide CRISPR screen. B, Volcano plot illustrating the genes whose knockout (KO) significantly upregulated (red) or downregulated (green) PD-L1 expression stimulated by IFNγ with top hits highlighted. C, Robust rank aggregation (RRA) of the top 7 hits from the screen whose KO increases PD-L1 expression. D, Enrichment of 10 individual sgRNAs against Trim28 in the PD-L1high population compared with controls. E, Expression of PD-L1 was determined by FACS analysis in ID8 cells expressing two individual shTRIM28 or control treated with or without 30 ng/ml IFNγ for 24 h. An isotype matched IgG was used as a negative control. F, Quantification of E, the mean fluorescence index of PD-L1 was determined from 3 biologically independent experiments. G, Significant correlation between TRIM28 and CD274 expression based on RNA-seq analysis from 24 TCGA cancer types with at least 100 patients. H, Expression of PD-L1 was determined by FACS analysis in control and Setdb1-knockout ID8 cells treated with or without 30 ng/ml IFNγ for 24 h. An isotype matched IgG was used as a negative control. I, Quantification of H, the mean fluorescence index of PD-L1 was determined from 3 biologically independent experiments. J, Correlation between SETDB1 and CD274 expression based on RNA-seq analysis in TCGA ovarian cancer dataset. K, Significant correlation between SETDB1 and CD274 expression based on RNA-seq analysis from 24 TCGA cancer types with at least 100 patients. Data represent mean ± SEM, n = 3 biologically independent experiments unless otherwise stated. P values were calculated using a two-tailed student t test except in B and D by Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) analysis, G, J and K by Pearson R analysis.
We focused on our analysis on genes whose targeting sgRNAs were enriched in PD-L1 expression because we hypothesized that their inhibition could be used to enhance the therapeutic response to anti–PD-1/PD-L1 (46,47). The list of top hits whose sgRNAs were enriched in the PD-L1high cell population included Leo1, Arid1a, Eed, Trim28, Ints12, Kdm1a and Sin3b (Fig. 1C). To prioritize our functional studies, we examined the correlation between these genes and CD274, which encodes PD-L1, in two publicly available databases, the Broad Institute Cancer Cell Line Encyclopedia (CCLE) and the TCGA ovarian serous cystadenocarcinoma. The analysis revealed that LEO1, ARID1A, TRIM28 and KDM1A negatively correlated with CD274 expression in the CCLE database (Supplementary Fig. S1A), and that ARID1A, TRIM28 and KDM1A negatively correlated with CD274 expression in the TCGA ovarian cancer database (Supplementary Fig. S1B). By overlapping these analyses, we identified three hits from the screen that negatively correlated with CD274 expression in both datasets: ARID1A, TRIM28 and KDM1A (Supplementary Fig. S1C). ARID1A inactivation is associated with high PD-L1 expression and better response to ICB, including as anti–PD-L1 treatment (48). Likewise, KDM1A ablation enables ICB by stimulating antitumor immunity (49). However, the role of TRIM28 in regulating PD-L1 expression and antitumor immunity has not been explored. Accordingly, we focused our validation and functional studies on the TRIM28 gene.
All 10 sgRNAs targeting Trim28 showed a consistent enrichment in PD-L1high cells (Fig. 1D). TRIM28 is a component of the HUSH complex with SETDB1 as its catalytic subunit (14,50). We validated that Trim28 knockdown in ID8 cells increased Cd274 expression both at the basal level and in response to IFNγ stimulation (Supplementary Fig. S1D, E). We also observed an increase in PD-L1 expression by flow cytometry analysis (Fig. 1E, F). Similar observations were made in the human OVCAR3 ovarian cancer cell line (Supplementary Fig. S1F). Indeed, the negative correlation between TRIM28 and CD274 was observed in a statistically significant majority of the cancer types in the 24 TCGA datasets with more than 100 patients (Fig. 1G). Since SETDB1 is the catalytic subunit of the HUSH complex, we examined whether SETDB1 loss phenocopies TRIM28 loss. Toward this goal, we knocked out Setdb1 in ID8 cells using CRISPR (Supplementary Fig. S1G). Indeed, Setdb1 knockout increased PD-L1 expression both at the basal level and in response to IFNγ stimulation (Fig. 1H, I). Similar observations were also made in human OVCAR3 cells (Supplementary Fig. S1H, I). In addition, SETDB1 expression negatively correlated with CD274 expression in the ovarian cancer TCGA dataset (Fig. 1J and Supplementary Fig. S1J). Finally, a negative correlation between SETDB1 and CD274 was observed in a statistically significant majority of the cancer types in the 24 TCGA datasets with more than 100 patients (Fig. 1K).
SETDB1–TRIM28 expression negatively correlates with effector CD8+ T–cell infiltration
We next sought to determine the correlation between SETDB1–TRIM28 and PD-L1 expression in a tumor microarray consisting of 131 cases of human HGSOC. Both SETDB1 and TRIM28 expression negatively correlated with PD-L1 expression (Fig. 2A–C and Supplementary Table S2). In addition, SETDB1 and TRIM28 expression positively correlated (Fig. 2D and Supplementary Table S2). Compared with normal fallopian tube epithelial cells, SETDB1 and TRIM28 were found to be overexpressed in several ovarian cancer cell lines and there was concordance between SETDB1 and TRIM28 expression in these cell lines (Supplementary Fig. S2A). This is consistent with previous reports that SETDB1 is often overexpressed in human cancers (51). Indeed, SETDB1 was amplified/overexpressed in ~26% of HGSOC in the TCGA dataset and its amplification correlated with a significantly higher mRNA expression (Supplementary Fig. S2B, C).
