

Summary
To accurately segment cell edges and quantify cellular morphodynamics from live-cell imaging data, we developed a deep learning-based pipeline termed MARS-Net (multiple-microscopy-type-based accurate and robust segmentation network). MARS-Net utilizes transfer learning and data from multiple types of microscopy to localize cell edges with high accuracy. For effective training on distinct types of live-cell microscopy, MARS-Net comprises a pretrained VGG19 encoder with U-Net decoder and dropout layers. We trained MARS-Net on movies from phase-contrast, spinning-disk confocal, and total internal reflection fluorescence microscopes. MARS-Net produced more accurate edge localization than the neural network models trained with single-microscopy-type datasets. We expect that MARS-Net can accelerate the studies of cellular morphodynamics by providing accurate pixel-level segmentation of complex live-cell datasets.
Keywords: deep learning, segmentation, cellular morphodynamics, machine learning, image analysis, live cell analysis, cell protrusion, cell motility
Graphical abstract
Highlights
-
•
A pipeline for segmentation of live cell imaging data using the MARS-Net neural network
-
•
MARS-Net utilizes transfer learning and is trained on multiple types of microscopy
-
•
Accurate, pixel-level segmentation allows quantitative profiling of cell-edge dynamics
Motivation
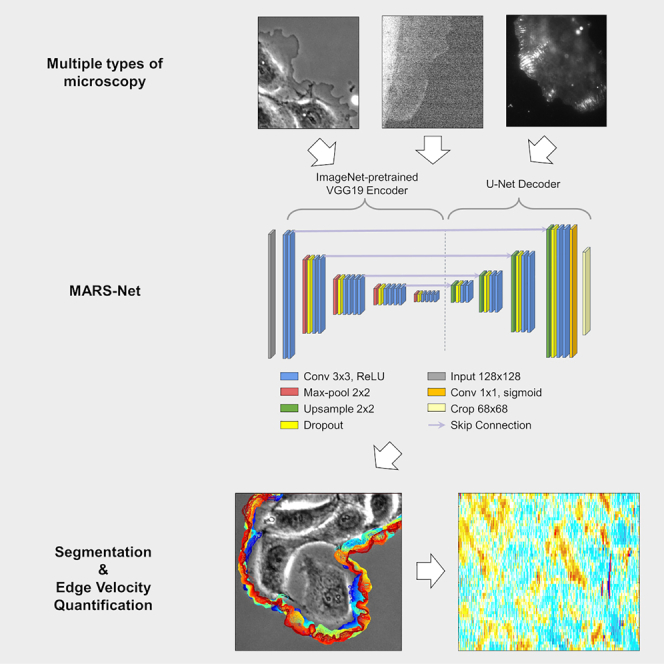
Quantitative studies of cellular morphodynamics rely on extracting leading-edge velocity time series based on accurate cell segmentation from live cell imaging. However, live cell imaging has numerous challenging issues regarding accurate edge localization. Fluorescence live cell imaging produces noisy and low-contrast images due to phototoxicity and photobleaching. While phase contrast microscopy is gentle to live cells, it suffers from the halo and shade-off artifacts that cannot be handled by conventional segmentation algorithms. Here, we present a deep learning-based pipeline, termed MARS-Net (Multiple-microscopy-type-based Accurate and Robust Segmentation Network), that utilizes transfer learning and data from multiple types of microscopy to localize cell edges with high accuracy, allowing quantitative profiling of cellular morphodynamics.
Jang et al. develop a deep learning pipeline for segmentation of live cell imaging data. The pipeline includes MARS-Net, a neural network that achieves higher accuracy through training on multiple types of microscopy data. Accurate cell edge identification in each movie frame allows quantification of edge velocities.
Introduction
Live cell imaging is a fundamental tool to study changes in cellular morphology (morphodynamics), which are involved in cancer metastasis, immune responses, and stem cell differentiation, among others (Buggenthin et al., 2017; Hermans et al., 2013; Leithner et al., 2016; Manak et al., 2018). Cellular morphodynamics is governed by protrusion and retraction of the leading edges of cells, driven by the cytoskeleton and adhesion processes (Lee et al., 2015; Machacek and Danuser, 2006). Due to their phenotypic heterogeneity, computational image analysis in conjunction with machine learning has been employed to understand and characterize cellular morphodynamics (Lee et al., 2015; Machacek and Danuser, 2006; Machacek et al., 2009; Wang et al., 2018, 2021).
Quantitative studies of cellular morphodynamics rely on extracting leading-edge velocity time series. Therefore, accurate and consistent edge segmentation at every frame of a live cell movie is necessary. Fluorescence microscopes can acquire high-contrast cellular images by introducing fluorescently tagged molecules, particularly for fixed cells. Fluorescence imaging, however, causes phototoxicity to live cells, which makes researchers limit light illumination and total image acquisition time. These make fluorescent live cell images noisy, low contrast, and low throughput. Selection of cells with low-level expression of fluorescent proteins and photobleaching further degrades the image quality (Stephens and Allan, 2003). Therefore, having reliable semantic segmentation from live cell images is a significant issue. The alternative to fluorescence microscopy is a label-free phase contrast microscopy that minimizes phototoxicity in the cell. However, phase contrast images contain halo and shade-off artifacts, incurring a significant challenge for reliable cell segmentation (Ambühl et al., 2012; Bensch and Ronneberger, 2015; Li and Kanade, 2009; Vicar et al., 2019).
Numerous conventional segmentation methods already exist, including the Otsu method (Otsu, 1979), the Canny detector (Canny, 1986), the active contour or snake-based method (Chan and Vese, 2001), and the pointwise mutual information (PMI) method (Isola et al., 2014), which rely on a few mathematical assumptions that tend to be broken in live cell imaging conditions. Previous studies on cellular morphodynamics used simple thresholding (Gonzalez et al., 2013) or thresholding followed by conventional image processing (Ma et al., 2018) to segment cells, but these methods did not accurately localize the cell edge for the analysis of cellular morphodynamics of our datasets. Supervised learning with a deep learning model can overcome such problems in conventional methods.
