
FIGURE 1.
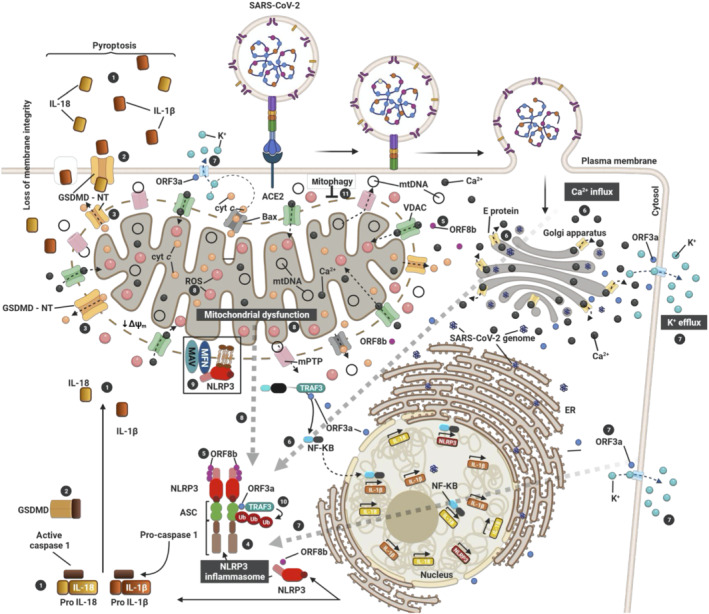
Activation of the NLRP3 inflammasome by SARS‐CoV‐2 via mitochondrial dysfunction. The three SARS‐CoV proteins (E, ORF3a and ORF8b) induce the activation of inflammasome (4). E protein (yellow) induces Ca2+ efflux through ERGIC/Golgi membranes to the cytosol (6). This induces influx into the mitochondria to generate mtROS (8). ORF3a (blue) induces K+ efflux to the extracellular space (7) and promotes inflammasome assembly (4) through TRAF3‐mediated ubiquitination of ASC (7; 10). On the other hand, TRAF3–ORF3a interaction is required for NF‐κβ activation, resulting in transcription of the pro–IL‐1β/IL‐18 and NLRP3 genes. ORF8b (violet) can interact directly with NLRP3 stimulating its activation (5). Consequent to inflammasome activation (4), gasdermin D (GSDMD) pores are formed on the plasma (2) and mitochondrial (3) membranes, causing IL‐1β/IL‐18 secretion (1), the cellular swelling associated with pyroptosis (1) and the induction of mitochondrial apoptotic pathway via Bax‐dependent release of cytochrome C into the cytosol. Additionally, activation of Bax can trigger NLRP3 activation via apoptotic caspases (dotted line) in a K+ efflux‐dependent manner (7). Thus, SARS‐CoV‐2 triggers NLRP3 inflammasome assembly and activation by damaging the mitochondria and inducing the production of mtROS (8) and the loss of mitochondrial membrane potential (ΔΨm) to release damaged mitochondrial DNA (mtDNA) in the cytosol through the mitochondrial pore transition (mPT). Therefore, mitophagy (11) stands as an important regulator of NLRP3 which is tethered on the mitochondria for activation in a mtROS‐dependent manner (black box)