Figure 2: SETDB1–TRIM28 negatively correlates with infiltration of effector CD8+ T cells.
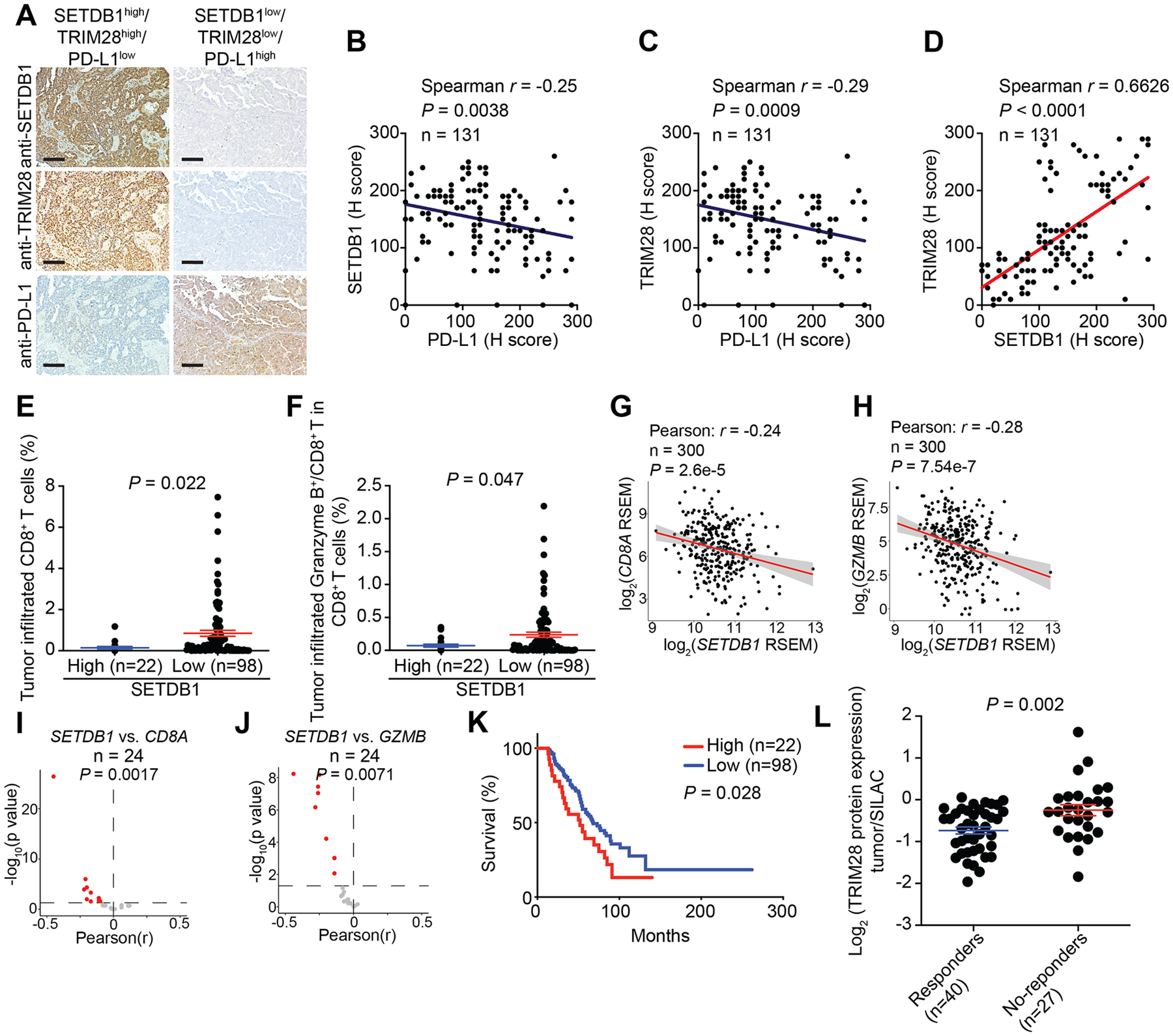
A, Representative staining of SETDB1, TRIM28 and PD-L1 in SETDB1–TRIM28 high or low expressing tumors from a TMA with 131 HGSOC cases. Scale bar = 100 μm. B, Negative correlation between SETDB1 and PD-L1 expression. C, Negative correlation between TRIM28 and PD-L1 expression. D, Positive correlation between SETDB1 and TRIM28 expression. E, F, High SETDB1 expression correlates with significantly lower tumor infiltrated CD8+ T cells (E) and Granzyme B+/CD8+ T cells (F). G, H, Negative correlation between SETDB1 and CD8A (G) or GZMB (H) in RNA-seq analysis using the TCGA ovarian cancer dataset. I, J, Significant negative correlation between SETDB1 and CD8A (I) or GZMB (J) expression based on RNA-seq analysis from 24 TCGA cancer types with at least 100 patients. K, Overall survival of patients with high or low SETDB1 from the TMA based on Kaplan-Meier analysis. L, TRIM28 protein is expressed at a significantly higher levels in melanoma patients who failed to respond to anti–PD-1 therapy compared with those responded to the therapy based on a publicly available proteomics dataset (35). Data represent mean ± SEM. P values were calculated using a two-tailed student t test except in B–D, G–J by Pearson R analysis, and K by log rank test.
