

Abstract
Background
PKARIα (protein kinase A type I‐α regulatory subunit) is redox‐active independent of its physiologic agonist cAMP. However, it is unknown whether this alternative mechanism of PKARIα activation may be of relevance to cardiac excitation–contraction coupling.
Methods and Results
We used a redox‐dead transgenic mouse model with homozygous knock‐in replacement of redox‐sensitive cysteine 17 with serine within the regulatory subunits of PKARIα (KI). Reactive oxygen species were acutely evoked by exposure of isolated cardiac myocytes to AngII (angiotensin II, 1 µmol/L). The long‐term relevance of oxidized PKARIα was investigated in KI mice and their wild‐type (WT) littermates following transverse aortic constriction (TAC). AngII increased reactive oxygen species in both groups but with RIα dimer formation in WT only. AngII induced translocation of PKARI to the cell membrane and resulted in protein kinase A–dependent stimulation of I Ca (L‐type Ca current) in WT with no effect in KI myocytes. Consequently, Ca transients were reduced in KI myocytes as compared with WT cells following acute AngII exposure. Transverse aortic constriction–related reactive oxygen species formation resulted in RIα oxidation in WT but not in KI mice. Within 6 weeks after TAC, KI mice showed an enhanced deterioration of contractile function and impaired survival compared with WT. In accordance, compared with WT, ventricular myocytes from failing KI mice displayed significantly reduced Ca transient amplitudes and lack of I Ca stimulation. Conversely, direct pharmacological stimulation of I Ca using Bay K8644 rescued Ca transients in AngII‐treated KI myocytes and contractile function in failing KI mice in vivo.
Conclusions
Oxidative activation of PKARIα with subsequent stimulation of I Ca preserves cardiac function in the setting of acute and chronic oxidative stress.
Keywords: heart failure, pressure overload, protein kinase A, redox
Subject Categories: Ion Channels/Membrane Transport, Oxidant Stress, Heart Failure
Nonstandard Abbreviations and Acronyms
- AngII
angiotensin II
- CaMKII
Ca/calmodulin‐dependent protein kinase II
- ECC
excitation–contraction coupling
- FRET
Förster‐resonance energy transfer
- I Ca
L‐Type Ca current, i.e. inward Ca current through the L‐type Ca channel
- KI
knock‐in
- LTCC
L‐type Ca channel
- Nox2
nicotinamide adenine dinucleotide phosphate oxidase 2
- Nox4
nicotinamide adenine dinucleotide phosphate oxidase 4
- PDGF
platelet‐derived growth factor
- PKA [C]
protein kinase A catalytic subunit
- PKA
protein kinase A
- PKARII
protein kinase A type II regulatory subunit
- PKARIα
protein kinase A type I‐α regulatory subunit
- PLB
phospholamban
- ROS
reactive oxygen species
- RyR2
ryanodine receptor type 2
- SERCA2a
SR Ca ATPase 2a
- SR
sarcoplasmic reticulum
- TAC
transverse aortic constriction
- WT
wild‐type
Clinical Perspective
What Is New?
The role of redox‐activated PKARIα in cardiac excitation–contraction coupling is largely unknown.
Oxidative activation of PKARIα with subsequent stimulation of I Ca preserves cardiac function in the setting of acute and chronic oxidative stress.
Conversely, enhanced heart failure is observed in redox‐dead Cys17Ser PKARIα knock‐in mice upon pressure overload as induced by transverse aortic constriction.
What Are the Clinical Implications?
Our study demonstrates an important compensatory function for redox‐activated PKARIα in the course of pressure overload–induced heart failure, which should be carefully considered when using antioxidant treatment for heart failure.
Heart failure (HF) is a state of impaired systolic and/or diastolic cardiac function (ie, reduced contractility and/or relaxation). At the cellular level, failing myocytes display a typical pattern of severely impaired intracellular Ca handling, pathologically increased reactive oxygen species (ROS), and altered activity of stress kinases such as CaMKII (Ca/calmodulin‐dependent protein kinase II) and cAMP (3',5'‐cyclic adenosine monophosphate)‐dependent PKA (protein kinase A). 1 , 2
Impaired contractile function in HF is a direct consequence of a reduction in the amplitude of the systolic Ca transient, which results from decreased transarcolemmal Ca influx (I Ca) through the LTCC (L‐type Ca channel), and reduced Ca release from the Ca‐depleted sarcoplasmic reticulum (SR). Disease‐related ROS may contribute to impaired Ca handling in HF, because ROS were shown to inhibit I Ca 3 and to decrease SR Ca content as a consequence of inhibited SR Ca reuptake 4 and enhanced diastolic SR Ca loss. 5 Furthermore, ROS may also indirectly aggravate SR Ca loss by activation of stress kinases such as CaMKII, 6 which results in a CaMKII‐dependent diastolic SR Ca leak. 7 The pathologic relevance of redox‐dependent CaMKII activation has been extensively investigated in the past few years and was identified to be involved in several cardiac diseases including myocardial injury and HF. 8
PKA is another important protein kinase, which shares many important targets with CaMKII in excitation–contraction coupling (ECC) (such as LTCC and PLB [phospholamban]). PKA is the major downstream effector of enhanced β‐adrenergic stimulation. 9 However, HF is characterized by a blunted response to β‐adrenergic signaling, 10 , 11 and there is evidence for an impaired β‐adrenergic reserve for I Ca stimulation in HF. 12 Thus, reduced PKA‐dependent stimulation of Ca handling proteins such as the LTCC or PLB (that regulates SR Ca reuptake) may contribute to contractile dysfunction in HF. PKA consists of 4 different types of regulatory (ie, RIα, RIβ, RIIα, and RIIβ) and 2 different types of catalytic subunits (PKA [C]). 13 Regulatory subunit RIα has been shown to be important for regulation of PKA activity in the brain, brown and white adipose tissue, skeletal muscle, and sperm. 14 Its essential requirement was further proven by a RIα knock‐out in mice, which results in early embryonic lethality because of aberrant cardiac morphogenesis. 14 RIα is unique among the regulatory subunits because it contains redox‐sensitive cysteine residues. Brennan et al found that oxidation of RIα can result in cAMP‐independent PKA activation, which results in a positive inotropic response in isolated cardiac myocytes. 15 This alternative oxidant‐dependent activation of PKARIα has been shown to be important for angiogenesis during tumor formation and ischemia, 16 as well as for PDGF (platelet‐derived growth factor)‐dependent signaling. 17 Recently, oxidative activation of RIα was shown to protect against myocardial ischemia‐reperfusion injury in the acute setting of global ischemia ex vivo, presumably through inhibition of lysosomal‐triggered Ca release. 18 This suggests that oxidative RIα activation may be an important acute regulator of cardiac inotropy. However, distinct targets of oxidized PKARIα in ECC that may underlie its inotropic potential as well as the long‐term consequences of oxidative RIα activation in vivo (ie, in cardiac pathology) have not been investigated. We have recently reported that oxidized PKARIα acutely regulates potassium currents in cardiac myocytes upon H2O2, thereby contributing to early afterdepolarizations in vitro. 19 However, this study did not investigate the role of oxidized PKA RIα for cardiac contractility (neither for the acute setting in vitro nor for the long‐term setting in vivo), nor did it investigate the distinct molecular targets of oxidized PKARIα in ECC that may underlie its inotropic relevance. Interestingly, AngII (angiotensin II)‐dependent regulation of I Ca was shown to require Nox2 (nicotinamide adenine dinucleotide phosphate oxidase 2)‐related ROS and PKA, 5 which suggests that oxidized PKARIα could be of particular relevance for I Ca gating and hence for cardiac inotropy.
We accordingly hypothesized that oxidative activation of PKARIα might be an important regulator of excitation–contraction coupling upon increased oxidative stress. We made use of a redox‐dead transgenic mouse model with homozygous knock‐in replacement of redox‐sensitive cysteine 17 with serine (within the regulatory subunits of PKARIα (KI).
As a model of acute oxidative stress, isolated cardiac myocytes from KI hearts were exposed to AngII. In addition, we tested the relevance of oxidized PKARIα under conditions of long‐term increased ROS formation and impaired β‐adrenergic signaling (ie, upon pressure overload–induced HF induced by transverse aortic constriction (TAC).
Our data show that oxidative activation of PKARIα is required to maintain I Ca both upon acute exposure to AngII and during pressure overload–induced HF, which identifies oxidized PKARIα as an important contributor of maintained Ca handling upon oxidative stress. Conversely, KI mice with absent stimulation of I Ca (and reduced Ca transients) developed aggravated heart failure and decreased survival following TAC that can be acutely rescued by cAMP‐independent stimulation of I Ca using Bay K8644.
Methods
All animal procedures were performed in accordance with the guide for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee. Data are available upon reasonable request to the authors. An extended description of materials and methods can be found in Data S1.
In Vivo Procedures
Generation of Cys17Ser PKARIα Mice
Mice that constitutively express PKARIα Cys17Ser (KI) were generated as previously described. 16 The point mutation Cys17Ser was introduced into exon 1 of the Prkar1a gene by site‐directed mutagenesis. These mice behave normally under physiologic conditions.
Transverse Aortic Constriction
Male mice at the age of 11 to 13 weeks (weight, 20–25 g) were subjected to transverse aortic constriction as previously described. 20 Briefly, a small incision was made into the skin above the upper thoracic inlet (~1 cm), and a 6‐0 nonabsorbable suture was used to form a knot around the transverse aorta between the right and left carotid artery. The knot was tightened against a 27‐gauge cannula. For control, a sham procedure was conducted applying a similar surgical procedure but without constriction of the aorta. Sham‐operated mice had normal survival and did not develop HF. Echocardiographic data of sham‐operated mice are given in Table 1.
Table 1.
Echocardiographic Parameters
|
WT basal |
KI basal |
WT sham, 1 wk | WT TAC, 1 wk | KI sham, 1 wk | KI TAC, 1 wk | WT sham, 6 wk | WT TAC, 6 wk | KI sham, 6 wk | KI TAC, 6 wk | P value for interaction | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of mice | 92 | 85 | 9 | 38 | 8 | 38 | 4 | 18 | 4 | 14 | |
| HR, bpm | 431±3 | 430±8 | 458±13 | 429±7 | 441±11 | 441±6 | 456±20 | 460±12 | 450±13 | 462±10 | 0.7776 |
| FS, % | 24.5±0.5 | 24.5±0.5 | 23.9±2.2 | 18.1±0.9 | 23.2±1.8 | 14.3±0.9 | 22.1±1.8 | 13.3±1.2 | 24.7±2.6 | 7.2±1.0 | 0.0014* |
| EF, % | 48.3±0.7 | 48.8±0.7 | 47.6±4.0 | 38.0±1.5 | 46.4±3.2 | 31.4±1.6 | 47.2±4.0 | 29.7±2.2 | 50.1±3.8 | 20.6±1.6 | 0.0004* |
| SV, µL | 39.9±0.6 | 39.2±0.7 | 34.9±2.9 | 30.7±1.2 | 37.5±2.2 | 26.2±1.4 | 42.2±2.5 | 31.6±1.7 | 38.0±2.7 | 26.8±1.7 | 0.0487* |
| CO, mL/min | 17.1±0.3 | 17.0±0.4 | 15.9±1.2 | 13.1±0.6 | 16.5±1.0 | 11.6±0.7 | 19.3±1.7 | 14.5±0.9 | 17.1±1.4 | 13.0±0.8 | 0.1922 |
| LVEDD, mm | 4.25±0.03 | 4.19±0.04 | 4.09±0.06 | 4.19±0.06 | 4.23±0.07 | 4.23±0.06 | 4.40±0.11 | 4.76±0.11 | 4.19±0.10 | 4.93±0.11 | 0.4503 |
| LVESD, mm | 3.32±0.04 | 3.32±0.04 | 3.20±0.11 | 3.46±0.08 | 3.37±0.12 | 3.72±0.08 | 3.44±0.17 | 4.16±0.14 | 3.19±0.15 | 4.61±0.13 | 0.0183* |
| LVEDV, µL | 82.9±1.5 | 81.0±1.4 | 74.1±2.4 | 83.1±3.7 | 81.6±3.2 | 86.6±4.6 | 87.5±5.4 | 112.2±6.6 | 76.2±4.1 | 134.3±8.2 | 0.0036* |
| LVESV, µL | 43.2±1.3 | 41.7±1.1 | 39.1±3.8 | 52.4±3.2 | 44.1±4.0 | 60.4±3.9 | 43.4±4.7 | 80.6±6.7 | 38.2±4.1 | 107.5±7.9 | 0.0002* |
| AWThd, µm | 0.79±0.02 | 0.80±0.02 | 0.79±0.03 | 1.02±0.04 | 0.80±0.04 | 1.00±0.03 | 0.73±0.03 | 0.92±0.03 | 0.77±0.08 | 0.93±0.03 | 0.8939 |
| PWThd, µm | 0.70±0.01 | 0.68±0.01 | 0.74±0.05 | 0.90±0.04 | 0.73±0.05 | 0.91±0.04 | 0.65±0.02 | 0.82±0.04 | 0.69±0.10 | 0.78±0.04 | 0.7879 |
AWThd indicates anterior wall thickness, diastolic; CO, cardiac output; EF, ejection fraction; FS, fractional shortening; HR, heart rate; KI, knock‐in; LVEDD, left ventricular end diastolic diameter; LVEDV, left ventricular end diastolic volume; LVESD, left ventricular end systolic diameter; LVESV, left ventricular end systolic volume; PWThd, posterior wall thickness, diastolic; SV, stroke volume; TAC, transverse aortic constriction; and WT, wild‐type.