Among deep learning models, convolutional neural network (CNN) excels in pattern recognition in images by learning complex features directly from the input images using its hierarchical structure (LeCun et al., 2015). CNN has achieved great success in image classification (He et al., 2016a, b; Krizhevsky et al., 2012; Simonyan and Zisserman, 2015) and segmentation (Ahmed et al., 2020; Badrinarayanan et al., 2017; Bertasius et al., 2015; Long et al., 2015; Ronneberger et al., 2015; Shen et al., 2015; Van Valen et al., 2016). In particular, U-Net (Ronneberger et al., 2015) is the most widely adopted CNN-based structure for image segmentation and has demonstrated promising segmentation results in static and live cell images (Al-Kofahi et al., 2018; Chai et al., 2018; Falk et al., 2019; Moen et al., 2019; Sadanandan et al., 2017). A U-Net (Ronneberger et al., 2015) is a CNN-based structure comprising an encoder, a decoder, and skip connections in between for segmentation. The architectural improvements of a U-Net-based structure yield even greater segmentation accuracy on microscopy images of cells or nuclei (Ali et al., 2021; Caicedo et al., 2019; Raza, 2019). For instance, U-Net-based models such as StarDist (Schmidt et al., 2018) and CellPose (Stringer et al., 2021) have additional structures or outputs to segment images of crowded cells and nuclei effectively. For the deep learning model's generalizability on various types of cell images, the generalist CellPose (Stringer et al., 2021) was trained on multiple types of cell images and showed superior generalizability compared with StarDist (Schmidt et al., 2018) or original U-Net (Ronneberger et al., 2015) models. Nowadays, deep learning-based segmentation models are accessible even for users without many computational resources or coding skills through image segmentation applications such as CellPose (Stringer et al., 2021), CellProfiler (McQuin et al., 2018), ZeroCostDL4Mic (Chamier et al., 2020), and DeepImageJ (Gómez-De-Mariscal et al., 2019).
Despite this progress, deep learning-based segmentation has not been tested for quantifying leading-edge velocities for morphodynamic profiling of live cells. Therefore, in this work, we focused on developing the deep learning model to increase the semantic segmentation accuracy for reliable estimation of the leading-edge velocities. Also, we investigated the relationship between the number of training frames and segmentation accuracy and discovered the efficient usage of labeled images to reduce labeling costs. Our deep learning framework, termed MARS-Net (Multiple-microscopy-type-based Accurate and Robust Segmentation Network), learns robust image features for accurate segmentation using the datasets from multiple types of microscopy. We reasoned that the cross-modal features learned from images of multiple types of microscopy could achieve more accurate and robust edge localization than the features from the single type of microscopy images. Therefore, we combined training data from live cell movies of migrating PtK1 cells independently taken by different microscopy techniques such as phase contrast, spinning disk confocal (SDC), and total internal reflection fluorescence (TIRF) microscopes.
In this pipeline, we used the U-Net (Ronneberger et al., 2015)-based structure and incorporated the transfer learning technique that initializes the weights of the network with those of the same network trained on ImageNet (Deng et al., 2009) for the image recognition task. Transfer learning has been applied to many deep learning segmentation models (FCN [Long et al., 2015], DeepEdge [Bertasius et al., 2015], TernausNetV2 [Iglovikov et al., 2018]) and classification tasks (Choi et al., 2017; Donahue et al., 2014; Kim et al., 2018; Oquab et al., 2014; Pratt, 1993; Razavian et al., 2014; Yosinski et al., 2014) to achieve high performance with a limited dataset. In addition, transfer learning allows our model to achieve higher edge-localization accuracy on multiple types of microscopy datasets. We replaced the U-Net encoder with one of the image classification networks, such as VGG16/VGG19 (Simonyan and Zisserman, 2015), ResNet50V2 (He et al., 2016b), and EfficientNetB7 (Tan and Le, 2020), and used the initial weights from the ImageNet (Deng et al., 2009) training. Among them, the pretrained VGG19 encoder coupled with U-Net decoder (VGG19-U-Net) segmented the boundary of the cell with the highest accuracy. Dropout (Srivastavanitish et al., 2014) layers were added to the model (VGG19D-U-Net) as a regularization method to prevent overfitting and boost the performance further. MARS-Net (VGG19D-U-Net trained on the images from multiple types of microscopy) was able to segment cell boundaries more accurately than the model trained on single-microscopy-type data, whereas U-Net could not gain significant performance benefit from training on the data from multiple types of microscopy. Also, we demonstrated that MARS-Net enables more reliable quantitative analyses of cellular morphodynamics compared to the single-microscopy-type model.
Results
Overview of the computational pipeline
We prepared the ground truth masks from live cell images semi-automatically using our labeling tool (Figure 1A). The images and the corresponding ground truth masks were preprocessed (see STAR Methods for details), and they were used to train the deep neural networks for segmentation (Figure 1B). The trained neural network generates a segmentation of the cell boundary, which can be used for morphodynamic profiling developed by Danuser's group (Machacek and Danuser, 2006) (Figure 1C). It measures local velocities of the cell boundary throughout the movie and summarizes local velocities for every probing spatial window and time frame. As this quantification method can be sensitive to pixel-level segmentation errors, accurate edge localization is necessary.
Figure 1.

Overview of the computational pipeline
(A) Labeling tool for the preparation of training sets.
(B) Deep learning for segmentation of live cell movies (MARS-Net).
(C) Quantification of cellular morphodynamics. PtK1 cell images acquired by a phase contrast microscope. Bars: 32.5 μm.
Deep learning requires large training datasets, and manual labeling of many frames per live cell movie can take several hours or days. Also, there is an inconsistency in the quality of labeled images depending on the labeler's experience. Therefore, we created the cell labeling tool to reduce manual labor by automating most labeling procedures and produce accurate and consistent labels. A systematic approach to create labels promotes reliable training and evaluation of the deep learning model (Bertram et al., 2020; Falk et al., 2019). The labeling tool takes the input image through a series of image processing operations as follows. In the edge extraction step (Figure 2A), the input image is blurred using Gaussian, bilateral, and guided blurring operations, and the Canny edge detector (Canny, 1986) extracts edges from the blurred images. Three extracted edge images are combined to one edge image by adding their pixel intensity values at each coordinate. The errors such as fragmented edges and incorrect edge detection are inherent problems of conventional segmentation methods, so the users must correct the output for further processing. In the post-processing step, edge images are converted into binarized segmented images, and any floating artifacts or noisy edges are removed from the edge images (Figure 2B). When running the labeling tool, users have to specify which side of the edge is foreground and which is background and adjust the hyperparameters based on the input image characteristics. The hyperparameters are kernel size for blurring operations and hysteresis thresholding min-max values for detecting edges.
Figure 2.

Deep learning architecture
(A and B) Workflow in the labeling tool comprising an edge detection step (A) and a post-processing step (B). A single frame from a phase contrast movie of a PtK1 cell and its edge images. Same representative images as shown in Figure 1A.