Immune-cell infiltration profiles are available on 120 of the 131 TMA cases we examined Supplementary Table S2). Thus, we correlated with SETDB1 and TRIM28 expression with immune-infiltration profile. SETDB1 expression negatively correlated with tumor infiltrated CD8+ T cells and activated CD8+ T cells, as evidenced by Granzyme B+/CD8+ T cells (Fig. 2E, F and Supplementary Table S2). Similar trends were also observed between TRIM28 expression and infiltration of CD8+ and Granzyme B+/CD8+ T cells (Supplementary Fig S2D–E and Supplementary Table 2). In contrast, there was no correlation between expression of SETDB1 or TRIM28 with immune suppressive populations such as regulatory T cells (Supplementary Table S2). Next, we sought to validate our findings in the ovarian cancer TCGA dataset. We observed a negative correlation between SETDB1 expression and CD8A expression and GZMB expression (Fig. 2G, H). Similar findings were also made between TRIM28 expression and CD8A expression and GZMB expression in the ovarian cancer TCGA dataset (Supplementary Fig S2F, G). Finally, the negative correlation between SETDB1 or TRIM28 expression and CD8A expression and GZMB expression was observed in a statistically significant majority of the cancer types in the 24 TCGA datasets with more than 100 patients (Fig. 2I, J, Supplementary Fig S2H, I).
We next sought to determine whether SETDB1 and/or TRIM28 expression predicted prognosis, as determined by overall survival in the 131 TMA cases examined. Consistent with previous reports that high CD8+ T–cell infiltration correlates with an increase in overall survival (52,53), we observed a negative correlation between SETDB1 expression and overall survival of ovarian cancer patients (Fig. 2K), which was validated in an independent, public ovarian-cancer microarray dataset from 1,287 patients (Supplementary Fig S2J) (54). A similar trend between TRIM28 expression and overall survival was observed, albeit the statistical analysis did not reach significance (Supplementary Fig S2K). Re-analysis of a publicly available melanoma proteomics dataset with clinical response to anti-PD1 ICB revealed that TRIM28 was expressed at significantly higher levels in patients who did not respond to anti-PD1 treatment compared with those who had a clinical response (Fig. 2L and Supplementary Table S3) (35). SETDB1 was not detected in the proteomics dataset. These findings are consistent with the notion that SETDB1–TRIM28 represses PD-L1 expression and inhibits the infiltration of effector CD8+ T cells.
SETDB1–TRIM28 loss activates the cGAS-STING pathway
We next sought to determine the mechanism by which SETDB1–TRIM28 regulates PD-L1 expression and immune cell infiltration. Toward this goal, we performed RNA-seq analysis in two independent Setdb1-knockout ID8 clones (Supplementary Fig S1G). Analysis of differentially expressed genes (DEG) revealed that 6,005 genes were significantly changed by Setdb1 knockout compared with controls (Supplementary Fig S3A). GSEA of the DEGs revealed that G2/M checkpoint and inflammatory response pathways were among the top pathways enriched by the analysis (Fig. 3A, B). Micronuclei-induced ISGs) such as Ccl5 and Cxcl10 were among the inflammatory genes upregulated by Setdb1 knockout (10) (Fig. 3C). We validated the upregulation of specific micronuclei-induced ISGs such as Ccl5 and Cxcl10 in both Setdb1- and Trim28-knockout ID8 cells (Fig. 3D and Supplementary Fig. S3B, C). Similar results were also obtained in Setdb1-knockout UPK10 mouse ovarian cancer cells (Supplementary Fig. S3D, E). Likewise, TRIM28 knockdown in OVCAR3 human ovarian cancer cells upregulated the expression of CCL5 and CXCL10 genes (Supplementary Fig. S3F). In addition, we validated that Setdb1-knockout increased G2/M phase of the cell cycle (Supplementary Fig. S3G, H). Given the fact that micronuclei are generated as a result of mitotic defects during the G2/M phase of the cell cycle (11) and Setdb1 knockout increased the expression of micronuclei-induced ISGs such as Ccl5 and Cxcl10, we examined whether Setdb1 loss induced micronuclei formation. Toward this goal, we stained control and Setdb1-knockout ID8 cells using markers of micronuclei such as cGAS and γH2AX (Fig. 3E). Indeed, Setdb1-knockout significantly increased micronuclei formation (Fig. 3F) (10,11). Similar observations were made in Setdb1knockout UPK10 mouse ovarian cancer cells (Supplementary Fig. S3I). Consistent with the notion that micronuclei resulted from damaged DNA (10,11), the marker of DNA damage γH2AX was increased in Setdb1-knockout ID8 cells (Supplementary Fig. S3J). Furthermore, Trim28-knockout induced a similar increase in micronuclei formation in ID8 cells (Fig. 3G, H), suggesting that the observed effects are dependent on the SETDB1–TRIM28 complex.
Figure 3: SETDB1–TRIM28 loss activates the cGAS-STING pathway.