P<0.05 in mixed‐effects model type III test WT vs. KI (without sham groups).
Echocardiography
Echocardiography was performed on anesthetized mice (isoflurane 1.5%, spontaneous respiration) at different time points (ie, at baseline, at 1 week after TAC, and at 6 weeks after TAC respectively) by experienced ultrasonographers who were unaware of the mice’s treatment details. In some experiments, I Ca was acutely stimulated by IP injection of 100 mg/kg Bay K8644 in anesthetized wild‐type (WT) and KI mice undergoing echocardiography at 1 week after TAC. The maximum of the acute inotropic response (ie, a Bay K8644–dependent change in left ventricular ejection fraction [LVEF]) was reached at ~6 minutes after Bay K8644 injection and was therefore taken for analysis.
Electrophysiological Studies
Mice were anesthetized with isoflurane and kept on a warm pad as mentioned above. A baseline surface ECG (lead II) was acquired using a multichannel data‐acquisition system (Powerlab 16/30; AD Instruments, Colorado Springs, CO). Data were stored using Labchart Pro software version 7 (AD Instruments). QT intervals were heart rate corrected (QTc) using Mitchell’s formula. 21 Afterward, a standard clinical protocol was used to determine electrophysiological parameters. In brief, right atrial or ventricular pacing was performed using 2‐millisecond current pulses delivered by an external stimulator (STG‐3008; Multi Channel Systems). Inducibility of ventricular arrhythmias was tested by decremental burst pacing. Burst pacing started at a 40‐millisecond cycle length, decreasing by 2 milliseconds every 2 seconds to a cycle length of 20 milliseconds. Burst pacing was repeated 1 minute after the previous burst concluded or the termination of arrhythmias. Burst pacing was performed for a total of 5 times for ventricular stimulation, respectively. Ventricular tachycardia was defined as rapid ventricular potentials that occurred independent from atrial potentials and displayed altered QRS morphology. If ventricular tachycardia was sustained for >1 second, it was considered as relevant.
Blood Pressure Measurements
AngII (1.0 mg/kg−1per d−1) was administered for 14 days via subcutaneous osmotic minipumps (model 1002; Alzet, Cupertino, CA) implanted under 2% isoflurane as described previously. 22 Mean blood pressure was assessed using the CODA mouse rat tail‐cuff system (Kent Scientific). We allowed mice to acclimatize to a clear acrylic tube with a nose cone holder (Kent Scientific) for at least half an hour before blood pressure recordings were started. Mean blood pressure was assessed at baseline and following 14 days of AngII treatment. A minimum of 20 blood pressure recordings were averaged for each mouse.
In Vitro Experiments
Isolation of Mouse Ventricular Myocytes
Ventricular cardiomyocytes were isolated according to established enzymatic procedure as previously described. 23 Only adult male mice aged 10 to 20 weeks were used.
Measurement of Intracellular Ca Transients, Ca Spark Frequency, and Cytosolic ROS
Intracellular Ca transients were measured as described previously using standard Tyrode's solution. 5 Briefly, myocytes on laminin‐coated recording chambers were loaded with 10 µmol/L Fura‐2‐AM, in the presence of 0.02% (w/v) pluronic acid (Molecular Probes, Eugene, OR) for 20 minutes at room temperature in the dark. All fluorescence emission was recorded using IonWizard software (IonOptix, Boston, MA). For some experiments, caffeine (10 mmol/L) was applied to induce rapid SR Ca release. Ca spark frequency was assessed in isolated ventricular myocytes loaded with 10 µmol/L Fluo‐4 AM (Molecular Probes) for 12 minutes at room temperature using an inverted laser scanning confocal microscope (Zeiss Pascal 5; Carl Zeiss) as described previously. 5 Cytosolic ROS production was measured in ventricular myocytes loaded with CellROX (Molecular Probes) at 5 µmol/L for 20 minutes at room temperature in the dark using an inverted laser scanning confocal microscope (Zeiss Pascal 5). CellROX was excited at 545 nm, and emission was collected at 565 nm. Once every minute, CellROX emission was measured and normalized to the basal fluorescence for further comparison. In some experiments, myocytes were exposed to AngII (1 µmol/L) for 15 minutes. PKA was inhibited using 5 µmol/L of H89. In another set of experiments, AngII pretreated cells were acutely exposed to 1 µmol/L of Bay K8644.
Patch‐Clamp Experiments
Ruptured‐patch whole‐cell voltage clamp was used to measure I Ca (voltage clamp configuration). In all experiments, myocytes were mounted on the stage of a microscope (Nikon Eclipse TE2000‐U). For I Ca measurements, microelectrodes (2–3 MΩ) were filled (in millimoles per liter) with 86 CsCl, 40 Cs‐glutamate, 0.92 MgCl2, 5 Mg‐ATP, 0.3 Li‐GTP, 10 HEPES, 5 EGTA, and 1.8 CaCl2 (free [Ca2+]i 100 nmol/L) (pH 7.2, CsOH). The bath solution contained (in millimoles per liter) 140 NaCl, 4 CsCl, 1 MgCl2, 10 glucose, 10 HEPES, 1 CaCl2 (pH 7.4, CsOH). For I Ca, access resistance was <8 MΩ. Signals were filtered with 2.9 and 10 kHz Bessel filters and recorded with an EPC10 amplifier (HEKA Elektronik). Recordings were started 2 to 3 minutes after rupture. All experiments were conducted at room temperature.
Measurement of cAMP Levels Using Förster Resonance Energy Transfer (FRET) with Fluorescence Lifetime Imaging Microscopy (FLIM)
Quantification of intracellular cAMP generation was performed in isolated adult ventricular Epac1‐camps‐transgenic mouse cardiomyocytes from sham versus TAC‐operated mice using Förster‐resonance energy transfer (FRET). As previously described, intracellular cAMP concentration can be traced back to a change in Epac1‐camps FRET signal. 24 , 25 However, the spectral separation of 2 fluorescence emissions cannot be performed with absolute precision when using the conventional ratiometric measurement of FRET on an inverted fluorescence microscope. Therefore, we applied a more accurate approach to measure FRET signal by fluorescence lifetime imaging microscopy (FLIM). 26 After isolation, myocytes were placed on a laminin‐coated recording chamber that was mounted on a specialized confocal microscope (DCS‐120; Becker & Hickl). Every 60 seconds an image of the chosen myocyte was recorded for a total of 14 minutes. Starting minute 3, forskolin (50 µmol/L) was applied to induce maximum stimulation of cAMP generation. Imaging data were collected and analyzed using SPCImage version 6.4 (Becker & Hickl).
Assessment of Cardiac cAMP Concentration and PKA Activity
To study the effect of TAC on cAMP concentration and PKA activity in cardiac tissue samples, a cAMP ELISA kit (Enzo Life Sciences, Lausen, Switzerland) and a PKA kinase activity assay (ab139435; Abcam, Berlin, Germany) were performed according to the manufacturer´s protocol.
Histology
Cardiac samples were immediately fixed in formalin and embedded in paraffin according to standard procedures. Then, 4‐µm sections were stained with antibodies directed against wheat germ agglutinin (at 1:300, Alexa Fluor 594; Life Technologies, Eugene, OR) and biotinylated Griffonia (Bandeiraea) Simplicifolia Lectin I (isolectin B4 at 1:200; Vector Laboratories, Burlingame, CA). To quantify cardiac fibrosis, the trichrome stain (Masson) kit was used (Sigma‐Aldrich, St. Louis, MO). All images were acquired on a Zeiss Axiostar plus microscope using the Axio Vision 4.9.1. software (Carl Zeiss Microscopy, Jena, Germany). Morphometric analysis was performed using Histo Quest software (TissueGnostics, Vienna, Austria). All data were analyzed by one observer blinded to mouse genotypes.
Western Blot Analysis
Both isolated cardiomyocytes and whole mouse heart samples were used for Western blot analysis. Immunoblots detecting the PKARIα dimer to monomer ratio were performed under nonreducing conditions. As recently described by Simon et al, 18 we also observed a nonspecific band above the PKARIα monomer band that was not used for analysis.
Statistical Analysis
All data are expressed as mean±SEM. Normality of data was tested by hierarchical application of D'Agostino‐Pearson, Shapiro‐Wilk, or Kolmogorov‐Smirnov test, respectively, depending on the number of experiments. For some experiments, data were transformed (log, reciprocal) to demask a normality distribution before normality and statistical testing. If data were not normally distributed or the sample size was too small for normality testing, nonparametric tests were used. For longitudinal data, 2‐way repeated‐measures ANOVA or mixed‐effects analysis was performed; where appropriate, a 1‐way ANOVA or Kruskal‐Wallis test with multiple comparison tests was used. For categorical data, Fisher's exact test was used. Otherwise, Student's unpaired t test was applied. Two‐sided P<0.05 was considered significant. The method of analysis for each data set is given in the figure legends. Statistics were performed using GraphPad Prism 9 (GraphPad Software, San Diego, CA) or Sigma Plot 12.5 (Systat Software, Palo Alto, CA). For analysis of I Ca steady‐state activation, data were fitted to a standard Boltzmann equation: h∞=1/{1 + exp [(V1/2 – V)/k∞]}, where V1/2 is the membrane potential for half‐maximal activation and k∞ is a slope factor indicating the slope in e‐folds per k∞ mV as described by Bers. 9
Results
Acute Exposure to AngII Results in ROS Formation, PKA RIα Dimerization, Translocation, and PKA‐Dependent Stimulation of I Ca
We have previously reported that AngII‐mediated ROS regulate LTCC through activated PKA. 5 Here, we acutely exposed isolated cardiomyocytes from KI and WT hearts to AngII to test whether PKA‐dependent regulation of I Ca upon increased ROS requires oxidized PKARIα.
As shown in Figure 1A and 1B, AngII (1 µmol/L) robustly increased intracellular ROS formation within a time frame of 14 minutes in both WT and KI myocytes without differences between groups. This demonstrates that AngII exposure can be used as an acute model of oxidative stress in isolated cardiac myocytes, and that KI replacement of Cys17 to Ser within the regulatory subunit of PKARIα does not affect ROS formation upon AngII. In contrast, AngII‐dependent ROS formation was associated with PKARIα dimer formation in WT myocytes only (Figure 1C).
Figure 1. Acute exposure to Angiotensin II stimulates reactive oxygen species (ROS) and PKARIα (protein kinase A type I‐α regulatory subunit) oxidation.