(C) The deep neural network, VGG19D-U-Net, for segmentation of cell edges. The number of filters in each convolutional block is shown underneath each convolutional block. Light violet lines indicate which features from the encoder are concatenated with which upsampled features in the decoder.
After the training sets were prepared, we trained the deep neural network, VGG19D-U-Net, which is a fully convolutional network with VGG19 encoder, U-Net decoder, and dropout layers (Figure 2C). VGG19 encoder contains five convolutional blocks, each of which contains one max-pooling layer and multiple convolutional layers with a depth of 64-128-256-512-512. The first convolutional block does not have a max-pooling layer. U-Net decoder has four deconvolutional blocks comprising one upsampling layer that concatenates with the encoded features and two convolutional layers with the depth of 512-256-128-64. Dropout layers are added after each max-pooling and upsampling layer. The first dropout layer is set to drop 25% of the incoming units, and the rest of the dropout layers are set to drop 50% of the incoming units.
Segmentation of phase contrast live cell movies using VGG19D-U-Net
We first tested VGG19D-U-Net with a dataset from a single type of microscopy, which contained five live cell movies of migrating PtK1 cells acquired by a phase contrast microscope for 200 frames at 5 s/frame. The segmentation accuracy was measured by precision, recall, and F1 score of edge localization (Arbelaez et al., 2010) (see STAR Methods for details) because edge localization is our main criterion for evaluation. The two-sided Wilcoxon signed-rank test was used to test the statistical significance of performance difference unless otherwise specified. We trained the models on six different numbers of training frames (1, 2, 6, 10, 22, 34) from each movie. The specified number of frames was randomly selected from each live cell movie as the training data. We used the leave-one-movie-out cross-validation, in which one unique movie was selected for testing, and the other movies were used for training in each validation step. As there were five phase contrast movies in total, there were five validation steps in leave-one-movie-out cross-validation.
We trained the segmentation architectures with various pretrained models integrated with the U-Net decoder: VGG16, VGG19, ResNet50V2, and EfficientNetB7 (Baheti et al., 2020). As demonstrated in the learning curve (Figure 3A), VGG19D-U-Net converged to a lower validation loss than U-Net, while the training losses were the same, suggesting less overfitting in VGG19D-U-Net than in U-Net. Overall, the F1 scores of all models tended to increase as more training frames were added, but their F1 scores plateaued as the number of training frames increased (Figure 3B). Among different encoder models, VGG16 and VGG19 performed the best compared to U-Net encoder, ResNet50V2, and EfficientNetB7. In particular, VGG19D-U-Net yielded the highest F1 score across the different numbers of training frames. ImageNet-pretrained U-Net (U-Net P, see STAR Methods for U-Net ImageNet training) consistently achieved a higher F1 score compared with non-pretrained U-Net as the number of training frames increased. However, U-Net P could not surpass any other models trained on the equivalent number of frames even with additional training frames. Notably, the F1 score of VGG19D-U-Net trained on one frame per movie is higher than U-Net P trained on 34 frames per movie by 0.007 (0.929 versus 0.922). When models were trained with 10 frames per movie (Figures 3D–3F), the F1 score of VGG19D-U-Net was significantly higher than the next-best model, VGG16D-U-Net, by 0.003 (0.937 versus 0.934) with p = 4.69 × 10−6. These results demonstrate the importance of transfer learning, network architecture, and dropout layers for accurate segmentation of the live cell image regardless of the size of the training dataset.
Figure 3.

Performance comparison of the models trained on the phase contrast microscopy dataset
(A) Average learning curves of U-Net and VGG19D-U-Net models trained on 10 frames per movie in leave-one-movie-out cross-validation. Solid lines are average training loss, and dotted lines are average validation loss.
(B) Average F1 scores of models trained on different numbers of frames per movie. Error bars: 95% confidence intervals of the mean.
(C) Training efficiency of models in terms of their model size, training time, and segmentation accuracy. The name of the model and number of parameters in italics are written on the bubble. The size of a bubble is proportional to the number of parameters in the model.
(D–F) Average F1, precision, and recall of models. For U-Net, suffix P denotes pretrained and no suffix P denotes non-pretrained model. Other models without suffix P are pretrained and have a U-Net decoder, same as the U-Net model. Suffix D denotes dropout layers added to the model. Significance was tested by the two-sided Wilcoxon signed-rank test. ns, p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.0001. Error bars: 95% confidence intervals of the bootstrap mean. For (B–F), the number of evaluated frames is n = 202, which is roughly 40 frames from each phase contrast live cell movie.
The size, training time, and performance of the models trained on 34 frames per movie were summarized (Figure 3C). The EfficientNetB7-U-Net was the deepest network with the most parameters (71.1M) and took the longest time (4.7 h) to train on average. ResNet50V2-U-Net took the least amount of time (0.81 h) to train but had a lower F1 than VGG16-U-Net and VGG19-U-Net. The training times among U-Net, VGG16-U-Net, and VGG19-U-Net were similar, but VGG16-U-Net and VGG19-U-Net had higher F1 than U-Net. Adding dropout layers to VGG16 or VGG19 encoders (VGG16D or VGG19D) made the model more accurate without additional parameters, but required longer training time. As our criterion for the best model is the high F1 score, not model size or training time, VGG19D-U-Net, with the highest F1 score (0.943), was chosen as the segmentation model in our pipeline.
We also visually confirmed that VGG19D-U-Net localized the cell boundary more accurately than U-Net (Figure 4). VGG19D-U-Net found the cell body regardless of the halo effect, unlike U-Net (Figure 4A, inset 3). Also, U-Net incorrectly segmented background as the cell body (Figure 4B, inset 1) or segmented cell body as the background (Figure 4B, inset 2). In the progression of the segmented cell boundary throughout the movie (Figure 4C), inaccurate segmentation by both models accumulated in multiple frames and became apparent. The white dashed boxes on the image indicate the regions where the cell boundary moves a far distance in a few frames. In Figure 4C (insets 1 and 2), U-Net incorrectly segmented the cell boundary for a few frames, while VGG19D-U-Net produced a smoother transition of the cell boundary. In Figure 4C (insets 3 and 4), both U-Net and VGG19D-U-Net incorrectly segmented a few frames. In Figure 4C (inset 5), U-Net produced a smoother transition than VGG19D-U-Net. Overall, both models can segment a few frames erroneously in various regions, but the size of the error made by U-Net was much greater than that of VGG19D-U-Net.
Figure 4.