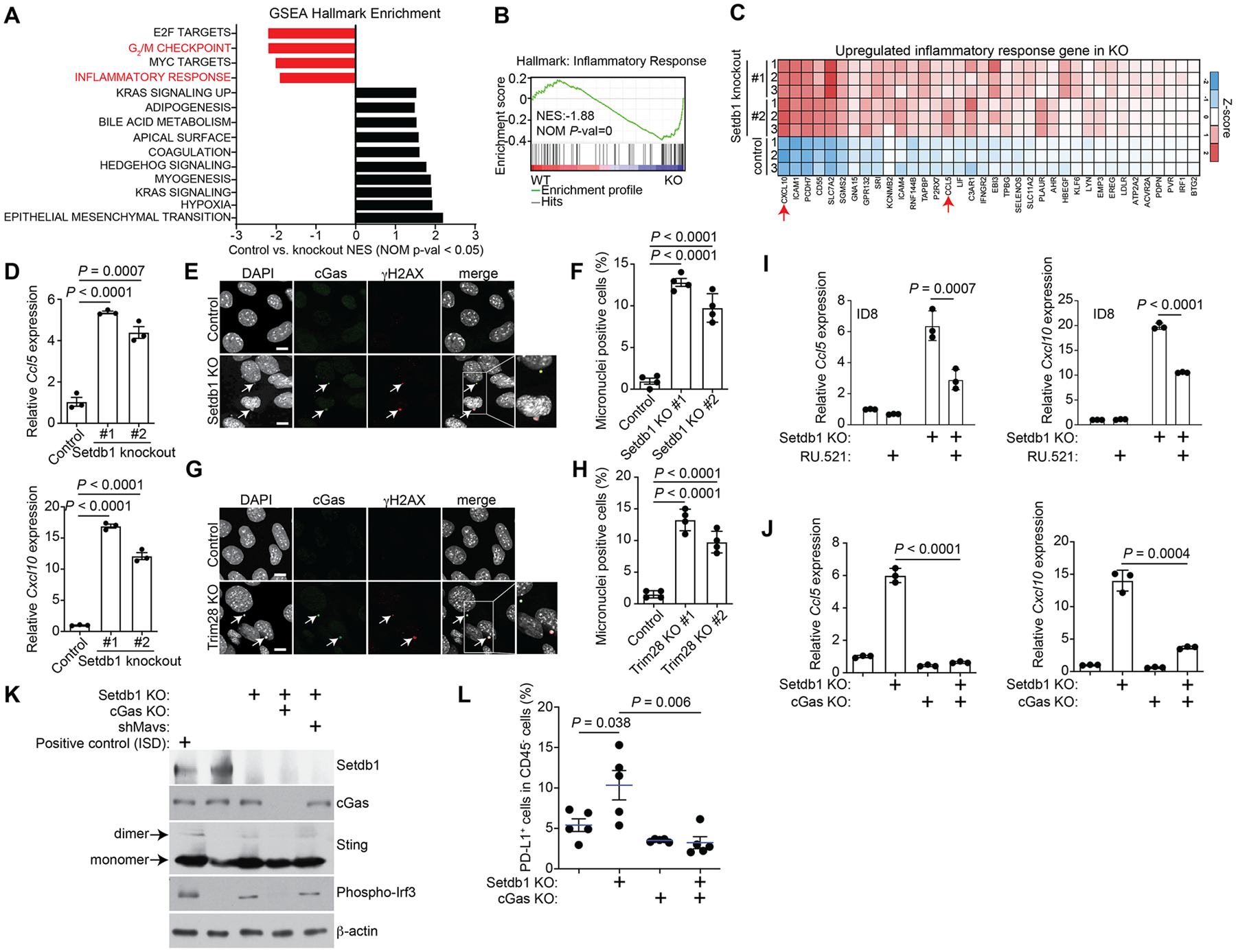
A, GSEA hallmark enrichment analysis of DEGs between control and Setdb1-knockout ID8 cells. B, Enrichment plot of GSEA for inflammatory response hallmark. C, Heatmap of the inflammatory response hallmark genes upregulated in Setdb1-knockout ID8 cells. D, Expression of Ccl5 and Cxcl10 in control and the indicated Setdb1-knockout ID8 cells determined by RT-qPCR analysis. E, Immunofluorescence staining of cGas and γ-H2AX in control and Setdb1-knockout ID8 cells. Arrows point to examples of cGas and γH2AX positive micronuclei. Scale bar = 10 μm. F, Quantification of micronuclei-positive cells in control and indicated Setdb1-knockout ID8 cells. G, H, Same as E, F but for Trim28-knockout ID8 cells. I, Expression of Ccl5 and Cxcl10 in control and Setdb1-knockout ID8 cells treated with or without the cGAS inhibitor RU.521 (2.5 μg/ml for 48 h), as determined by RT-qPCR analysis. J, Expression of Ccl5 and Cxcl10 in control, Setdb1-knockout, cGas-knockout and cGas/Setdb1-double knockout ID8 cells determined by RT-qPCR analysis. K, Expression of Setdb1, cGas, Sting, phospho-Irf3 and a loading control β-actin in the indicated cells determined by immunoblot. L, Percentage of PD-L1+ tumor cells in tumors formed by orthotopic injection of the indicated ID8 cell lines into C57BL/6J mice. Data represent mean ± SEM, n = 3 biologically independent experiments unless otherwise stated. P values were calculated using a two-tailed student t test except in B by GSEA analysis.