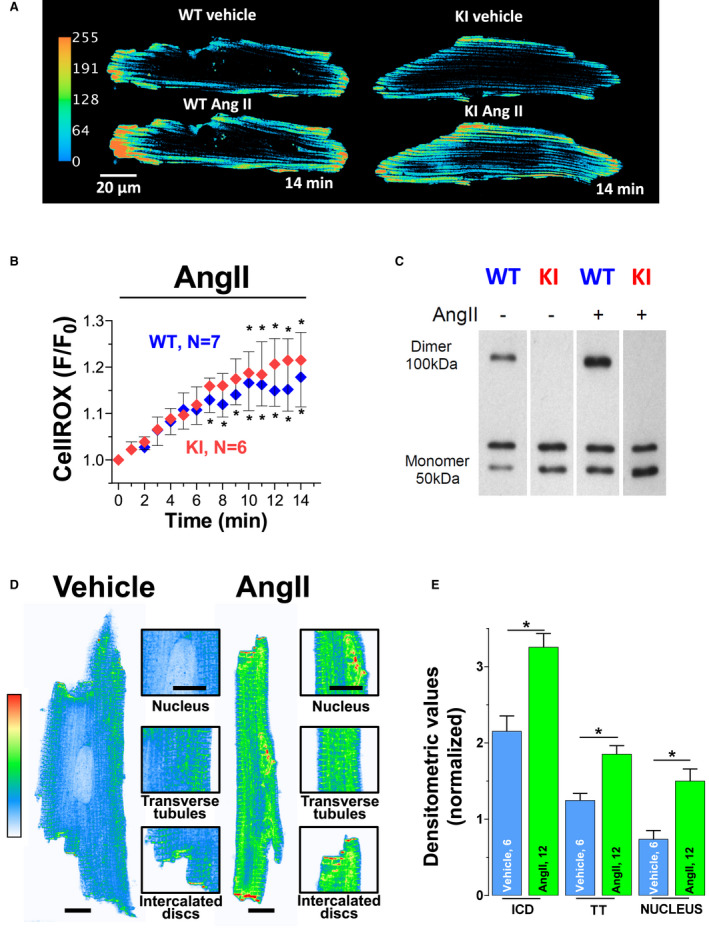
Original traces (A) and mean data (B) of isolated ventricular myocytes from wild‐type (WT) and knock‐in (KI) mice loaded with the ROS sensor CellROX that were acutely exposed to AngII (angiotensin II). For improved visualization, gray scale values were converted to color using the depicted calibration bar. F/F0 indicates fluorescence intensity normalized to baseline fluorescence. AngII induced cytosolic ROS to a similar extent in WT and KI myocytes. Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs baseline using mixed‐effects analysis with Holm‐Sidak post‐test. C, An original Western blot of PKARIα dimer formation (ie, oxidation) in isolated WT and KI hearts perfused with AngII (1 µmol/L, 10 minutes). Upon nonreducing conditions, monomeric PKARIα subunits form a band at around 50 kDa, whereas oxidized PKARIα dimers have twice the molecular weight (at around 100 KDa). Compared with vehicle (AngII–), AngII increased PKARIα oxidation in WT only. Original traces (D) and mean values (E) for immunocytochemical analysis of PKARI (protein kinase A type I regulatory subunit) localization in isolated mouse ventricular myocytes. Scale bar in (D)=10 μm. Insets show magnification of subcellular localization at nucleus, transverse tubules (TT), and intercalated discs (ICD). For improved visualization, grayscale values were converted to color using the depicted calibration bar. Interestingly, exposure to AngII resulted in significantly enhanced PKARIα translocation to these specific regions of interest. At least 3 independent mice were used per group. Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs vehicle using 1‐way ANOVA with Holm‐Sidak post‐test.
We next investigated PKARIα localization and subsequent functional alterations in excitation–contraction coupling including I Ca gating upon acute AngII exposure. AngII treatment resulted in PKARIα translocation to subcellular compartments, which are of great relevance for excitation–contraction coupling, including the transverse tubules, intercalated discs, and the nucleus (Figure 1D and 1E). Functionally, AngII‐mediated PKARIα activation significantly increased peak I Ca in WT cells, whereas it had no effect on peak I Ca in KI cells (Figure 2A and 2B). This stimulatory effect of AngII on I Ca was PKA dependent, because the PKA inhibitor H89 (5 µmol/L) completely prevented the increase in peak I Ca in the presence of AngII in WT cells. As a potential mechanism of oxidized PKARIα, serine 1928 phosphorylation at LTCC was found to be increased in WT but not in KI myocytes (Figure 2C and 2D).
Figure 2. AngII acutely regulates L‐type Ca current (I Ca) gating by oxidized PKARIα (protein kinase A type I‐α regulatory subunit).
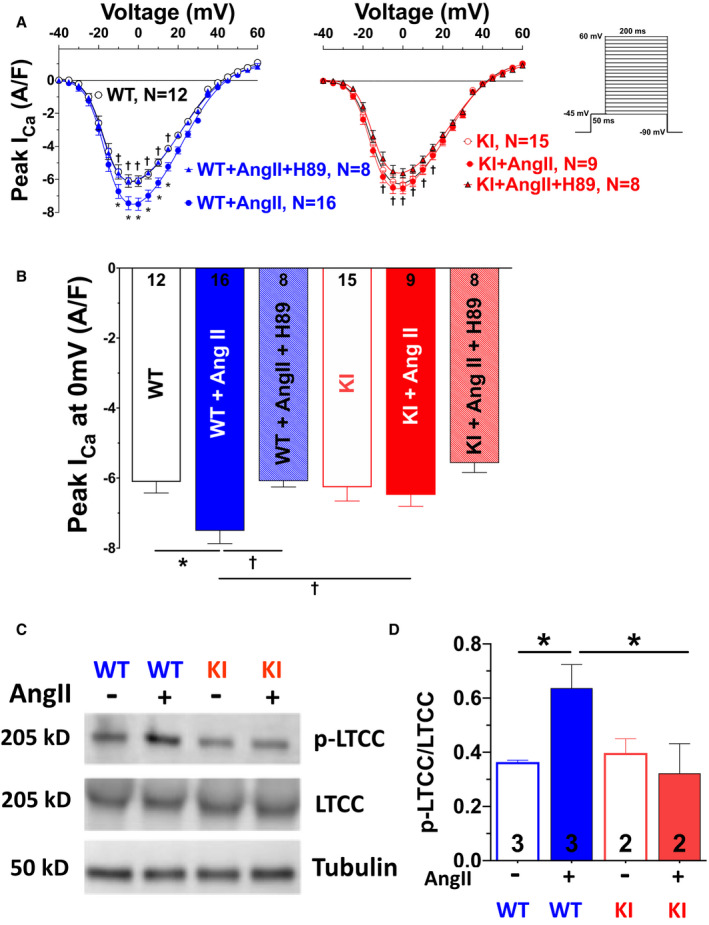
Mean data for peak I Ca voltage relationship (A) and peak I Ca at 0 mV (B) measured by whole‐cell patch‐clamp technique in isolated mouse ventricular myocytes (protocol depicted on the right in A). Acute exposure to AngII (angiotensin II; 1 μmol/L, 10 minutes) significantly enhances peak I Ca, which could be blocked by inhibition of protein kinase A (PKA) using the PKA inhibitor H89. This AngII‐PKA–dependent enhancement of I Ca was completely absent in knock‐in (KI) myocytes. Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs wild‐type (WT). †Indicates significance vs WT+AngII (2‐way repeated‐measures ANOVA, mixed‐effects model with Tukey post‐test). C and D, Original scans (C) and mean densitometric values (D) for Western blot analysis of L‐type Ca channel, alpha 1C subunit (CaV1.2) expression and serine 1928 phosphorylation (p‐LTCC [L‐type Ca channel]) in isolated hearts perfused with AngII (1 μmol/L, 10 minutes). Exposure to AngII significantly enhanced LTCC phosphorylation in WT but not KI mice. Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs WT+AngII using 1‐way ANOVA with Holm‐Sidak post‐test.
The functional relevance of PKARIα‐dependent I Ca regulation for ECC, respectively, for the systolic Ca transient was further corroborated by the fact that AngII‐treated KI myocytes (that lack AngII‐dependent stimulation of peak I Ca) revealed dramatically decreased Ca transient amplitudes upon AngII treatment at all stimulation frequencies from 0.5 to 3 Hz (Figure 3A and 3B). In contrast, Ca transient decay kinetics that can be taken as an approximate for SR Ca reuptake were found to be unaffected (Figure 3C). AngII exposure further resulted in a similar increase in diastolic Ca leak from the SR (Figure 3D and 3E), and a similarly decreased SR Ca content in KI and WT myocytes (Figure 3F). Notably, PKA inhibition using H89 did not prevent the AngII‐mediated increase in SR Ca leak, suggesting a PKA‐independent mechanism of AngII‐mediated SR Ca leak.
Figure 3. Lack of oxidized PKARIα (protein kinase A type I‐α regulatory subunit) impairs Ca handling upon acute AngII (angiotensin II) exposure.
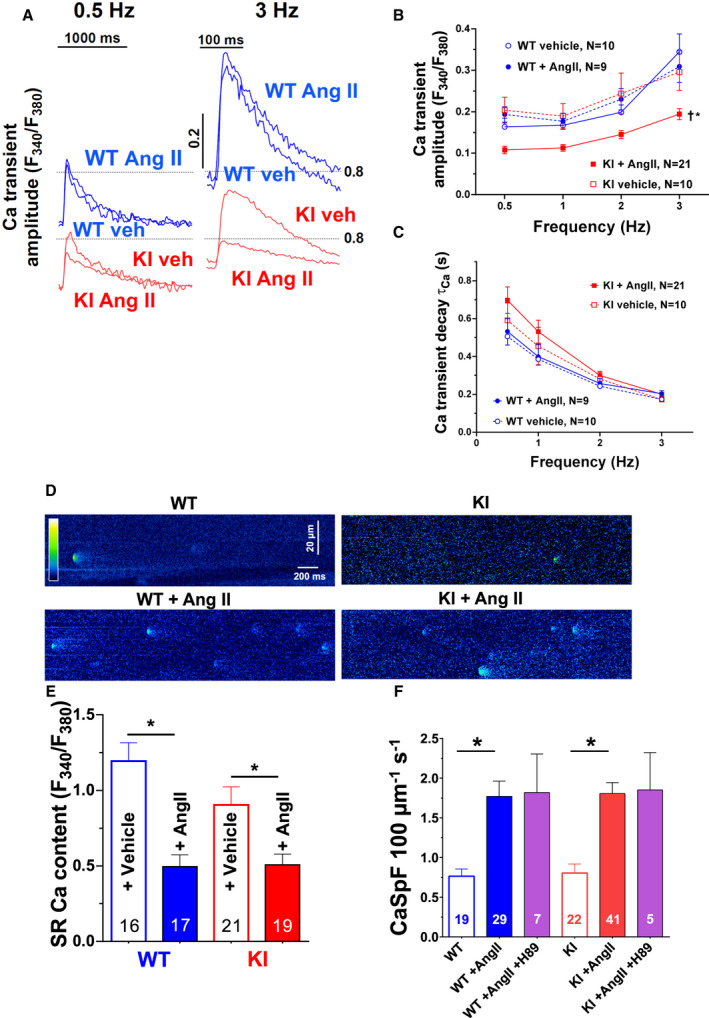
Original traces of intracellular Ca transients at frequencies of 0.5 Hz (left) and 3 Hz (right) are shown in (A). Mean data for Ca transient amplitude upon a force‐frequency protocol (B) and the respective time constant (τ) of Ca transient decay (C) as measured in Fura‐2 loaded ventricular myocytes exposed to AngII or vehicle (veh). In wild‐type (WT) cardiomyocytes, exposure to AngII (1 μmol/L) does not affect Ca transient amplitude nor Ca transient decay. In contrast, AngII exposure significantly reduced Ca transient amplitude but did not alter Ca transient decay in myocytes lacking oxidative activation of PKRIα (knock‐in [KI]). Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs WTAngII. †Indicates significance vs KI vehicle (2‐way repeated‐measures ANOVA, mixed‐effects model). D, Original confocal line scan images of isolated ventricular myocytes loaded with Fluo‐4 from WT (left panel) and KI (right panel) hearts. E, Sarcoplasmic reticulum (SR) Ca content as assessed by caffeine‐induced Ca transients (10 mmol/L) were evoked in ventricular myocytes loaded with Fura‐2. Exposure to AngII significantly reduced SR Ca content in both WT and KI myocytes. Data are normally distributed (Shapiro‐Wilk test). *Indicates significance using 1‐way ANOVA with Holm‐Sidak post‐test. F, Mean data for diastolic Ca spark frequency (CaSpF). Acute exposure to AngII (1 μmol/L) significantly enhanced CaSpF in both WT and KI myocytes to a similar extent. This increase was not abolished by PKA (protein kinase A) inhibition using the pharmacological PKA inhibitor H89. At least 3 independent mice were used per group. Data are not normally distributed. *Indicates significance using Kruskal‐Wallis test with Dunn post‐test. F340/F380 indicates the fluorescence intensity ratio measured with Fura‐2.
These observations collectively indicate that oxidized PKARIα is critical for the maintenance of the systolic Ca transient through stimulation of I Ca in the context of acute oxidative stress.