Visualization of segmentation results from U-Net and VGG19D-U-Net trained on the phase contrast microscopy dataset
(A and B) Edges extracted from the ground truth mask and predictions from U-Net and VGG19D-U-Net are overlaid on the first frame of the movie of a PtK1 cell acquired by a phase contrast microscope. Each edge is represented by one of three primary colors. The overlap of two or more edges is represented by the combination of those colors.
(C) Progression of cell edges segmented by U-Net and VGG19D-U-Net overlaid on the first frame of the movie of a PtK1 cell acquired by a phase contrast microscope (blue, 0 s; red, 1,000 s time points). Same representative image as shown in Figure 1B. White dashed boxes indicate regions that either U-Net or VGG19D-U-Net segmented incorrectly, or both models segmented incorrectly. Bars: 32.5 μm.
We further investigated the roles of individual components in our VGG19D-U-Net structure (Figures S1A–S1C). The segmentation accuracy of models in terms of F1, precision, and recall has similar trends, so we refer to them collectively as the performance. When encoders of U-Net, VGG16-U-Net, and VGG19-U-Net are ImageNet pretrained, their performance significantly improved compared to their non-pretrained counterparts. The performance of pretrained VGG16/VGG19 was significantly better than that of pretrained U-Net, even though non-pretrained U-Net was significantly better than non-pretrained VGG16-U-Net/VGG19-U-Net. Also, the largest model, VGG19-U-Net, had the most performance boost from ImageNet pretraining compared with VGG16-U-Net. This suggests that ImageNet pretraining may contribute to the improvement of performance differentially, depending on the model size. Adding dropout layers to pretrained VGG16-U-Net and VGG19-U-Net models further increased their performance, but adding batch normalization layers to them reduced their performance. Also, combining a structured form of dropout for convolutional networks, DropBlock (Ghiasi et al., 2018), and batch normalization layers (VGG19DB-U-Net), which resemble SD-UNet (Guo et al., 2019), resulted in a significantly lower performance than that of VGG19D-U-Net. The performance of VGG19DB-U-Net might have been low due to the variance shift that occurs when using both dropout and batch normalization layers (Li et al., 2019).
Different sizes of the cropped patch were also investigated (Figures S1D–S1F). The size indicated here is the size of the image patches, so the size of the ground truth mask patches is smaller because our network crops out the output image (see STAR Methods for details). The model trained on patches of size 96 × 96 had similar performance compared with other models trained on bigger patches. The models trained on 128 × 128, 192 × 192, and 256 × 256 patches had almost identical precision (0.969). However, if the size of a patch was reduced to 80 × 80 or 64 × 64, the performance of the model decreased significantly. These results suggest that the features relevant to detecting cellular boundaries exist in the patch size of 128 × 128 even though it lacks contextual information of the entire cell body. As training on smaller patches has the benefits of reducing memory usage and training on more diverse patches using the same computational resources, we used the patch size of 128 × 128 in our pipeline.
Segmentation of live cell movies from a single type of fluorescence microscopy using VGG19D-U-Net
In this section, we tested the segmentation accuracy of VGG19D-U-Net using fluorescence live cell movies. The training sets consisted of five live cell movies of PtK1 cells expressing GFP-mDia1 using a spinning disk confocal (SDC) microscope for 200 frames at 5 s/frame, and six live cell movies of PtK1 cells expressing paxillin-HaloTag-TMR acquired by a total internal reflection fluorescence (TIRF) microscope for 200 frames at 5 s/frame. These live cell images are very challenging for segmentation using conventional intensity-thresholding methods. The SDC images are highly noisy and low contrast because the cells expressed low levels of GFP-mDia1. Although the TIRF images have higher contrast and less noise than SDC images, they have other technical challenges as follows: (1) high-intensity signals of paxillin accumulated in focal adhesions make edge segmentation difficult, particularly for intensity threshold-based methods; (2) the non-uniform light illumination of a TIRF microscope incurs additional issues for the segmentation; and (3) the leading edge of cells could be transiently lifted and leave the thin TIRF illumination, resulting in less visible cell edges.
To prepare reliable segmentation training sets for the SDC images, we also expressed SNAP-tag-actin and labeled it using TMR (SNAP-tag-TMR-actin) and performed the multiplexed imaging together with GFP-mDia1. The images in the channel of SNAP-tag-TMR-actin have good contrast along the cell boundary. Therefore, conventional image thresholding was applied to SNAP-tag-TMR-actin images, and the resulting binary masks were used as ground truth labels for the SDC datasets. For the preparation of more reliable ground truth masks for the TIRF images, fluorescence images of the same cells were also taken using standard wide-field illumination. We used our labeling tool to label the cell edges in the wide-field images, which served as ground truth labels for the TIRF datasets.
We trained U-Net and VGG19D-U-Net on the fluorescence SDC and TIRF datasets separately and evaluated their performance by leave-one-movie-out cross-validation. During training (Figures 5A and 5D), VGG19D-U-Net converged to a lower validation loss than U-Net, while U-Net overfitted as the epoch increased, as demonstrated by the increase of difference between training and validation loss. Across the different numbers of training frames, VGG19D-U-Net yielded a higher F1 score than U-Net on SDC datasets (Figure 5B). For one of the movies, U-Net performed considerably worse than VGG19D-U-Net, indicated by the faint line with the smallest F1 score of about 0.2. Even though the average F1 score seems to stay consistent as the number of training frames increases in the graph, VGG19D-U-Net trained on 34 frames per movie had a greater average F1 score than the same model trained on one frame per movie by 0.028 (0.891 versus 0.863). When models were trained on 10 training frames per movie, VGG19D-U-Net had a greater F1 than U-Net by 0.115 (0.866 versus 0.751) with p = 2.95 × 10−16. Also, the distribution of evaluated frames in the F1 score (Figure 5C) showed that all frames that were evaluated as 0 in F1 score, when segmented by U-Net, had higher F1 scores when segmented by VGG19D. The superior performance of VGG19D-U-Net compared to U-Net is consistent with the results on phase contrast datasets.
Figure 5.