Since upregulation of micronuclei-induced ISGs depends on cGAS signaling (10,11), we sought to determine whether inhibition of cGAS ws sufficient to block the observed increase in micronuclei-induced ISGs such as Ccl5 and Cxcl10. Toward this goal, we inhibited cGAS activity either by a small molecule inhibitor RU.521 (55) or genetically by knocking out cGas in Setdb1-knockout ID8 cells (Supplementary Fig. S3K). Indeed, both RU.521 and cGas knockout significantly suppressed the upregulation of Ccl5 and Cxcl10 observed in Setdb1-knockout cells (Fig. 3I, J). Similar findings were also made in UPK10 Setdb1-knockout cells (Supplementary Fig. S3L). Consistently, Sting dimerization and the increase in phosphor-Irf3 induced by Setdb1 knockout cells were both blocked by cGas knockout (Fig. 3K). Furthermore, the upregulation of PD-L1 induced by Setdb1 loss was completely eradicated by cGas knockout in Setdb1-knockout cells both in vitro and in vivo in an orthotopic mouse model (Fig. 3L and Supplementary Fig. 3M, N). This suggests that upregulation of PD-L1 induced by Setdb1 loss is mediated by cGas signaling. Consistently, it has previously been shown that upregulating the cGas-Sting pathway in PD-L1 low mouse cells is sufficient to upregulate PD-L1 expression (56). These findings suggest a model whereby SETDB1 or TRIM28 loss activates the cGAS–STING pathway to simultaneously upregulate PD-L1 expression and increase immune cell infiltration through micronuclei-induced ISGs.
The best characterized function of the SETDB1–TRIM28 complex is in silencing TEs (57). Indeed, a very recent study shows that SETDB1 loss derepresses latent TE-derived regulatory elements to trigger TE-specific cytotoxic T–cell responses (29). Consist with these data, we observed an extensive upregulation of TEs in Setdb1-knockout ID8 cells (Supplementary Fig. S4A, B). To determine whether TEs contribute to the observed upregulation of inflammatory genes such as Ccl5 and Cxcl10, we knocked down Mavs expression in Setdb1-knockout cells (Supplementary Fig. S4C). Mavs functions downstream of the essential adaptor proteins Rig-I and Mad-5, which mediate the immune response, such as induction of ISGs, triggered by double stranded RNAs in response to derepressed TEs in Setdb1-knockout cells (57,58). In contrast to cGas knockout, Mavs knockdown did not affect the expression of Ccl5 or Cxcl10 (Supplementary Fig. S4D). Likewise, Mavs knockdown did not affect either Sting dimerization or phosphor-Irf3 expression in Setdb1 knockout cells (Fig. 3K). Together, we conclude that Setdb1-induced micronuclei formation and the associated cGas–Sting signaling are independent of the depression of TEs that is induced by Setdb1 knockout.
Setdb1 loss enhances the antitumor effects of anti–PD-L1
Setdb1 loss simultaneously upregulates PD-L1 expression and increases infiltration of CD8+ T cells whose presence in the tumor microenvironment is a prerequisite for response to ICBs such as anti–PD1/PD-L1. Thus, we hypothesized that Setdb1 loss could enhance the antitumor effects of anti–PD-L1 treatment. To test this hypothesis, we utilized a mouse orthotopic syngeneic ID8 ovarian cancer model (Fig. 4A). Mice receiving Setdb1-knockout ID8 cells had significantly reduced tumor burden, as determined by a decrease in tumor weight, compared with mice receiving control ID8 cells (Fig. 4B, C). Setdb1 knockout did not significantly affect the growth of cancer cells in vitro (Supplementary Fig. S5). Consistent with these observations, Setdb1 knockout was shown to reduce tumor burden in immunocompetent syngeneic, but not in immunocompromised xenograft, mouse models of both melanoma and lung cancer (29). Together, these findings suggest the reduction in tumor burden observed as a result of Setdb1 knockout in the immunocompetent syngeneic mouse model was due to Setdb1 induced antitumor immunity, rather than intrinsic growth inhibition of cancer cells. Anti–PD-L1 and Setdb1 loss were synergistic in reducing the burden of established orthotopic tumors (Fig. 4C). To determine whether the observed synergy depended on cGas signaling, we performed the same experiments in parallel using Setdb1 and cGas double knockout cells. Indeed, cGas knockout completely eradicated the antitumor effects of anti–PD-L1 and the synergy between anti–PD-L1 and Setdb1 loss (Fig. 4B, C).
Figure 4: Setdb1 loss synergizes with anti–PD-L1 in reducing tumor burden.