Decreased Survival and Enhanced Heart Failure in KI Mice Following TAC
We next aimed to investigate the pathophysiologic relevance of PKARIα‐dependent I Ca regulation in the context of long‐term cardiac pathology in vivo by performing TAC surgery leading to HF, which is a well‐established model of chronic oxidative stress. As recently reported, 18 , 27 WT and KI mice did not show structural or functional cardiac abnormalities at baseline as revealed by in vivo echocardiography (Table 1). However, compared with WT, KI mice had decreased survival following TAC (Kaplan‐Meier analysis in Figure 4A). This was associated with aggravated left ventricular (LV) dysfunction already manifesting as early as 1 week after TAC when LVEF was reduced by ~6% (Figure 4B and 4C and Table 1). Interestingly, features of structural LV remodeling like LV hypertrophy (Figure 4D, Figure S1A and S1B), fibrosis (Figure S1C and S1D), or reduced capillary density (Figure S1E and S1F) were not different at 1 week after TAC. This finding suggests that a pathomechanism affecting the contractile function of cardiomyocytes may be present in KI mice in the first days after TAC. In contrast, at 6 weeks after TAC, cellular hypertrophy was significantly more pronounced in KI hearts, and fibrosis slightly increased in trend (P=0.075, see Figure S1C and S1D). Consistent with aggravated HF development in KI mice, LVEF deteriorated even more in KI mice (by ~9% as compared with WT) (Figure 4C), and LV dilatation was more evident late after TAC (Figure 4E). Likewise, wet weight of excised hearts, which integrates LV dilatation (ie, volume) and LV mass, was significantly more increased in KI mice 6 weeks after TAC. Heart weight normalized to body weight was 16.63±1.01 versus 13.57±1.05 mg/g (KI versus WT, P<0.05) indicating functionally and structurally enhanced HF in KI late after TAC. In contrast, long‐term treatment with pressor doses of AngII (at 1 mg/kg−1 per d−1) 22 resulted in a similar hypertensive response in WT and KI mice at comparable heart rates (Figure S2), which suggests that oxidized PKARIα may not be of major relevance to blood pressure regulation in the context of chronic oxidative stress.
Figure 4. Enhanced heart failure development and increased mortality in PKARIα knock‐in (KI) mice.
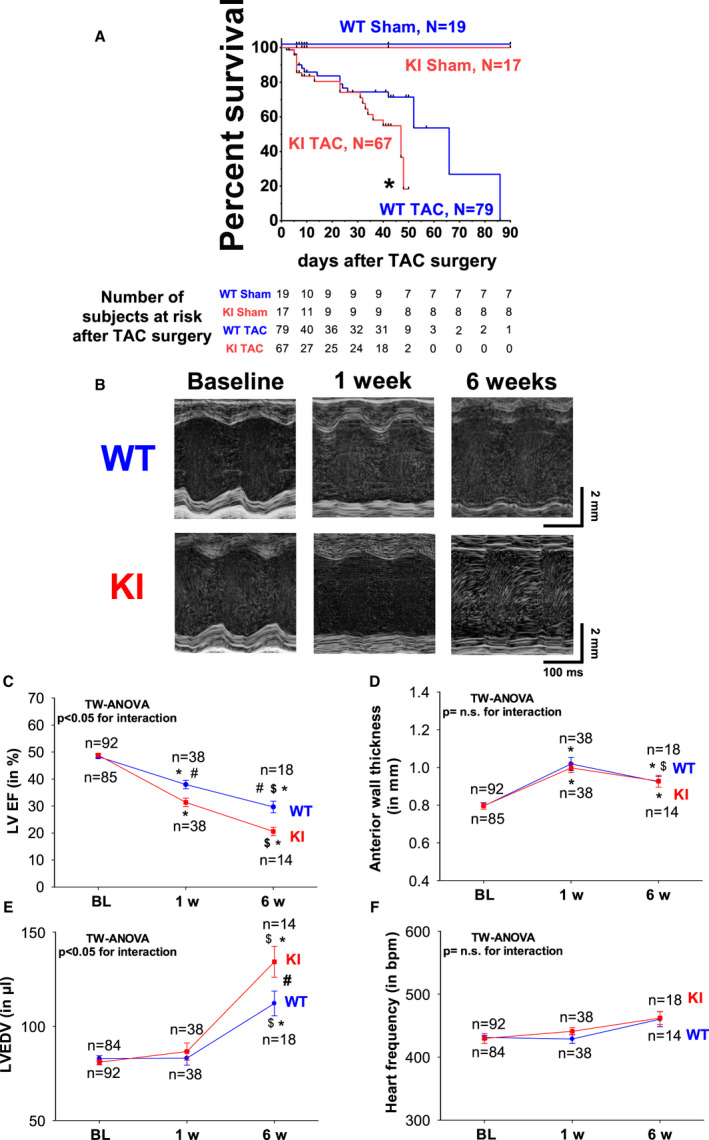
Kaplan‐Meier analysis as depicted in (A) revealed that the survival of KI mice after transverse aortic constriction (TAC) (KI TAC) was dramatically reduced compared with wild‐type (WT) mice (WT TAC) *Indicates significance vs WT TAC). Original M‐mode traces (B) at baseline (BL), 1 week, and 6 weeks after TAC in WT (upper panel) and KI mice (lower panel). KI mice developed aggravated heart failure indicated by reduced left ventricular ejection fraction (LVEF) (C) in the face of comparable left ventricular hypertrophy (indicated by anterior wall thickness) (D) and initially compensated left ventricular end diastolic volume (LVEDV) (E). Heart frequency (F) was comparable between groups. Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs baseline. #Indicates significance vs WT. $Indicates significance vs previous phase using 2‐way (TW) ANOVA mixed‐effects model with Holm‐Sidak post‐test. n.s. indicates not significant.
Impaired Systolic Ca Transients and Decreased I Ca in KI Myocytes Following TAC
To test if impaired intracellular Ca handling might contribute to contractile dysfunction and aggravated HF development in KI mice following TAC, Ca handling was assessed in failing myocytes from both genotypes. Consistent with the reduced LVEF that we observed as early as 1 week after TAC in KI mice, we found significantly decreased Ca transient amplitudes (by ~18%) in KI myocytes compared with WT cells at 1 week after TAC (Figure 5A and 5B). In line with this, Ca transient decay was also impaired in KI at 1 week after TAC (Figure 5C). However, the reduction in Ca transient amplitude was not accompanied by reduced SR Ca content or enhanced diastolic SR Ca leak (Figure 5D through 5F), suggesting that the SR Ca leak may not be directly regulated by oxidative‐activated PKARIα and does not underlie reduced Ca transients in the early stage after TAC. Moreover, this observation points to reduced Ca entry through the LTCC as the underlying cause of decreased Ca transients in KI myocytes following TAC. Original traces and quantitative data for the current–voltage relationship revealed that TAC led to a failure of peak I Ca stimulation in KI myocytes at 1 week after TAC, a phenomenon that was present in WT myocytes only (Figure 6). Consistent with the progressive deterioration of LVEF at 6 weeks after TAC, peak I Ca was significantly decreased in KI as compared with baseline, whereas peak I Ca of WT cells was found to be normalized to baseline conditions only (Figure 6A through 6C). Importantly, peak I Ca was persistently reduced in KI myocytes as compared with WT cells following TAC (P<0.05 for interaction using 2‐way ANOVA). These functional observations suggest that a reduction in transsarcolemmal Ca influx via the LTCC may causally underlie decreased Ca transients in KI myocytes in vitro and presumably decreased cardiac function in vivo.
Figure 5. Lack of oxidized PKARIα (protein kinase A type I‐α regulatory subunit) impairs Ca handling as early as 1 week after transverse aortic constriction (TAC).
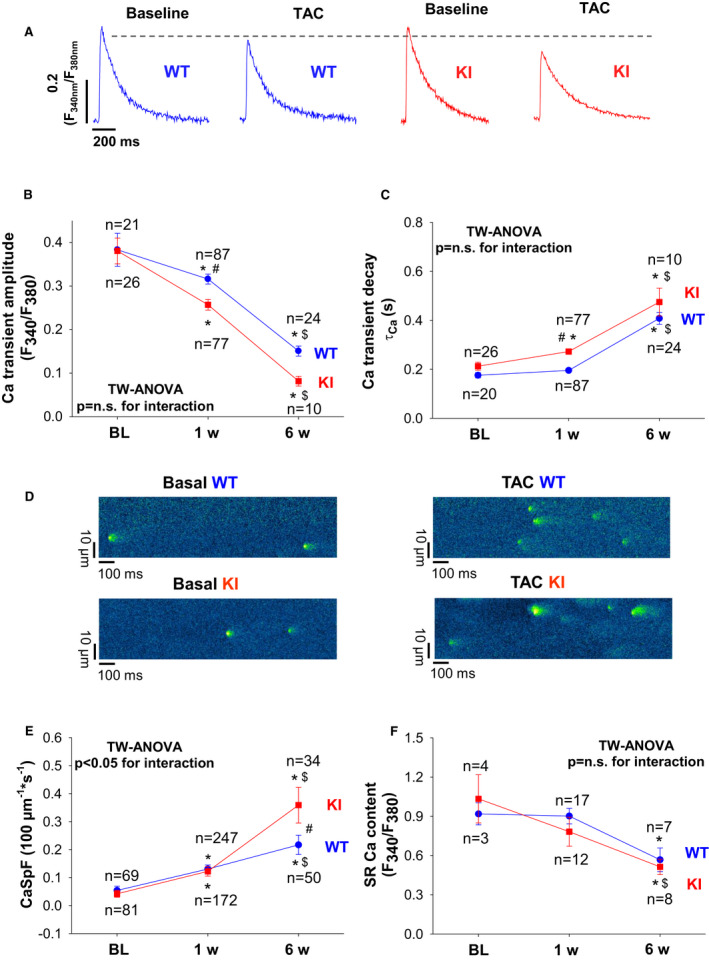
Original traces (A) of intracellular Ca transients measured in Fura‐2–loaded ventricular myocytes from wild‐type (WT) (left panel) and knock‐in (KI) mice (right panel). Consistent with impaired ejection fraction, TAC resulted in significantly reduced Ca transient amplitude. However, this reduction was significantly more pronounced in KI mice. B, Mean data for Ca transient amplitudes during the time course of 6 weeks after TAC illustrate that Ca transient amplitudes were significantly decreased in KI as early as 1 week after TAC already. Data are normally distributed (Kolmogorov‐Smirnov test). Two‐way (TW) ANOVA with Holm‐Sidak post‐test. C, Sarcoplasmic reticulum (SR) Ca reuptake as approximated by Ca transient decay (τCa) was significantly increased in KI vs WT cells after TAC. Data are normally distributed (D'Agostino‐Pearson test). Two‐way ANOVA with Holm‐Sidak post‐test. D, Original confocal line scan images of isolated ventricular myocytes from WT and KI hearts loaded with Fluo‐4 at baseline (left panel) and following TAC (right panel). E, Mean data for diastolic Ca spark frequency (CaSpF) illustrate that CaSpF is increased in KI late after TAC (ie, after 6 weeks), but largely comparable at 1 week after TAC. Data are normally distributed (Shapiro‐Wilk test). Two‐way ANOVA with Holm‐Sidak post‐test. F, Mean data for SR Ca content as assessed by caffeine‐induced Ca transients. Data are normally distributed (Shapiro‐Wilk‐test). Two‐way ANOVA with Holm‐Sidak post‐test. *Indicates significance vs baseline (BL). #Indicates significance vs WT. $Indicates significance vs previous phase. n.s. indicates not significant. F340/F380 indicates the fluorescence intensity ratio measured with Fura‐2.
Figure 6. Oxidized PKARIα (protein kinase A type I‐α regulatory subunit) is required to stimulate I Ca upon transverse aortic constriction (TAC).