Performance comparison of U-Net and VGG19D-U-Net trained on fluorescence microscopy datasets
(A–H) (A–C and G) Models trained on SDC datasets and (D–F and H) models trained on TIRF datasets. (A and D) Average learning curves of U-Net and VGG19-U-Net models trained on 10 frames per movie in leave-one-movie-out cross-validation. Solid lines are average training loss, and dotted lines are average validation loss. (B and E) Average F1 scores of models trained on varying numbers of training frames. Lighter lines represent individual test set results in the leave-one-movie-out cross-validation, and darker and thicker lines represent the average of all test set results. Error bars: 95% confidence intervals of the bootstrap mean. (C and F) The distribution of F1 score in violin plot and boxplot in black with the median indicated by the white circle. Significance was tested by the two-sided Wilcoxon signed-rank test. ∗p < 0.05 and ∗∗p < 0.0001. The numbers of evaluated frames are (B and C) n = 1,000 and (E and F) n = 132. (G and H) Visualization of edges extracted from ground truth masks and predictions from U-Net and VGG19D-U-Net overlaid on the first frame of the movies of PtK1 cells acquired by an SDC (G) and a TIRF (H) microscope. Each edge is represented by one of three primary colors. The overlap of two or more edges is represented by the combination of those colors. Bars: 7.2 μm (G) and 6.5 μm (H).
On TIRF datasets (Figure 5E), U-Net initially surpassed VGG19D-U-Net when trained on one or two frames per movie, but they converged to similar average F1 scores as the number of training frames increased. When models were trained on two frames per movie, VGG19D-U-Net had a lower average F1 score than U-Net by 0.02 (0.840 versus 0.860) with p = 2.88 × 10−7. But when models were trained on 10 training frames per movie, VGG19D-U-Net had a marginally lower average F1 score than U-Net by only 0.002 (0.871–-0.873) with p = 0.002. This similarity is also reflected in the distributions of the evaluated frames of both models (Figure 5F).
The visual inspection of edges segmented by U-Net and VGG19D-U-Net demonstrated that VGG19D-U-Net performed well on all SDC datasets, while U-Net failed to segment one of the SDC datasets (Figure 5G). Both U-Net and VGG19D-U-Net did not perform well on one of the TIRF datasets, shown by the mismatch between the ground truth edge and the segmented edges from U-Net and VGG19D-U-Net (Figure 5H). The mismatch is because of the limited illumination of the cell boundary that is hard to detect even with the human eye. The ground truth mask was created using the same cell images by wide-field illumination, so a portion of the cell edge may be lifted from the surface and away from the thin illumination of the TIRF microscope.
Training of VGG19D-U-Net on the datasets from multiple types of microscopy
We established that VGG19D-U-Net outperformed U-Net when they were both trained with either the phase contrast or the SDC dataset. We then prepared the training set from multiple types of microscopy by combining the previous data (phase contrast, SDC, and TIRF microscopy) and trained VGG19D-U-Net on them to create MARS-Net. As in the evaluation for the single-microscopy-type models, leave-one-movie-out cross-validation was used. When this training strategy was applied, multiple-microscopy-type VGG19D-U-Net (VGG19D-U-NetM, MARS-Net) converged to lower validation loss than multiple-microscopy-type U-Net (U-NetM) without overfitting (Figure 6A). VGG19D-U-NetM had a significantly higher F1 than single-microscopy-type VGG19D-U-Net (VGG19D-U-NetS), by 0.028 (0.904 versus 0.876), whereas there was not a significant difference in F1 between U-NetM and single-microscopy-type U-Net (U-NetS) (Figure 6B). The distribution of the evaluated frames (Figure 6C) from VGG19D-U-NetM also contained fewer outliers than for other models.
Figure 6.

Comparison of single-microscopy-type and multiple-microscopy-type training using U-Net and VGG19D-U-Net
(A) Average learning curves of U-NetM and VGG19-U-NetM models trained on two frames per movie in leave-one-movie-out cross-validation. Solid lines are average training loss, and dotted lines are average validation loss.
(B) Average F1 scores across all datasets. “Single” represents single-microscopy type, “Multi” represents multiple-microscopy type, and “VGG19D” represents the VGG19D-U-Net. Statistical significance was tested by two-sided Wilcoxon signed-rank test. ns, p ≥ 0.05; ∗p < 0.05; and ∗∗p < 0.001. Error bars: 95% confidence intervals of the bootstrap mean.
(C) The distribution of F1 scores in the violin plot and the boxplot in black with the median indicated by the white circle. The statistical significances are not shown because they are indicated in (B). The number of evaluated frames is n = 335. Every fifth frame in the movie is sampled to gather about 21 frames from each movie.
(D) Close-up views of the segmentation results of PtK1 cells on all three microscopes: phase contrast (insets 1 and 2), SDC (insets 3 and 4), and TIRF (insets 5 and 6). Edges extracted from ground truth masks and predictions are overlaid on their corresponding original image. Each edge is represented by one of three primary colors. Overlap of two or more edges is represented by the combination of those colors.
(E–G) Class activation map of PtK1 cells from the single-microscopy-type and multiple-microscopy-type VGG19D-U-Net with respect to the ground truth edge. (E) The last layer in the third and fourth block of the encoder is visualized for one randomly chosen frame in the phase contrast live cell movie in order from left to right. The last layer in the fifth block of the encoder is visualized for one randomly chosen frame in two of the (F) SDC and (G) TIRF live cell movies. In the heatmap, the value 0 means no activation, and 1 means the highest activation. The green line represents the ground truth edge of the cell body.
When the performance was averaged per microscopy type (Figures S2A–S2C), VGG19D-U-NetM had a higher F1 than VGG19D-U-NetS for every microscopy type. It had significantly higher F1 in the phase contrast dataset by 0.003 (0.933 versus 0.930) with p = 0.039 (the paired sample t-test was used because their differences in F1 are normally distributed according to the Lilliefors test [p = 0.062]). Also, VGG19D-U-NetM significantly improved F1 more than VGG19D-U-NetS in the SDC dataset by 0.05 (0.911 versus 0.861) with p = 2.61 × 10−51 and in the TIRF dataset by 0.029 (0.878 versus 0.849), with p = 0.012 by a two-sided Wilcoxon signed-rank test. While U-NetM marginally improved F1 better than U-NetS in the SDC dataset by 0.012 (0.767 versus 0.755) with p = 1.39 × 10−16 and did not significantly improve F1 in the TIRF datasets with p = 0.068, U-NetM significantly reduced F1 for the phase contrast dataset by 0.014 (0.884 versus 0.898) with p = 1.43 × 10−5. The distributions of the evaluated frames (Figures S2D–S2F and S3) show that MARS-Net can accurately segment many frames that VGG19D-U-NetS could not handle in the SDC and TIRF datasets.