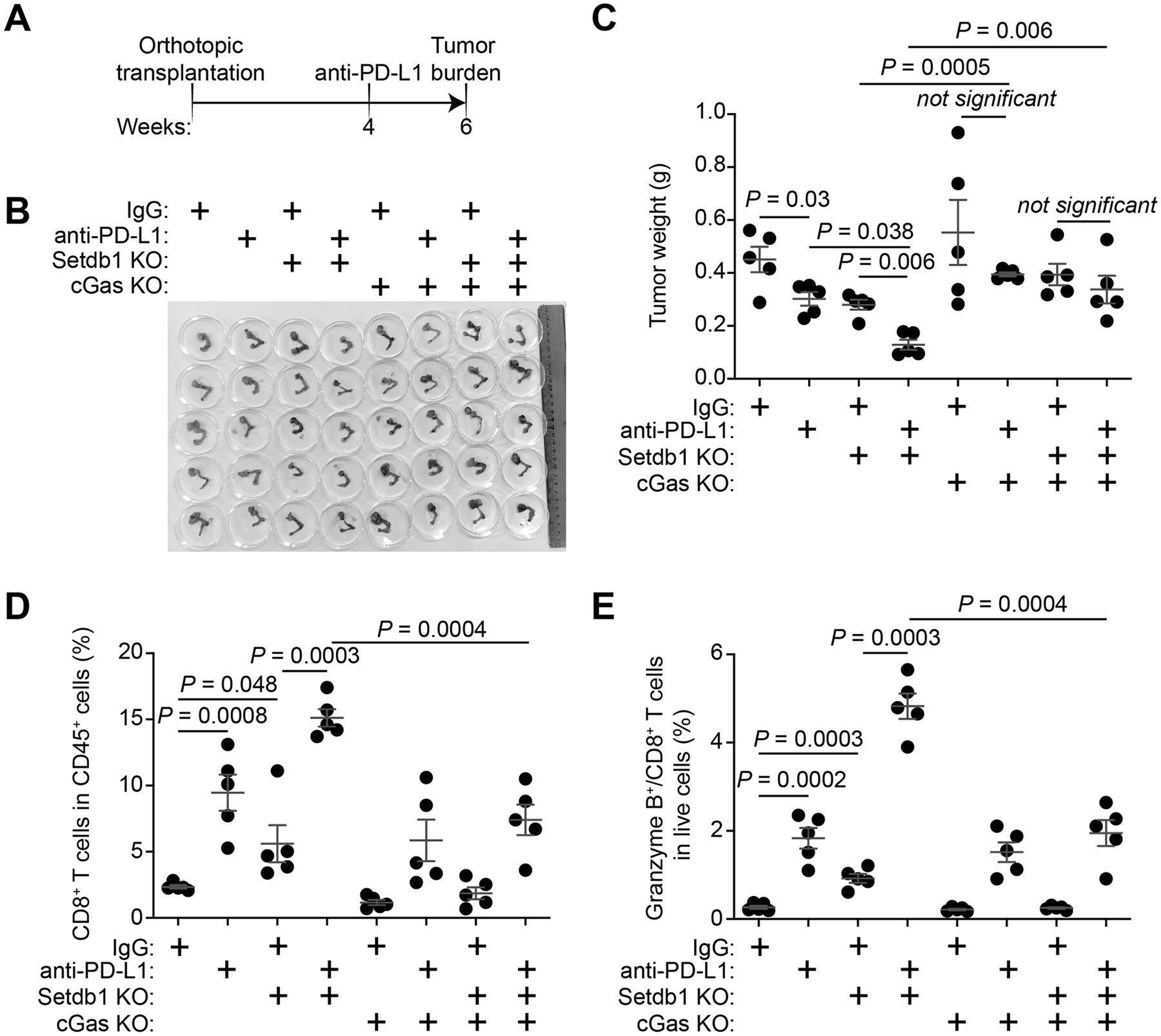
A, Schematic of the experimental design using the ID8 syngeneic orthotopic mouse ovarian cancer model. B, Reproductive tracts with tumors from the indicated treatment groups were dissected at the end of treatment (n=5 mice per group). C, The weights of tumors dissected from the indicated groups were measured as a surrogate for tumor burden. P-values were calculated using a two-tailed t test. Combination index (CI) for Setdb1 loss and anti–PD-L1 is 0.82 (<1), which indicates synergy between Setdb1 loss and anti-PD1 combination. D, E, Infiltration of CD8+ T cells (D) and Granzyme B+/CD8+ T cells (E) in the tumors dissected from the indicated treatment groups were analyzed by flow cytometry (n = 5 mice per group). Data represent mean ± SEM, P values were calculated using a two-tailed t test.
We next profiled changes in immune-cell infiltration to correlate the observed antitumor effects with the mechanisms we have revealed. Indeed, both Setdb1 loss and anti–PD-L1 treatment increased the infiltration of CD8+ and Granzyme B+ CD8+ T cells (Fig. 4D, E). Moreover, a combination of anti–PD-L1 and Setdb1 loss were synergistic in increasing the infiltration of CD8+ T cells (Fig. 4D, E). Although cGas loss completely eradiated the antitumor effects of anti–PD-L1, Setdb1 loss and their combination, its loss only partially blocked the infiltration of CD8+ and Granzyme B+ CD8+ T cells (Fig. 4D, E). Together, we conclude that Setdb1 loss and anti–PD-L1 are synergistic in reducing tumor burden, and this correlates with changes in infiltration of CD8+ and CD8+/Granzyme B+ T cells. In addition, cGas is required for the observed synergy, but only partially contributes to the observed changes in immune infiltration.