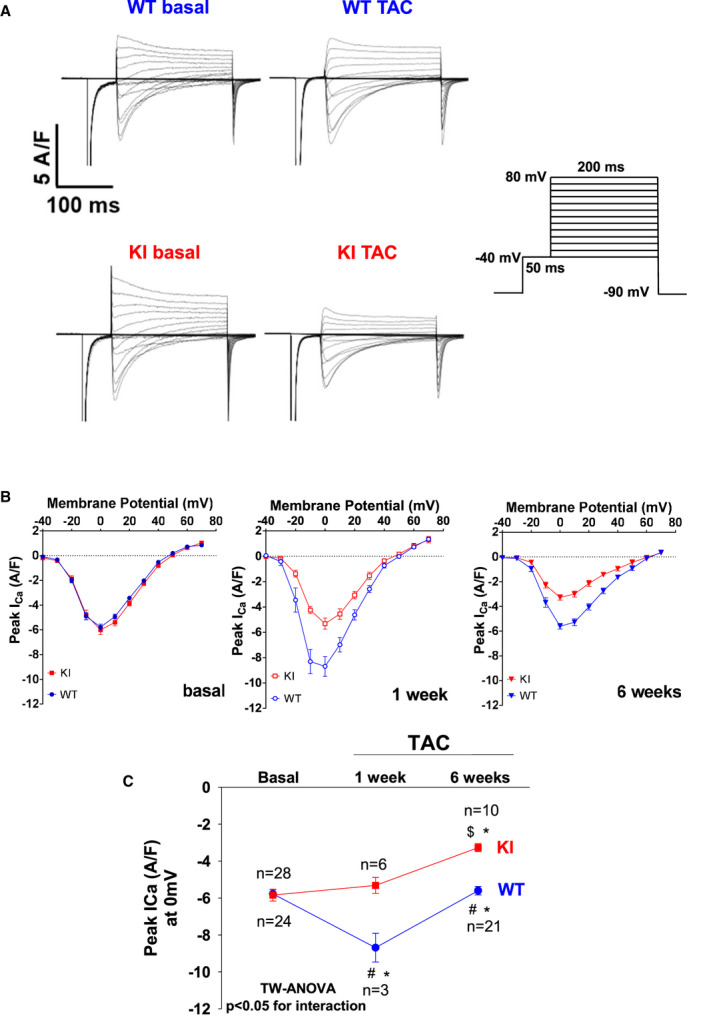
A, Original traces for I Ca measured by whole‐cell rupture‐patch clamp technique (protocol in the right panel) in isolated ventricular myocytes at baseline and late after TAC (ie, at 6 weeks). B, Mean data for peak I Ca voltage relationship at baseline (left panel), at 1 week after TAC (middle panel), and at 6 weeks after TAC (right panel). Peak I Ca at 0 mV is shown in (C). While I Ca density was transiently increased in wild‐type (WT) at 1 week after TAC and was still maintained at 6 weeks after TAC as compared with baseline, knock‐in (KI) cells displayed a lack of I Ca stimulation at 1 week and a reduction of peak I Ca at 6 weeks after TAC. Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs baseline. #Indicates significance vs WT. $Indicates significance vs previous phase using 2‐way (TW) ANOVA with Holm‐Sidak post‐test.
PKA Activation and Signaling During TAC‐Induced HF Development in KI Hearts
The enhanced HF development in KI mice after TAC suggests that oxidative activation of PKARIα may be required for the heart to adapt to pressure overload. We therefore aimed to investigate potential upstream signals and downstream targets of oxidized PKARIα to gain further insights into the presumably adaptive PKARIα signaling following TAC. Consistent with previously reported oxidative stress after TAC, 27 isolated ventricular myocytes exhibited an enhanced CellROX fluorescence signal as a measure of intracellular ROS production (Figure 7A and 7B). Importantly, there was no difference in ROS production between WT and KI mice after TAC, which implies that the lack of PKARIα oxidation in KI mice is not because of different levels of ROS production following TAC. On the protein level, PKARIα dimerization was completely absent in KI hearts both at baseline and following TAC, with a transient increase in WT at 1 week after TAC (P<0.05 for interaction using 2‐way ANOVA (Figure 7C and 7D and Tables 2, 3, 4). However, expression and PKA‐dependent phosphorylation of important SR Ca handling proteins such as the RyR2a (ryanodine receptor 2) (at serine 2809) and PLB (at serine 16) were largely comparable between groups (Figure 7C and Tables 2, 3, 4), which was in line with our observation of largely unaffected SR Ca handling. We found robust cAMP generation in our TAC model as indicated by the representative FLIM FRET recordings in Figure S3A through S3C, yet intracellular cAMP levels as well as global PKA activity were not different between groups following TAC (Figure 7E and 7F). This again points to the fact that oxidized PKARIα may rather act in distinct intracellular compartments such as within the proximity of the LTCC. We therefore assessed the phosphorylation site serine 1928 at the LTCC at baseline and following TAC (see Figure S4A and S4B) and observed increased serine 1928 phosphorylation following TAC in WT hearts only. As with acute AngII exposure, this finding suggests that oxidized PKARIα may selectively act in close proximity to LTCC, possibly through direct target phosphorylation.
Figure 7. Absent PKARIα (protein kinase A type I‐α regulatory subunit) oxidation in PKARIα knock‐in (KI) following transverse aortic constriction (TAC).
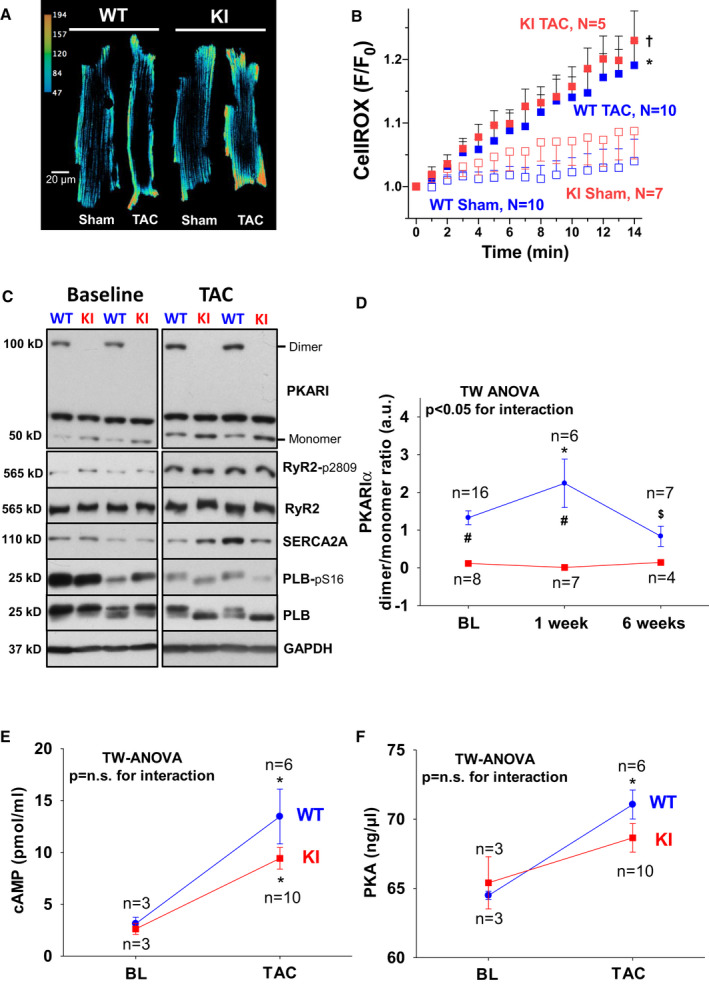
Original traces (A) and mean data (B) of CellROX‐loaded isolated ventricular myocytes from wild‐type (WT) (left panel) and KI (right panel) following sham and TAC surgery imaged every minute for 13 minutes by confocal microscopy. F/F0 indicates the fluorescence intensity normalized to baseline fluorescence. For improved visualization, gray scale values were converted to color using the depicted calibration bar. TAC induced cytosolic ROS to a similar extent in WT and KI myocytes as depicted in (B). Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs KI sham. †Indicates significance vs WT sham (using 2‐way [TW] ANOVA mixed‐effects model with Tukey post‐test). C, Original Western blots depict PKARIα dimer formation in WT hearts at baseline (BL) (upper left panel) and following TAC (upper right panel) that is completely absent in KI samples. Bottom panels in (C) depict representative Western blots of important protein kinase A (PKA)‐dependent target proteins including the RyR2 (ryanodine receptor type 2) (and PKA‐specific phosphorylation at serine 2809), SERCA2a (SR Ca ATPase 2a), and PLB (phospholamban) (and PKA‐specific phosphorylation at serine 16). Mean data for PKARIα oxidation (ie, dimer to monomer ratio) are given in (D). E, A TAC‐related increase in cAMP (3',5'‐cyclic adenosine monophosphate) was observed to a similar extent in WT and KI hearts. F, Comparable activity of PKA following TAC between groups. D, Data are normally distributed (D'Agostino‐Pearson test). Two‐way ANOVA with Holm–Sidak post‐test. E and F, Data are normally distributed (Shapiro‐Wilk test). Two‐way ANOVA with Holm‐Sidak post‐test. *Indicates significance vs baseline. #Indicates significance vs WT. $Indicates significance vs previous phase. GAPDH indicates glyceraldehyde‐3‐phosphate dehydrogenase; and n.s., not significant.
Table 2.
Baseline
| Protein | WT | n | KI | n | P value |
|---|---|---|---|---|---|
| SERCA2a | 1.00±0.44 | 3 | 1.49±0.35 | 3 | 0.44 |
| LTCC | 1.00±0.22 | 3 | 1.19±0.06 | 3 | 0.46 |
| PLB | 1.00±0.10 | 3 | 1.10±0.11 | 3 | 0.56 |
| pS16/PLB | 1.00±0.30 | 3 | 0.77±0.28 | 3 | 0.61 |
| pT17/PLB | 1.00±0.13 | 3 | 0.69±0.21 | 3 | 0.28 |
| RyR2 | 1.00±0.06 | 3 | 1.05±0.12 | 3 | 0.70 |
| pS2809/RyR2 | 1.00±0.03 | 3 | 1.95±0.30 | 3 | 0.03 |
| pS2814/RyR2 | 1.00±0.06 | 3 | 1.17±0.17 | 3 | 0.40 |
| PKA [C] | 1.00±0.04 | 3 | 1.11±0.00 | 3 | 0.05 |
| PKARIα dimer/monomer | 1.000±0.038 | 3 | 0.005±0.001 | 3 | <0.05* |
| CaMKII | 1.00±0.15 | 3 | 1.01±0.05 | 3 | 0.95 |
| pCaMKII | 1.00±0.07 | 3 | 1.09±0.21 | 3 | 0.70 |
| oxCaMKII | 1.00±0.08 | 3 | 0.97±0.05 | 3 | 0.78 |
CaMKII indicates Ca/calmodulin‐dependent protein kinase II; KI, knock‐in; LTCC, L‐type calcium channel; oxCaMKII, oxidized CaMKII; pCaMKII, phosphorylated CaMKII; PKA [C], protein kinase A, catalytic subunit; PKARIα, protein kinase A type I‐α regulatory subunit; PLB, phospholamban; pS16, phosphorylation at serine 16); pS2809, phosphorylation at serine 2809; pS2814, phosphorylation at serine 2814; pT17, phosphorylation at threonine 17; RyR2, ryanodine receptor 2; SERCA2a, sarcoplasmic reticulum Ca‐ATPase 2a; and WT, wild‐type.
Indicates significance using Student’s unpaired t‐test.
Table 3.