In addition, the performance of MARS-Net was similar to or greater than that of U-NetM on each of the microscopy types. Unlike VGG19D-U-NetS, which had a significantly lower F1 than U-NetS in the TIRF dataset, the difference of F1 between MARS-Net and U-NetM was not significant (p = 0.637). The distributions of the evaluated frames (Figures S2D–S2F and S3) show that MARS-Net can accurately segment outlier frames that U-NetM could not handle in the phase contrast and SDC datasets. These results demonstrate that our deep learning architecture, VGG19D-U-Net, was more effective in learning the cross-modal features from the datasets of multiple types of microscopy and generalized to unseen datasets than U-Net.
We also visually confirmed the performance between VGG19D-U-NetS and MARS-Net in edge localization of all three microscopies. In most cases, both accurately localize the edge and overlaps with the ground truth illustrated by white lines (Figures 6D and S2G–S2I). Also, the edge progressions produced by the two models look almost identical (Figure S4). However, in the cases where VGG19D-NetS made inaccurate edge localization, MARS-Net accurately localized the ground truth edge, shown as pink lines. Even for one of the TIRF movies that VGG19D-U-NetS struggled with in the previous section (Figure 5H, inset 2), MARS-Net can localize edges more accurately (Figure 6D, inset 5). Taken together, VGG19D-U-Net can be trained with live cell images from multiple types of microscopy and produce more accurate and robust segmentation than the single-microscopy-type model.
To understand the effect of multiple-microscopy-type training on VGG19D-U-Net, we made the class activation maps of the convolutional layers in the encoder using SEG-GRAD-CAM (Vinogradova et al., 2020) (Figures 6E–6G). The class activation map shows which pixels in the original image positively influence the feature maps in the convolutional layer to segment the cell boundary pixels. In the encoder of VGG19D-U-Net comprising five blocks of convolutional layers, the last layers in each block are visualized. In the phase contrast images, the activation map from the third and fourth block showed consistent differences in activated features between single- and multiple-microscopy-type models (Figure 6E). In the third block, the multiple-microscopy-type model utilized features on both the outside and the inside of the edge, while the multiple-microscopy-type model mainly utilized features on the outside of the edge exclusively for segmenting cell boundary. The activated regions from the multiple-microscopy-type model are associated with the brightness outside the cell boundary due to the halo effect in phase contrast microscopy. Also, in the fourth block, the multiple-microscopy-type model mainly utilized features inside the cell boundary, while the multiple-microscopy-type model utilized features along the cell boundary. These results illustrate how changing the training dataset influences the same deep learning model to utilize different features in the image for segmentation.
Complete arrangement of class activation maps from first to last blocks in the encoder (Figures S5A–S5C) demonstrates that the earlier block spots low-level, fine-grained features, and the last block spots the entire cell body. Based on this observation, the multiple-microscopy-type models could spot the whole cell body in fluorescence images (SDC and TIRF), while the single-microscopy-type model could find only a portion of the cell body for some of the movies (Figures 6F and 6G). In total, the single-microscopy-type model could not find the entire cell body for 4 of 11 fluorescence movies, while the multiple-microscopy-type model correctly found cell bodies in all fluorescence movies. These results suggest that the cross-modal features learned from multiple-microscopy-type dataset are more effective than the single-modal features from single-microscopy-type dataset.
Quantitative profiling of cellular morphodynamics
After segmenting phase contrast, SDC, and TIRF live cell movies by U-NetS and MARS-Net, we quantified local protrusion velocities to see how MARS-Net improves cellular morphodynamics profiling (Machacek and Danuser, 2006) over the standard U-Net. The protrusion maps of the phase contrast movies segmented by MARS-Net contained fewer errors or noise than the protrusion maps by U-NetS (Figure 7A). We defined the velocity errors as the dramatic change in protrusion or retraction velocity within a few frames due to the segmentation error, indicated by alternating red and blue color. To corroborate these visual observations, we located the erroneous regions in the protrusion map by thresholding the noise images from each protrusion map (Figures 7B and S6) (see STAR Methods for details). For the phase contrast movie, when pixels in large erroneous regions are counted, U-NetS produced more errors than MARS-Net (567 versus 307). In the SDC movie, U-NetS not only produced more erroneous regions than MARS-Net, but also produced more background noise (Figure 7C). When all magnitudes of noise were counted and plotted against their frequency, U-NetS produced more noise than MARS-Net (Figure 7D). For the TIRF movie, instead of reducing the noise, MARS-Net exhibited stronger patterns of protruding cell boundary than U-NetS, illustrated by long columns of red in the black ellipses (Figure 7E). For a quantitative comparison, we thresholded the noise-filtered protrusion maps (Figure 7F). When the pixels in large regions of the protrusive regions were counted, MARS-Net produced more protrusion than U-NetS (1,013 versus 582). Strongly protruding edges have low contrast, as they are lifted upward and away from the TIRF illumination. Our analysis demonstrates that MARS-Net is capable of detecting these edges, allowing more accurate morphodynamic profiling. Taken together, considering that protrusion maps can be used to identify phenotypes from subcellular movement (Wang et al., 2018), both error/noise reduction and protrusion enhancement of the morphodynamics pattern from the accurate segmentation of MARS-Net will benefit further analysis.
Figure 7.

Morphodynamic profiling of the segmented movies by single-microscopy-type U-Net and MARS-Net
(A–E) (A, C, and E) Protrusion velocity maps made from U-NetS and MARS-Net. The black ellipses emphasize (A and C) some of the erroneous regions from U-NetS and (E) the improved protrusion patterns from MARS-Net. (B) Overlay of the large regions of error/noise containing at least 10 pixels in the protrusion maps of the phase contrast movie (A) from U-NetS and MARS-Net. (D) Comparison of overall noise present in protrusion maps of the SDC movie (C) from U-NetS and MARS-Net.
(F) Overlay of the large protrusion areas containing at least 10 pixels in the protrusion map of the TIRF movie (E) from U-NetS and MARS-Net.
Discussion
Our transfer learning approach employing VGG19 and dropout layers is shown to be superior to conventional U-Net for segmenting live cell time-lapse images in both single-microscopy-type and multiple-microscopy-type training. ImageNet-pretrained VGG19-U-Net has been effective in the segmentation of medical images (Conze et al., 2020; Jha et al., 2020). Here, we showed that the VGG19 encoder was significantly better than other encoders with deeper layers, Res50V2 and EfficientNetB7. This may be because ResNetV2 and EfficientNetB7 reduce the input spatial dimension by half in their first convolutional layers, while the VGG19 encoder preserves the input spatial dimension with convolutional layers that perform convolution with padding. As our objective is to segment the cell boundary accurately, retaining low-level features in the first convolutional layers that can identify edges (Zeiler and Fergus, 2014) is crucial for the localization of cell boundary.