Discussion
Here we identify Trim28 as a regulator of PD-L1 expression in an unbiased CRISPR-Cas9 epigenome library screen. Although Setdb1 sgRNAs were included in the library, Setdb1 was not among the top hits (Supplementary Table S1). A possible reason for this might be variation in knockout efficacy by these sgRNAs and/or low infection efficacy by some of the sgRNAs. Consistent with this hypothesis, although 6 out of 10 individual Setdb1 sgRNAs were scored as positive hits, Setdb1 as a gene did not reach statistical significance in our screen. Regardless, our validation studies clearly show that Setdb1 loss phenocopies Trim28 loss in regulating PD-L1 expression.
We show that the synergy in reducing tumor burden observed by combining anti–PD-L1 and Setdb1 knockout is cGas dependent because cGas knockout blocked the observed antitumor effects. However, cGas knockout was not sufficient to block the infiltration of CD8+ T cells induced by either anti–PD-L1 alone or a combination of Setdb1 loss and anti–PD-L1. This is consistent with the notion that immune infiltration induced by a cell-intrinsic mechanism caused by Setdb1 loss is cGas dependent, whereas infiltration of CD8+ T cells induced by anti–PD-L1 occurs through a non-cell-intrinsic mechanism and is independent of cGas. In addition, while we were preparing this manuscript, it was reported that Setdb1 loss triggers TE-specific cytotoxic T–cell responses (29). In addition, it was shown that disruption of Setdb1 or its interacting partner Atf7ip augments tumor immunogenicity concomitant with elevated endogenous retroviral antigens and message RNA intron retention (59). Thus, the role of Setdb1 in regulating CD8+ T cells is likely multifaceted and context dependent.
Our results show that SETDB1–TRIM28 loss simultaneously increases PD-L1 expression and activates cGas–Sting signaling via micronuclei formation. In addition, our results further support a model whereby cGas activation mediates PD-L1 upregulation induced by SETDB1–TRIM28 loss. These results are important because SETDB1–TRIM28 loss bridges innate immunity and adaptive immunity to synergize with ICB in reducing tumor burden. The synergy is achieved by at least two mechanisms. First, Setdb1 loss increases PD-L1 expression, the target of anti–PD-L1 whose upregulation by both genetic alterations and therapeutic treatment has been linked to improved response to ICB. Consistent with this, we show that in a cohort of anti-PD1 treated melanoma patients, TRIM28 is expressed at significantly lower levels in responders compared with non-responders. Second, Setdb1 loss increases infiltration of effector CD8+ T cells in the tumor microenvironment, a prerequisite for response to ICB. Our findings raise the possibility that SETDB1–TRIM28 complex may serve as a biomarker to predict the therapeutic response to ICB. In addition, they suggest a need to develop therapeutic approaches to target the complex to boost antitumor immunity. A limitation of our study is the fact that the ID8 cells that we used lack a TP53 genomic alteration. However, we show comparable results using TP53 inactivated UPK10 cells and TP53 mutant human ovarian cancer cells. In summary, our results establish the SETDB1–TRIM28 complex as an attractive therapeutic target to boost antitumor immunity alone or in combination with ICB such as anti–PD-L1 treatment.
Supplementary Material
Synopsis.
Using a CRISPR-Cas9 screen, the authors identify the SETDB1–TRIM28 complex as a promising epigenetic target to simultaneously activate cGAS-STING signaling and upregulate PD-L1 expression to enhance the antitumor effects of anti-PD1 immune checkpoint blockade.
Financial support:
This work was supported by US National Institutes of Health grants (R01CA202919, R01CA239128 and P50CA228991 to R.Z.), US Department of Defense (OC180109 and OC190181 to R.Z.), the Honorable Tina Brozman Foundation for Ovarian Cancer Research and the Tina Brozman Ovarian Cancer Research Consortium 2.0 (to R.Z.), and Ovarian Cancer Research Alliance (Collaborative Research Development Grant #596552 to R.Z. and Ann and Sol Schreiber Mentored Investigator Award #649658 to J.L.). Support of Core Facilities was provided by Cancer Centre Support Grant (CCSG) CA010815 to The Wistar Institute.
Footnotes
Declaration of Interests: The Authors declare no competing interests.