Protein levels at 1 week
| Protein | WT | n | KI | n | P value |
|---|---|---|---|---|---|
| Sham (1 wk) | |||||
| SERCA2a | 1.00±0.15 | 3 | 0.76±0.19 | 3 | 0.37 |
| LTCC | 1.00±0.29 | 3 | 0.89±0.35 | 3 | 0.82 |
| PLB | 1.00±0.41 | 3 | 1.50±0.58 | 3 | 0.52 |
| pS16/PLB | 1.00±0.39 | 3 | 0.88±0.40 | 3 | 0.84 |
| pT17/PLB | 1.00±0.28 | 3 | 0.78±0.45 | 3 | 0.70 |
| RyR2 | 1.00±0.03 | 3 | 1.10±0.04 | 3 | 0.12 |
| pS2809/RyR2 | 1.00±0.39 | 3 | 0.99±0.17 | 3 | 0.98 |
| pS2814/RyR2 | 1.00±0.21 | 3 | 0.95±0.18 | 3 | 0.86 |
| PKA [C] | 1 00±0.04 | 3 | 1.22±0.08 | 3 | 0.08 |
| PKARIα dimer/monomer | 1.000±0.197 | 3 | 0.002±0.000 | 3 | <0.05* |
| CaMKII | 1.00±0.23 | 3 | 0.96±0.11 | 3 | 0.90 |
| pCaMKII | 1.00±0.23 | 3 | 0.58±0.19 | 3 | 0.23 |
| oxCaMKII | 1.00±0.23 | 3 | 1.04±0.16 | 3 | 0.89 |
| TAC, 1 wk | |||||
| SERCA2a | 1.00±0.23 | 6 | 0.92±0.10 | 7 | 0.74 |
| LTCC | 1.00±0.14 | 6 | 1.36±0.21 | 7 | 0.18 |
| PLB | 1.00±0.09 | 6 | 1.26±0.16 | 7 | 0.21 |
| pS16/PLB | 1.00±0.19 | 6 | 1.02±0.31 | 7 | 0.97 |
| pT17/PLB | 1.00±0.14 | 6 | 0.64±0.14 | 7 | 0.10 |
| RyR2 | 1.00±0.07 | 6 | 1.05±0.06 | 7 | 0.57 |
| pS2809/RyR2 | 1.00±0.20 | 6 | 1.05±0.12 | 7 | 0.84 |
| pS2814/RyR2 | 1.00±0.12 | 6 | 0.70±0.08 | 7 | 0.06 |
| PKA [C] | 1 00±0.03 | 6 | 1.00±0.02 | 7 | 0.99 |
| CaMKII | 1.00±0.06 | 6 | 1.03±0.05 | 7 | 0.71 |
| pCaMKII | 1.00±0.36 | 6 | 1.07±0.47 | 7 | 0.91 |
| oxCaMKII | 1.00±0.10 | 6 | 1.03±0.11 | 7 | 0.83 |
| Troponin | 1.00±0.34 | 6 | 0.69±0.21 | 7 | 0.43 |
| pTroponin | 1.00±0.30 | 6 | 0.92±0.26 | 7 | 0.84 |
CaMKII indicates Ca/calmodulin‐dependent protein kinase II; KI, knock‐in; LTCC, L‐type calcium channel; oxCaMKII, oxidized CaMKII; pCaMKII, phosphorylated CaMKII; PKA [C], protein kinase A, catalytic subunit; PKARIα, protein kinase A type I‐α regulatory subunit; PLB, phospholamban; pS16, phosphorylation at serine 16; pS2809, phosphorylation at serine 2809; pS2814, phosphorylation at serine 2814; pT17, phosphorylation at threonine 17; pTroponin, phosphorylated troponin; RyR2, ryanodine receptor 2; SERCA2a, sarcoplasmic reticulum Ca‐ATPase 2a; TAC, transverse aortic constriction; and WT, wild‐type
Indicates significance using Student’s unpaired t‐test.
Table 4.
Protein levels at 6 weeks
| Protein | WT | n | KI | n | P value |
|---|---|---|---|---|---|
| Sham (6 wk) | |||||
| SERCA2a | 1.00±0.28 | 4 | 1.25±0.48 | 3 | 0.41 |
| PLB | 1.00±0.34 | 4 | 0.96±0.55 | 3 | 0.91 |
| pS16/PLB | 1.00±0.34 | 3 | 0.57±0.22 | 3 | 0.14 |
| RyR2 | 1.00±0.21 | 3 | 0.98±0.20 | 3 | 0.91 |
| pS2809/RyR2 | 1.00±0.23 | 3 | 1.36±0.05 | 2 | 0.13 |
| PKA [C] | 1.00±0.11 | 3 | 0.93±0.04 | 2 | 0.46 |
| PKARIα dimer/monomer | 1.00±0.37 | 4 | 0.05±0.02 | 4 | <0.05* |
| CaMKII | 1.00±0.22 | 3 | 1.02±0.13 | 2 | 0.91 |
| pCaMKII | 1.00±0.42 | 3 | 1.68±0.56 | 2 | 0.21 |
| TAC (6 wk) | |||||
| SERCA2a | 1.00±0.26 | 4 | 1.37±0.12 | 5 | <0.05* |
| PLB | 1.00±0.54 | 4 | 0.81±0.21 | 5 | 0.48 |
| pS16/PLB | 1.00±0.16 | 3 | 0.91±0.27 | 4 | 0.64 |
| RyR2 | 1.00±0.48 | 7 | 0.65±0.25 | 5 | 0.17 |
| pS2809/RyR2 | 1.00±0.10 | 3 | 0.98±0.08 | 3 | 0.79 |
| PKA [C] | 1.00±0.08 | 3 | 0.89±0.12 | 3 | 0.25 |
| CaMKII | 1.00±0.29 | 4 | 0.97±0.14 | 3 | 0.89 |
| pCaMKII | 1.00±0.19 | 4 | 1.06±0.26 | 3 | 0.74 |
CaMKII indicates Ca/calmodulin‐dependent protein kinase II; KI, knock‐in; pCaMKII, phosphorylated CaMKII; PKA [C], protein kinase A, catalytic subunit; PKARIα, protein kinase A type I‐α regulatory subunit; PLB, phospholamban; pS16, phosphorylation at serine 16; pS2809, phosphorylation at serine 2809; RyR2, ryanodine receptor 2; SERCA2a, sarcoplasmic reticulum Ca‐ATPase 2a; TAC, transverse aortic constriction; and WT, wild‐type.
Indicates significance using Student’s unpaired t‐test.
Because Nox4 (nicotinamide adenine dinucleotide phosphate oxidase 4) has been shown to act as an adaptive ROS source upon pressure overload 28 and because Nox4‐dependent ROS formation was shown to be involved in PKARIα oxidation, 16 we tested whether Nox4 may be required for PKARIα oxidation in the context of pressure overload as induced by abdominal aortic banding for 6 weeks. 28 As shown in Figure S4C and S4D, the PKARIα dimer‐to‐monomer ratio was decreased in Nox4 knock‐out hearts, which suggests that Nox4 may contribute to PKARIα oxidation in vivo.
Stimulation of I Ca Using Bay K8644 Rescues Ca Transients in AngII‐Treated KI Myocytes and LV Function in Failing KI Mice Following TAC
We further aimed to investigate the underlying cause for the observed reduction in survival of KI mice following TAC, which in principle may be a consequence of increased arrhythmogenicity and/or decreased LV contractility (ie, pump failure). As shown in Figure S5A and S5B, HF‐related electric remodeling was evident in both groups following TAC in terms of significantly prolonged corrected QT intervals (see also Table S1). Interestingly, ventricular arrhythmias (ie, ventricular tachycardia) could be induced to a similarly increased extent in WT and KI hearts following TAC (Figure S5C and S5D), which speaks against an increased arrhythmogenicity in KI mice as the prime cause of decreased survival following TAC. We therefore tested in a next step whether stimulation of the presumably causative reduction in I Ca in KI (using Bay K8644 in vivo) would rescue reduced contractility in KI. Bay K8644 significantly increased Ca transient amplitudes in AngII‐treated WT and KI myocytes, thereby normalizing the difference between WT and KI cells that was present in the absence of I Ca stimulation using Bay K8644 (Figure 8A and 8B). Similarly, Bay K8644 led to a significantly increased inotropic response in TAC‐operated KI mice (relative increase in ejection fraction by 56%±12%, n=10) as compared with WT mice (by 12%±16%, n=8) at 6 minutes after IP injection in vivo (see Figure 8C and 8D). Importantly, this occurred in the face of unaffected heart rates upon Bay K8644 in both groups (heart rate was 452±16 bpm in WT mice and 460±10 bpm in KI mice following Bay K8644, P=not significant), which in summary underscores the pathologic relevance of reduced I Ca for impaired cardiac contractility in KI mice in HF.
Figure 8. Pharmacological stimulation of I Ca (L‐type Ca current) using Bay K8644 restores Ca transients in AngII (angiotensin II)‐treated knock‐in (KI) myocytes and rescues left ventricular dysfunction in failing KI mice following transverse aortic constriction (TAC).
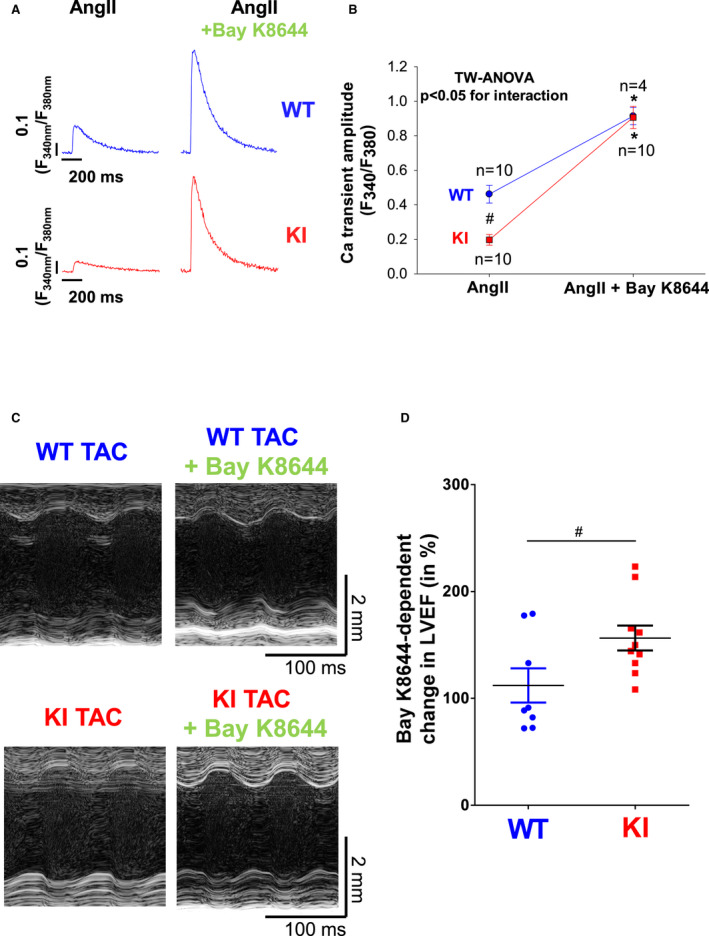
A, Original traces (A) of intracellular Ca transients measured in Fura‐2 loaded ventricular myocytes from wild‐type (WT) (upper panel) and KI mice (lower panel) in the absence (left) and presence of 1 µmol/L Bay K8644 (right). F340/F380 indicates the fluorescence intensity ratio measured with Fura‐2. Although Ca transients were significantly reduced in KI cells upon sole AngII treatment (left), addition of Bay K8644 induced a significant increase in Ca transient amplitude in both groups that was significantly more pronounced in KI cells (right). Average data for (A) are depicted in (B). Data are normally distributed (Shapiro‐Wilk test). *Indicates significance vs AngII control using 2‐way (TW) ANOVA with Holm‐Sidak post‐test. C, Original M‐mode traces of WT (upper panel) and KI mice (lower panel) after TAC and following acute stimulation of I Ca using Bay K8644. Average data in (D) demonstrate a significantly enhanced inotropic response in KI mice following acute stimulation of I Ca using Bay K8644. Data are normally distributed (D'Agostino‐Pearson test). #Indicates significance vs WT using unpaired t test. LVEF indicates left ventricular ejection fraction.
Discussion
Our study demonstrates that myocardial PKARIα becomes oxidized upon acute and chronic oxidative stress, induced by acute AngII treatment and in the context of long‐term pressure overload, respectively. Myocytes from KI hearts that lack oxidative activation of PKARIα fail to increase I Ca upon acute stimulation with AngII, whereas KI mice develop enhanced LV dysfunction and have decreased survival upon long‐term pressure overload in the face of persistently reduced I Ca. Conversely, pharmacological stimulation of I Ca using Bay K8644 is capable of restoring Ca transients in AngII‐treated KI myocytes and LV function in failing KI mice.
Our study indicates that oxidized PKARIα may act as an adaptive signal in the heart through stimulation of I Ca, both in the context of acute and chronic myocardial oxidative stress. This should be carefully considered when using antioxidant treatment for HF.
ROS Are Signaling Molecules That Activate PKARIα in Specific Compartments of the Cardiomyocyte
HF is characterized by increased myocardial ROS production. 29 However, large clinical trials of antioxidant therapies failed to show protection against cardiovascular diseases. This may be because of the fact that ROS have also been considered as important physiological signaling molecules. 1 In that regard, cAMP‐independent activation of PKARIα by oxidation 15 has been described to be required for ischemia‐dependent angiogenesis 16 as well as for PDGF‐dependent signaling. 17 Recently, oxidation of PKARIα was further shown to protect against acute ischemia/reperfusion injury ex vivo, most likely through negative regulation of 2‐pore channel‐dependent excess Ca release from the lysosome. 18 In line with this, we observed acute AngII/ROS‐mediated oxidation and translocation of PKARIα to distinct cellular compartments, which included transverse tubules, intercalated discs, as well as the nucleus. This corresponds well with our functional observation that oxidized PKARIα specifically regulates LTCC at the sarcolemmal membrane. It should be noted that Simon et al reported distinct localization of PKARIα to the lysosomes in their model of untreated cell culture that per se resulted in near‐complete PKARIα disulfide formation. However, distinct stimuli and sources of ROS may differentially influence localization of PKARIα. Here, we used external application of AngII or performed TAC surgery, both of which are known to activate Nox2, which is localized at the cell membrane. 27 , 30 Nox2‐dependent ROS could have thereby contributed to oxidize PKARIα at the cell membrane/transverse tubules (ie, in the proximity of the LTCC). In addition, we found evidence that Nox4 may contribute to PKARIα oxidation following pressure overload as well, because Nox4‐deficient hearts subjected to abdominal aortic banding had less dimerized PKARIα and more monomeric PKARIα as compared with WT following abdominal aortic banding. This suggests that different sources and species of ROS (ie, Nox2 and O2 −, respectively, Nox4 and H2O2) may be capable of oxidizing PKARIα upon pressure overload. Because Nox4 is known to exert adaptive signaling upon pressure overload, 28 oxidized PKARIα could represent a likely downstream signal for Nox4, which warrants further investigation.