Cell images from different types of microscopy can have drastically different image qualities and distributions of intensity, so training on them together could have degraded the performance by confusing the model instead. In CellPose (Stringer et al., 2021), multiple types of cell images were combined to train one generalist model, but the generalist model had a segmentation accuracy similar to that of the specialist model trained on one type of cell image when they were evaluated on the specialist dataset. In contrast, MARS-Net trained on live cell movies of three different microscopes (phase contrast, SDC, TIRF) significantly enhanced the segmentation accuracy on each type of microscopy by extracting effective features for cell edges across different microscopy data instead of overfitting on a single microscopy’s data. Remarkably, although three types of live cell images employed in this study are very difficult to segment by conventional algorithms, the cross-modal features synergistically learned by MARS-Net were able to successfully detect extremely low-contrast cell edges that could not be detected by the single-microscopy-type model due to the noise and the limited TIRF illumination (Figure 6D, insets 5 and 6).
Through transfer learning with ImageNet-pretrained weights, the deep learning model reuses diverse features learned from millions of images on the Internet (Deng et al., 2009). This benefits the model to become invariant to various imaging conditions such as brightness, contrast, and camera resolution. Similarly, multiple-microscopy-type training benefits VGG19D-U-Net to become invariant to the imaging modality and attempt to create a robust model that identifies cell boundaries with semantic understanding, as shown by our activation maps. Another benefit of multiple-microscopy-type training is that it reduces the need to create new training datasets, because the training dataset in one microscopy can be reused to analyze the dataset in another microscopy. This is consistent with the previous study demonstrating that multifidelity data were used to increase the size of the training set and improved the performance of the deep learning model in material science research (Chen et al., 2021).
Morphodynamic profiling has been usually undertaken with high-contrast fluorescence live cell images amenable for standard threshold-based segmentation methods, limiting the throughput of the analysis pipeline. Particularly, phase contrast microscopy images have not been used due to the segmentation issues. As phase contrast microscopy does not require expensive optical components and fluorescence labeling, MARS-Net, in conjunction with phase contrast microscopy, can substantially accelerate quantitative studies of cellular morphodynamics.
Limitations of the study
The presented method is limited to the segmentation of cells on a relatively simple background, and was not tested for the cases where multiple cells touch or overlap one another. Also, it can take long time to train MARS-Net for leave-one-movie-out cross-validation. Considering that we have 16 movies in total, training 16 models for leave-one-movie-out cross-validation takes about a week, making it challenging to train MARS-Net with large datasets. In this case, using simpler testing procedures could reduce the training time.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Cell Lines | ||
| PtK1 cell lines | Gaudenz Danuser Lab | https://www.utsouthwestern.edu/labs/danuser/ |
| Chemicals, peptides, and recombinant proteins | ||
| HaloTag-TMR | Promega | G8251 |
| SNAP-tag-TMR | New England Biolab | S9105S |
| Recombinant DNA | ||
| GFP-mDia1 | Lee et al., 2015 | NA |
| SNAP-tag-actin | Lee et al., 2015 | NA |
| paxillin-HaloTag | Lee et al., 2015 | NA |
| Software and Algorithms | ||
| MARS-Net | This paper | Zenodo: https://doi.org/10.5281/zenodo.5541392 |
| Tensorflow v2.3 | Tensorflow | https://www.tensorflow.org/ |
| Matplotlib v3.3.4 | Matplotlib | https://matplotlib.org/ |
| Numpy v1.18.5 | Numpy | https://numpy.org/ |
| Anaconda v4.5.11 | Anaconda | https://www.anaconda.com/ |
| CUDA v11.0 | NVIDIA | https://developer.nvidia.com/cuda-toolkit |
| Python 3.6.8 | Python Software Foundation | https://www.python.org/ |
| MATLAB 2019b | MathWorks | https://www.mathworks.com/products/matlab.html |
| Extended Berkeley segmentation Benchmark | David Stutz | https://github.com/davidstutz/extended-berkeley-segmentation-benchmark |
| Windowing and protrusion package | Gaudenz Danuser lab | https://github.com/DanuserLab/Windowing-Protrusion |
| npy-matlab | Kwik Team | https://github.com/kwikteam/npy-matlab |
| Other | ||
| Phase contrast microscope | This paper | NA |
| Spinning disk confocal microscope | This paper | NA |
| Total internal reflection fluorescence microscope | Lee et al., 2015 | NA |
Resource availability
Lead contact
Correspondence and requests for materials, data, and code should be addressed to the lead contact, Dr. Kwonmoo Lee (kwonmoo.lee@childrens.harvard.edu).
Materials availability
We did not generate any new unique reagents in this study.
Experiment model and subject details
Cell culture
PtK1 cells were cultured in Ham’s F12 medium (Invitrogen) supplemented with 10% FBS, 0.1 mg ml−1 streptomycin, and 100 U ml−1 penicillin. PtK1 cells were acquired from Gaudenz Danuser lab. They were routinely tested for mycoplasma contamination.
Transfection
PtK1 cells were transfected with the DNA constructs of GFP-mDia1 and SNAP-tag-actin or paxilin-HaloTag using Neon transfection system (Invitrogen) according to the manufacturer’s instructions (1 pulse, 1400 V, 20 ms) and were grown on acid-washed glass #1.5 coverslips for 2 days before imaging.
Method details
Data collection
Prior to imaging, expressed SNAP-tag-actin or paxillin-HaloTag proteins were labeled with SNAP-tag-TMR (New England BioLabs) or HaloTag-TMR (Promega) ligands, respectively according to the manufacturers’ instructions. All imaging was performed in imaging medium (Leibovitz’s L-15 without phenol red, Invitrogen) supplemented with 10% fetal bovine serum (FBS), 0.1 mg ml−1 streptomycin, 100 U ml−1 penicillin, 0.45% glucose, 1.0 U ml−1 Oxyrase (Oxyrase Inc.) and 10 mM Lactate. Cells were then imaged at 5 second intervals for 1000 seconds using 0.45 NA Super Plan Fluor ELWD 20X ADM objective for phase contrast imaging and 60X, 1.4 NA Plan Apochromat objective for fluorescence spinning disk confocal imaging, 1.49NA Apochromat TIRF 100X for total internal reflection fluorescence imaging.