References
- 1.O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 2017;52:71–81 [DOI] [PubMed] [Google Scholar]
- 2.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 2016;8:328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao J, Yan Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020;6:580–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018;553:91–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol 2020;17:75–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer 2019;19:151–61 [DOI] [PubMed] [Google Scholar]
- 7.Pilger D, Seymour LW, Jackson SP. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev 2021;35:602–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339:786–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013;153:1094–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017;548:461–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548:466–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013;498:380–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Xia L, Wu DY, Wang H, Chansky HA, Schubach WH, et al. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 2002;21:148–52 [DOI] [PubMed] [Google Scholar]
- 14.Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Gottgens B, et al. GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 2015;348:1481–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lazaro-Camp VJ, Salari K, Meng X, Yang S. SETDB1 in cancer: overexpression and its therapeutic implications. Am J Cancer Res 2021;11:1803–27 [PMC free article] [PubMed] [Google Scholar]
- 16.Czerwinska P, Mazurek S, Wiznerowicz M. The complexity of TRIM28 contribution to cancer. J Biomed Sci 2017;24:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inoue Y, Matsuura S, Kurabe N, Kahyo T, Mori H, Kawase A, et al. Clinicopathological and Survival Analysis of Japanese Patients with Resected Non-Small-Cell Lung Cancer Harboring NKX2–1, SETDB1, MET, HER2, SOX2, FGFR1, or PIK3CA Gene Amplification. J Thorac Oncol 2015;10:1590–600 [DOI] [PubMed] [Google Scholar]
- 18.Lafuente-Sanchis A, Zuniga A, Galbis JM, Cremades A, Estors M, Martinez-Hernandez NJ, et al. Prognostic value of ERCC1, RRM1, BRCA1 and SETDB1 in early stage of non-small cell lung cancer. Clin Transl Oncol 2016;18:798–804 [DOI] [PubMed] [Google Scholar]
- 19.Ho YJ, Lin YM, Huang YC, Chang J, Yeh KT, Lin LI, et al. Significance of histone methyltransferase SETDB1 expression in colon adenocarcinoma. APMIS 2017;125:985–95 [DOI] [PubMed] [Google Scholar]
- 20.Hou Z, Sun L, Xu F, Hu F, Lan J, Song D, et al. Blocking histone methyltransferase SETDB1 inhibits tumorigenesis and enhances cetuximab sensitivity in colorectal cancer. Cancer Lett 2020;487:63–73 [DOI] [PubMed] [Google Scholar]
- 21.Ropa J, Saha N, Hu H, Peterson LF, Talpaz M, Muntean AG. SETDB1 mediated histone H3 lysine 9 methylation suppresses MLL-fusion target expression and leukemic transformation. Haematologica 2020;105:2273–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orouji E, Federico A, Larribere L, Novak D, Lipka DB, Assenov Y, et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int J Cancer 2019;145:3462–77 [DOI] [PubMed] [Google Scholar]
- 23.Yokoe T, Toiyama Y, Okugawa Y, Tanaka K, Ohi M, Inoue Y, et al. KAP1 is associated with peritoneal carcinomatosis in gastric cancer. Ann Surg Oncol 2010;17:821–8 [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Jiang J, Li Q, Ma H, Xu Z, Gao Y. KAP1 is overexpressed in hepatocellular carcinoma and its clinical significance. Int J Clin Oncol 2016;21:927–33 [DOI] [PubMed] [Google Scholar]
- 25.Wang YY, Li L, Zhao ZS, Wang HJ. Clinical utility of measuring expression levels of KAP1, TIMP1 and STC2 in peripheral blood of patients with gastric cancer. World J Surg Oncol 2013;11:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu M, Fu X, Cui Y, Xu S, Xu Y, Dong Q, et al. Expression of KAP1 in epithelial ovarian cancer and its correlation with drug-resistance. Int J Clin Exp Med 2015;8:17308–20 [PMC free article] [PubMed] [Google Scholar]
- 27.Cui Y, Yang S, Fu X, Feng J, Xu S, Ying G. High levels of KAP1 expression are associated with aggressive clinical features in ovarian cancer. Int J Mol Sci 2014;16:363–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi ZX, Cai JJ, Chen LC, Yue Q, Gong Y, Yao Y, et al. TRIM28 as an independent prognostic marker plays critical roles in glioma progression. J Neurooncol 2016;126:19–26 [DOI] [PubMed] [Google Scholar]
- 29.Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021;595:309–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karst AM, Drapkin R. Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat Protoc 2012;7:1755–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller I, Moroni AS, Shlyueva D, Sahadevan S, Schoof EM, Radzisheuskaya A, et al. MPP8 is essential for sustaining self-renewal of ground-state pluripotent stem cells. Nat Commun 2021;12:3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jordan KR, Sikora MJ, Slansky JE, Minic A, Richer JK, Moroney MR, et al. The Capacity of the Ovarian Cancer Tumor Microenvironment to Integrate Inflammation Signaling Conveys a Shorter Disease-free Interval. Clin Cancer Res 2020;26:6362–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watson ZL, Yamamoto TM, McMellen A, Kim H, Hughes CJ, Wheeler LJ, et al. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin Epigenetics 2019;11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018;173:291–304 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, et al. Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 2019;179:236–50 e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin Y, Hammell M. Analysis of RNA-Seq Data Using TEtranscripts . Methods Mol Biol 2018;1751:153–67 [DOI] [PubMed] [Google Scholar]
- 40.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011;27:1739–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984;22:27–55 [DOI] [PubMed] [Google Scholar]
- 44.Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, et al. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015;4:e1008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiem A, Hesbacher S, Kneitz H, di Primio T, Heppt MV, Hermanns HM, et al. IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression. J Exp Clin Cancer Res 2019;38:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002;99:12293–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med 2018;24:556–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018;174:549–63 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ 3rd. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 2002;16:919–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strepkos D, Markouli M, Klonou A, Papavassiliou AG, Piperi C. Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential. Cancer Res 2021;81:525–34 [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003;348:203–13 [DOI] [PubMed] [Google Scholar]
- 53.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005;102:18538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer 2012;19:197–208 [DOI] [PubMed] [Google Scholar]
- 55.Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun 2017;8:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grabosch S, Bulatovic M, Zeng F, Ma T, Zhang L, Ross M, et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019;38:2380–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol 2017;216:3535–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005;122:669–82 [DOI] [PubMed] [Google Scholar]
- 59.Hu H, Khodadadi-Jamayran A, Dolgalev I, Cho H, Badri S, Chiriboga LA, et al. Targeting the Atf7ip-Setdb1 Complex Augments Antitumor Immunity by Boosting Tumor Immunogenicity. Cancer Immunol Res 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.