Importantly, PKARIα translocation and activation occurred despite comparably elevated levels of ROS and cAMP following TAC. Furthermore, despite absent dimerization of PKARIα in KI mice, global PKA activity was found to be unaffected and the PKA‐dependent phosphorylation status of important Ca handling proteins such as at the RyR2 and PLB (at serine 2809 and serine 16, respectively) was not affected. Likewise, we did not observe different expression levels of important Ca handling proteins in WT versus KI hearts during the development of HF that included SERCA2a (SR Ca ATPase 2a), RyR2, PLB, CaMKII, and PKA [C] (protein kinase A, catalytic subunit). These observations collectively indicate that oxidized PKARIα translocates to distinct cellular compartments upon disease‐related ROS, where it differentially regulates Ca handling proteins including LTCC.
Oxidized PKARIα Maintains Systolic Ca Transients Through Stimulation of L‐Type Ca Current Upon Acute Oxidative Stress as Induced by AngII
Although Simon et al found no relevant changes in ECC in untreated KI myocytes under basal conditions, 18 we used AngII treatment to investigate the acute consequences of oxidized PKARIα for excitation–contraction coupling in the absence of an HF phenotype that becomes relevant following TAC. The addition of AngII was needed to unmask the ECC phenotype of KI cells that did not show relevant differences in ECC upon vehicle treatment in our hands as well. AngII acutely increased peak I Ca in a PKA‐dependent manner in WT cells, and the PKA inhibitor H89 blocked this stimulatory effect. Even more importantly, AngII‐dependent stimulation of peak I Ca was completely abolished in KI cells. The functional relevance of defective peak I Ca stimulation for intracellular Ca handling in KI cells became evident in terms of acutely reduced Ca transient amplitudes that we observed at all stimulation frequencies in our force‐frequency protocol. The causative role of reduced transsarcolemmal Ca influx through the LTCC for impaired systolic Ca transients in KI cells may be further derived from the fact that SR Ca content was comparably impaired in WT and KI cells upon AngII. Although Ca spark frequency as a measure of diastolic SR Ca loss was clearly increased upon AngII, this occurred to a similar extent in both groups and in a manner independent from PKA, because H89 failed to prevent SR Ca leakage. Other triggers of SR Ca loss such as direct ROS‐dependent oxidation of RyR2 31 or CaMKII‐dependent RyR2 regulation may have been relevant for SR Ca leakage in the context of AngII exposure as published previously. 5
Enhanced HF Upon Chronic Pressure Overload Is Attributable to Reduced Peak I Ca in KI Mice
To further investigate the role of oxidized PKARIα in cardiac pathology in vivo, WT and KI mice underwent TAC surgery to induce long‐term pressure overload as a model of chronic oxidative stress. In the course of HF development following TAC, KI mice showed significantly impaired LV function as compared with WT as early as 1 week after TAC. This early contractile dysfunction in KI mice was evident in the absence of signs of enhanced structural remodeling such as LV or cellular hypertrophy, fibrosis, and reduced capillary density. Hence, we interpret this finding in a way that functional remodeling precedes structural remodeling in KI mice that ultimately appeared late after TAC (ie, at 6 weeks after TAC). On the cellular level, KI myocytes had reduced Ca transient amplitudes as compared with WT cells at 1 week after TAC, which should have contributed to reduced LV function as observed in vivo. It is well documented that Ca transients in failing myocytes exhibit slowed decay kinetics. 32 , 33 Significant prolongation of Ca transient decay upon TAC was observed in our model as well, but with only minor difference between WT and KI and in the face of comparable PLB phosphorylation at serine 16. As already mentioned above, no slowing of Ca transient decay was observed upon acute exposure to AngII, which suggests that PKARIα is not critically involved in the regulation of PLB and SR Ca reuptake in the context of increased ROS formation. As with acute AngII treatment, Ca spark frequency was similarly increased in the face of a comparable SR Ca content following TAC. This suggests that unlike PKARII (protein kinase A type II regulatory subunit), 9 oxidized PKARIα may not be involved in the regulation of SR Ca content and release. Accordingly, PKA‐dependent phosphorylation at the RyR2 was similar between groups following TAC. In sharp contrast, peak I Ca was greatly and persistently reduced in KI as compared with WT cells throughout TAC‐induced HF development. Early after TAC (ie, at 1 week), TAC‐related stimulation of peak I Ca was present in WT only, which mimics the situation upon acute AngII exposure. Late after TAC (ie, at 6 weeks), peak I Ca was normalized to baseline conditions in WT cells, whereas at this point in time, KI cells had significantly reduced peak I Ca as compared with baseline. Because SR Ca handling was largely comparable between groups, this observation implies that reduced peak I Ca may be the causative mechanism of reduced Ca transient amplitudes and contractility in KI mice following TAC.
Although it remains controversial whether I Ca current is reduced in cardiomyocytes from animal models or humans in heart failure, 34 there is general agreement that β‐adrenergic I Ca reserve is impaired in HF. 12 Our data are in contrast to previous studies in animal models of heart disease showing that inhibition of LTCC activity may prevent or even reverse pathological cardiac remodeling and hypertrophy. 35 , 36 Moreover, upregulation of I Ca by enhanced expression of the β subunit β2a has been shown to result in cardiac damage. 37 The latter, however, resulted in an artificial basal increase in I Ca by 50% with enhanced contractility, mitochondrial Ca overload, and cellular necrosis. 37
Clinical trials, on the other hand, have not only failed to show a clinical benefit of Ca channel blockers but also resulted in worsening HF and increased mortality. 38
The mechanistic relationship between impaired L‐type Ca current and HF has recently been investigated in more detail. 39 Heterologous knock‐out of CaV1.2 in mice resulted in a 40% reduction in peak I Ca density, which led to an age‐dependent progressive decline of fractional shortening and LV enlargement assessed by echocardiography in vivo. 39 This effect correlated with the magnitude of reduction in CaV1.2 expression. The smaller the expression of CaV1.2, the more severe was the reduction in fractional shortening, the more pronounced was the enlargement of LV volume and the higher was the mortality in these mice. 39 Moreover, mice with heterologous knock‐out of CaV1.2 exhibited enhanced HF development upon TAC, 39 which strikingly resembles the data from the present study. Thus, our study is consistent with clinical data showing that inhibition of LTCC may be detrimental in HF and adds to the growing body of evidence that L‐type Ca current is a major regulator of excitation–contraction coupling not only in healthy myocardium but also in HF.
Pharmacological Stimulation of Peak I Ca Using Bay K8644 Acutely Rescues Ca Transients Amplitudes in AngII‐Treated Cardiomyocytes and LV Function in Failing KI Mice
The crucial relevance of I Ca for ECC in KI is further supported by our observation that acute pharmacological stimulation of I Ca with Bay K8644 restored Ca transients in AngII‐treated KI myocytes and rescued LV function in failing KI mice after TAC. This corroborates the importance of I Ca to maintain cardiac contractility upon oxidative stress and underscores the crucial role of oxidized PKARα to stabilize Ca transients and thereby cardiac contractility via I Ca stimulation. Pump failure may also be the leading cause of increased mortality in KI mice that dramatically increased from 4 weeks after TAC, when structural remodeling also became manifest. Ventricular arrhythmias, on the other hand, were induced to a similar extent in WT and KI mice following TAC. In that regard, the activity and expression of CaMKII, a key regulator of arrhythmogenesis in HF, 23 , 40 was similar in WT and KI mice.
Increased Phosphorylation of the α Subunit of LTCC CaV1.2 as a Potential Mechanism of I Ca Regulation by Oxidized PKARIα
To date, the role for PKARIα in the heart is poorly described. In contrast, PKARII, which is known to bind to A‐kinase anchoring proteins that determine its substrate specificity, has been shown to be involved in phosphorylation and regulation of important cardiac ion channels and transporters. Among them are LTCC and PLB. 9 PKA‐dependent phosphorylation of the α subunit of LTCC CaV1.2 has been shown to stimulate I Ca. 41 , 42 , 43 , 44 , 45 , 46 , 47 PKA‐dependent phosphorylation of PLB at serine 16 results in dissociation of this repressor from SR Ca ATPase, which stimulates SR Ca reuptake. 9 Our study indicates that enhanced ROS production leads to PKARIα oxidation and PKARIα‐dependent stimulation of I Ca. Interestingly, CaV1.2 expression levels did not change; thus, the stimulation of peak I Ca was most likely because of altered I Ca gating, which is consistent with the observed increase in phosphorylation of CaV1.2 at serine 1928. The role of PKARIα for the regulation of I Ca gating was further corroborated by our observation that acute AngII exposure increased peak I Ca density in WT myocytes in parallel to PKARIα dimerization (no masking hypertrophy), which was completely absent in KI myocytes.
In contrast, mice lacking oxidative activation of PKARIα had no stimulation of I Ca early after TAC and showed dramatically reduced I Ca density late after TAC, which was in line with reduced phosphorylation of CaV1.2 at serine 1928.
Interestingly, the involvement of serine 1928 in the PKA‐dependent regulation of LTCC has been suggested before. 42 Many authors have reported that serine 1928 is readily phosphorylated by PKA and can be used as a measure of PKA‐dependent CaV1.2 phosphorylation. 43 However, other reports have questioned the role of serine 1928 but confirmed the importance of the C‐terminal region of CaV1.2. 41 Importantly, all these reports have investigated I Ca stimulation by either direct or indirect increase of cytoplasmic cAMP (forskolin, isoproterenol) or application of cAMP mimetics (8‐Br‐cAMP). Thus, they have investigated the cAMP‐dependent and not the ROS‐dependent stimulation of I Ca. Beside serine 1928, other PKA phosphorylation sites even outside of the C‐terminus (ie, serine 1142, 44 serine 1700, 45 and serine 1829 46 ) have also been suggested. Thus, identification of the exact PKARIα‐specific phosphorylation site that is responsible for the functional alteration of I Ca warrants further investigation but is beyond the scope of the present study.
Limitations
Our study has several important limitations that need to be considered. First, although our study adds evidence that PKARIα acutely translocates to the sarcolemmal membrane upon oxidative stress, which is associated with increased phosphorylation of the LTCC and augmented I Ca (see above), we did not establish a clear protein–protein interaction here, and did not investigate the association of oxidized PKARIα with other compartments such as the lysosomes and lysosomal Ca handling. Second, we cannot exclude that oxidative activation of PKARIα is actually because of a thiol/disulfide exchange reaction only remotely associated with ROS. Third, although it appears tempting to speculate that stimulation of I Ca could serve as a treatment option for HF associated with increased oxidative stress, the long‐term effects of inotropic stimulation using Bay K8644, which would likely increase potentially harmful oxygen consumption, have not been subjects of the current study. Finally, although we present clear evidence of an acute relevance of oxidized PKARIα for I Ca gating (such as upon AngII), we cannot exclude that there may be only a narrow time window during which oxidative PKARIα activation would be relevant, with all long‐term effects following TAC being secondary to this acute regulation. We observed a biphasic behavior of the PKARIα dimer to monomer ratio in WT mice following TAC with a transient increase during the early stage after TAC (ie, at 1 week) and a normalization to baseline conditions late after TAC (ie, at 6 weeks). However, this may be also influenced by altered antioxidants levels in HF that were not investigated here.
Conclusions
Our study demonstrates the following: (1) PKARIα is oxidized in the myocardium of mice subjected to TAC or in myocytes acutely exposed to AngII, both conditions of increased ROS production. (2) Oxidation of PKARIα is critically involved in the regulation of L‐type Ca current possibly by phosphorylation. (3) Mice that lack oxidative activation of PKARIα develop enhanced LV contractile dysfunction because of reduced Ca transient amplitude and have increased mortality upon TAC. Thus, oxidative activation of PKA may be required for the heart to adapt to increased workload. This should be carefully considered when using antioxidant treatment for heart failure.
Sources of Funding
This work was supported by International Research Training Group GRK 1816 of the Deutsche Forschungsgemeinschaft (DFG) to S.W. and L.S.M. C.M.S. is funded by the DFG (SA 3282/1‐1). S.W. is funded by DFG grants WA 2539/4‐1, 5‐1, 7‐1 and 8‐1, and Deutsche Stiftung für Herzforschung grant (F/35/15). M.Z. is funded by the British Heart Foundation (PG/17/39/33027). A.M.S. is supported by the British Heart Foundation (RG/20/3/34823, CH/1999001/11735) and the Fondation Leducq (17CVD04). L.S.M. is funded by DFG grants MA 1981/5‐1 and 7‐1. S.W. and L.S.M. are also funded by the DFG SFB grant 1350 and the ReForM program of the faculty.
Disclosures
None.
Supporting information
Acknowledgments
The authors acknowledge the expert technical assistance of T. Sowa, F. Radtke, and T. Schulte. The authors also thank Drs Schäfer and Gogiraju for their cooperation during the study.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.021985
For Sources of Funding and Disclosures, see pages 20 and 21.
References
- 1. Wagner S, Rokita AG, Anderson ME, Maier LS. Redox regulation of sodium and calcium handling. Antioxid Redox Signal. 2013;18:1063–1077. DOI: 10.1089/ars.2012.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sag CM, Wagner S, Maier LS. Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic Biol Med. 2013;63:338–349. DOI: 10.1016/j.freeradbiomed.2013.05.035. [DOI] [PubMed] [Google Scholar]
- 3. Gill JS, McKenna WJ, Camm AJ. Free radicals irreversibly decrease Ca2+ currents in isolated guinea‐pig ventricular myocytes. Eur J Pharmacol. 1995;292:337–340. [DOI] [PubMed] [Google Scholar]
- 4. Morris TE, Sulakhe PV. Sarcoplasmic reticulum Ca(2+)‐pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic Biol Med. 1997;22:37–47. [DOI] [PubMed] [Google Scholar]
- 5. Wagner S, Dantz C, Flebbe H, Azizian A, Sag CM, Engels S, Möllencamp J, Dybkova N, Islam T, Shah AM, et al. NADPH oxidase 2 mediates angiotensin II‐dependent cellular arrhythmias via PKA and CaMKII. J Mol Cell Cardiol. 2014;75:206–215. DOI: 10.1016/j.yjmcc.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 6. Erickson JR, Joiner M‐L, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin‐Burns N, et al. A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. DOI: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. [DOI] [PubMed] [Google Scholar]
- 8. Anderson ME. Oxidant stress promotes disease by activating CaMKII. J Mol Cell Cardiol. 2015;89:160–167. DOI: 10.1016/j.yjmcc.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bers DM. Excitation‐Contraction Coupling and Cardiac Contractile Force, 2nd ed. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2001. [Google Scholar]
- 10. Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J. Beta2‐adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–1657. DOI: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 11. Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta‐adrenergic receptor kinase and beta 1‐adrenergic receptors in the failing human heart. Circulation. 1993;87:454–463. DOI: 10.1161/01.CIR.87.2.454. [DOI] [PubMed] [Google Scholar]
- 12. Chen X, Piacentino V 3rd, Furukawa S, Goldman B, Margulies KB, Houser SR. L‐type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91:517–524. [DOI] [PubMed] [Google Scholar]
- 13. Han YS, Arroyo J, Ogut O. Human heart failure is accompanied by altered protein kinase A subunit expression and post‐translational state. Arch Biochem Biophys. 2013;538:25–33. DOI: 10.1016/j.abb.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amieux PS, McKnight GS. The essential role of RI alpha in the maintenance of regulated PKA activity. Ann NY Acad Sci. 2002;968:75–95. [DOI] [PubMed] [Google Scholar]
- 15. Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, Begum S, Kentish JC, Eaton P. Oxidant‐induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem. 2006;281:21827–21836. DOI: 10.1074/jbc.M603952200. [DOI] [PubMed] [Google Scholar]
- 16. Burgoyne JR, Rudyk O, Cho HJ, Prysyazhna O, Hathaway N, Weeks A, Evans R, Ng T, Schroder K, Brandes RP, et al. Deficient angiogenesis in redox‐dead Cys17Ser PKARIalpha knock‐in mice. Nat Commun. 2015;6:7920. [DOI] [PubMed] [Google Scholar]
- 17. Eisel F, Boosen M, Beck M, Heide H, Wittig I, Beck KF, Pfeilschifter J. Platelet‐derived growth factor triggers PKA‐mediated signalling by a redox‐dependent mechanism in rat renal mesangial cells. Biochem Pharmacol. 2013;85:101–108. DOI: 10.1016/j.bcp.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 18. Simon JN, Vrellaku B, Monterisi S, Chu SM, Rawlings N, Lomas O, Marchal GA, Waithe D, Syeda F, Gajendragadkar PR, et al. Oxidation of protein kinase A regulatory subunit PKARIα protects against myocardial ischemia‐reperfusion injury by inhibiting lysosomal‐triggered calcium release. Circulation. 2021;143:449–465. DOI: 10.1161/CIRCULATIONAHA.120.046761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trum M, Islam MMT, Lebek S, Baier M, Hegner P, Eaton P, Maier LS, Wagner S. Inhibition of cardiac potassium currents by oxidation‐activated protein kinase A contributes to early afterdepolarizations in the heart. Am J Physiol Heart Circ Physiol. 2020;319:H1347–H1357. DOI: 10.1152/ajpheart.00182.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Toischer K, Rokita AG, Unsöld B, Zhu W, Kararigas G, Sossalla S, Reuter SP, Becker A, Teucher N, Seidler T, et al. Differential cardiac remodeling in preload versus afterload. Circulation. 2010;122:993–1003. DOI: 10.1161/CIRCULATIONAHA.110.943431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q‐T interval in the conscious mouse. Am J Physiol. 1998;274:H747–H751. DOI: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- 22. Sag CM, Schnelle M, Zhang J, Murdoch CE, Kossmann S, Protti A, Santos CXC, Sawyer G, Zhang X, Mongue‐Din H, et al. Distinct regulatory effects of myeloid cell and endothelial cell NAPDH oxidase 2 on blood pressure. Circulation. 2017;135:2163–2177. DOI: 10.1161/CIRCULATIONAHA.116.023877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wagner S, Dybkova N, Rasenack ECL, Jacobshagen C, Fabritz L, Kirchhof P, Maier SKG, Zhang T, Hasenfuss G, Brown JH, et al. Ca2+/calmodulin‐dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. DOI: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Börner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ, Nikolaev VO. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat Protoc. 2011;6:427–438. DOI: 10.1038/nprot.2010.198. [DOI] [PubMed] [Google Scholar]
- 25. Dybkova N, Wagner S, Backs J, Hund TJ, Mohler PJ, Sowa T, Nikolaev VO, Maier LS. Tubulin polymerization disrupts cardiac beta‐adrenergic regulation of late INa. Cardiovasc Res. 2014;103:168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y, Saulnier JL, Yellen G, Sabatini BL. A PKA activity sensor for quantitative analysis of endogenous GPCR signaling via 2‐photon FRET‐FLIM imaging. Front Pharmacol. 2014;5:56. DOI: 10.3389/fphar.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, Shah AM. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J Am Coll Cardiol. 2006;47:817–826. DOI: 10.1016/j.jacc.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 28. Zhang M, Brewer AC, Schroder K, Santos CXC, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, et al. NADPH oxidase‐4 mediates protection against chronic load‐induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA. 2010;107:18121–18126. DOI: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–H2190. DOI: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 30. Zhang M, Prosser BL, Bamboye MA, Gondim ANS, Santos CX, Martin D, Ghigo A, Perino A, Brewer AC, Ward CW, et al. Contractile function during angiotensin‐II activation: increased Nox2 activity modulates cardiac calcium handling via phospholamban phosphorylation. J Am Coll Cardiol. 2015;66:261–272. DOI: 10.1016/j.jacc.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Terentyev D, Györke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. DOI: 10.1161/01.CIR.85.3.1046. [DOI] [PubMed] [Google Scholar]
- 33. O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation‐contraction coupling in canine tachycardia‐induced heart failure, I: experimental studies. Circ Res. 1999;84:562–570. DOI: 10.1161/01.RES.84.5.562. [DOI] [PubMed] [Google Scholar]
- 34. Richard S, Leclercq F, Lemaire S, Piot C, Nargeot J. Ca2+ currents in compensated hypertrophy and heart failure. Cardiovasc Res. 1998;37:300–311. DOI: 10.1016/S0008-6363(97)00273-3. [DOI] [PubMed] [Google Scholar]
- 35. Liao Y, Asakura M, Takashima S, Ogai A, Asano Y, Asanuma H, Minamino T, Tomoike H, Hori M, Kitakaze M. Benidipine, a long‐acting calcium channel blocker, inhibits cardiac remodeling in pressure‐overloaded mice. Cardiovasc Res. 2005;65:879–888. DOI: 10.1016/j.cardiores.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 36. Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, et al. The L‐type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. DOI: 10.1172/JCI200214677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BHL, Hewett TE, Robbins J, et al. Ca2+‐ and mitochondrial‐dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. DOI: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Multicenter Diltiazem Postinfarction Trial Research Group . The effect of diltiazem on mortality and reinfarction after myocardial infarction. N Engl J Med. 1988;319:385–392. [DOI] [PubMed] [Google Scholar]
- 39. Goonasekera SA, Hammer K, Auger‐Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, et al. Decreased cardiac L‐type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012;122:280–290. DOI: 10.1172/JCI58227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, et al. Reactive oxygen species‐activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ganesan AN, Maack C, Johns DC, Sidor A, O'Rourke B. Beta‐adrenergic stimulation of L‐type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of alpha1C but not serine 1928. Circ Res. 2006;98:e11–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. cAMP‐dependent regulation of cardiac L‐type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. DOI: 10.1016/S0896-6273(00)80358-X. [DOI] [PubMed] [Google Scholar]
- 43. De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. Specific phosphorylation of a site in the full‐length form of the alpha 1 subunit of the cardiac L‐type calcium channel by adenosine 3',5'‐cyclic monophosphate‐dependent protein kinase. Biochemistry. 1996;35:10392–10402. [DOI] [PubMed] [Google Scholar]
- 44. Erxleben C, Gomez‐Alegria C, Darden T, Mori Y, Birnbaumer L, Armstrong DL. Modulation of cardiac Ca(V)1.2 channels by dihydropyridine and phosphatase inhibitor requires Ser‐1142 in the domain III pore loop. Proc Natl Acad Sci USA. 2003;100:2929–2934. DOI: 10.1073/pnas.2628046100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight‐or‐flight response. Science Signal. 2010;3:ra70. DOI: 10.1126/scisignal.2001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takahashi E, Fukuda K, Miyoshi S, Murata M, Kato T, Ita M, Tanabe T, Ogawa S. Leukemia inhibitory factor activates cardiac L‐Type Ca2+ channels via phosphorylation of serine 1829 in the rabbit Cav1.2 subunit. Circ Res. 2004;94:1242–1248. [DOI] [PubMed] [Google Scholar]
- 47. Weiss S, Oz S, Benmocha A, Dascal N. Regulation of cardiac L‐type Ca(2)(+) channel CaV1.2 via the beta‐adrenergic‐cAMP‐protein kinase A pathway: old dogmas, advances, and new uncertainties. Circ Res. 2013;113:617–631. [DOI] [PubMed] [Google Scholar]
- 48. Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor‐induced signal propagation. J Biol Chem. 2004;279:37215–37218. DOI: 10.1074/jbc.C400302200. [DOI] [PubMed] [Google Scholar]
- 49. Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ. Persistent cAMP‐signals triggered by internalized G‐protein‐coupled receptors. PLoS Biol. 2009;7:e1000172. DOI: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tournoux F, Petersen B, Thibault H, Zou L, Raher MJ, Kurtz B, Halpern EF, Chaput M, Chao W, Picard MH, et al. Validation of noninvasive measurements of cardiac output in mice using echocardiography. J Am Soc Echocardiogr. 2011;24:465–470. DOI: 10.1016/j.echo.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, et al. Oxidized Ca(2+)/calmodulin‐dependent protein kinase II triggers atrial fibrillation. Circulation. 2013;128:1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–C1081. DOI: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.