Phase contrast and SDC microscopy was performed using the set up as follows: Nikon Ti-E inverted motorized microscope (including motorized focus, objective nosepiece, fluorescence filter turret, and condenser turret) with integrated Perfect Focus System, Yokogawa CSU-X1 spinning disk confocal head with a manual emission filter wheel with Spectral Applied Research Borealis modification, Spectral Applied Research custom laser merge module (LMM-7) with AOTF and solid state 445 nm (200 mW), 488 nm (200 mW), 514 nm (150 mW), 561 nm (200 mW), and 637 nm (140 mW) lasers, Semrock 405/488/561/647 and 442/514/647 dichroic mirrors, Ludl encoded XY stage, Ludl piezo Z sample holder for high speed optical sectioning, Prior fast transmitted and epi-fluorescence light path shutters, Hamamatsu Flash 4.0 LT sCMOS camera, 37°C microscope incubator enclosure with 5% CO2 delivery (in vivo), Molecular Devices MetaMorph v7.7, TMC vibration-isolation table.
TIRF microscopy was performed using the set up as follows (Lee et al., 2015): Nikon Ti-E inverted motorized microscope with integrated Perfect Focus System, Nikon 100x 1.49 NA TIRF DIC objective lens, and Nikon dual-port TIRF/Epi illuminator with motorized laser incident angle adjustment. Spectral Applied Research laser launch with 401nm (100mW), 442nm (40mW), 491nm (50mW), 515nm (50mW), 561nm (50mW) and 640nm (100mW) solid state lasers with a fiber-optic delivery system and AOTF. A Prior Proscan III controller for fast excitation and emission filter wheels, fast transmitted and epifluorescence light path shutters, and a linear-encoded motorized stage. A Chroma 405/491/561/638 dichroic mirror with a 561 laser line and 600/50 emission filter for HaloTag-TMR. A custom Chroma laser notch filter in the emission path to further block the illumination light from reaching the camera and to minimize interference patterns, a Hamamatsu ORCA R2 CCD camera, MetaMorph v7.7 (Molecular Devices). Exposure times were typically 500 ms using 30∼50% laser power.
Dataset
The live cell movies used for training and evaluation of our pipeline are as follows.
-
•
Five movies of label-free migrating PtK1 cells by a phase contrast microscope
-
•
Five dual-color movies of PtK1 cells expressing GFP-mDia1 and SNAP-tag-actin by a spinning disk confocal (SDC) microscope.
-
•
Six movies of PtK1 cells expressing paxillin-HaloTag-TMR, a marker of cell-matrix adhesions by a Total Internal Reflection Fluorescence (TIRF) microscope
Each live cell movie contains 200 frames, and about 40 frames per movie were labeled by our labeling tool for each phase contrast movie. Every five frames starting from the first frame was labeled, so ideally, there would be 41 labeled frames from each movie. However, the first frame or the last frame was not labeled for some movies, so some movies have 40 labeled frames. For each SDC movie, all 200 frames were labeled by thresholding actin images. For each TIRF movie, 22 frames were labeled using the images from standard wide-field fluorescence microscopy images and our labeling tool. Overall, 202 frames from phase contrast, 1000 frames from SDC, and 132 frames from TIRF movies are labeled to train and test our pipeline. The pixel size is 325nm for the phase contrast datasets, 72nm for the SDC datasets, and 65nm for the TIRF datasets.
The ground truth masks for phase contrast and TIRF images are labeled using our labeling tool (Figures 2A and 2B). SDC images have the corresponding high contrast images of SNAP-tag-TMR-actin with good contrast along the cell boundary. Therefore, ground truth masks for SDC images can be labeled by applying denoising and thresholding. The non-local means method implemented in ImageJ for denoising (sigma=15 and smoothing_factor=1) was applied to each SNAP-tag-TMR-actin image. Then, thresholding was applied to all frames with an optimal threshold determined by visually checking the generated masks and re-adjusting the threshold until the generated masks align the best with the cell boundary. The generated binary masks were used as the ground truth labels for SDC datasets.
Training dataset preparation
Before training the deep learning model, frames and their ground truth mask are processed for training and testing. Six different numbers of training frames (1,2,6,10,22,34) are used as a sample size for randomly sampling a set of training frames from each live cell movie except for the test set movie. The chosen frames become part of the training/validation set. Then, 200 grayscale patches of 128X128 pixels are randomly cropped from each frame and its corresponding ground-truth mask. The cropping is necessary to reduce memory and computational requirement Because only patch sizes in multiple of 16 can be handled by our models, other input sizes cause a mismatch of spatial size between encoded features and decoded features when concatenating them in U-Net structure. 60% of the cropped images are from the boundary of cytoplasm illustrated by red boxes, and 20% are from inside, and the other 20% are from the outside of the cytoplasm illustrated by blue boxes in Figure 1B.
Patches are augmented to negate the effect of small training size and improve performance. The augmentation methods include random rotation within 50 degrees, width, and height shift within 10% of the image’s width and height, shear in counter-clockwise direction within 36 degrees, zoom in or out randomly within 10% of the image size, and horizontal and vertical flips of the image. The original image’s reflection is used to replace the portion of the augmented images outside the boundary of the original image. The default number of augmented patches is 6400, and they are augmented before training so that the same augmented images are used for each training iteration. For instance, the total number of patches in training and validation sets from two frames of each live cell movie in leave-one-movie-out cross-validation is 8000 (2 × 4 × 200 + 6400). Then, patches are randomly split into training and validation sets in the ratio of 8:2.
Image patches are preprocessed to facilitate the deep learning model training. For the phase contrast and the SDC datasets, all image patches from one movie are standardized based on the mean and the standard deviation of pixel values of the cropped and augmented patches in that movie. In this way, the distribution of pixel values per movie has the mean and standard deviation equal to zero and one, respectively. Image patches from the TIRF dataset have poor contrast, so they are preprocessed differently from phase contrast or SDC datasets. After the mean and the standard deviation of pixel values of a TIRF movie are calculated, the pixel values are replaced with the following values when they are less than or greater than .
Then, the min-max normalization was applied to rescale the pixel ranges to [0, 1].
For prediction, images and masks are not cropped or augmented, but the same standardization or preprocessing steps are applied based on the microscopy type.
Neural network architecture
All models mentioned in this paper are based on the same U-Net structure comprised of encoder and decoder. We replaced the original encoder in U-Net with other encoders such as VGG16, VGG19, ResNet50V2, and EfficientNetB7 and prefixed model names by their encoder names. All models had the same decoder structure, including the last crop layer and four skip connections that concatenate encoded features with decoded features, as shown in Figure 2. However, models did not have dropout layers except for MARS-Net and model names specified by the letter B or D, which represents batch normalization and dropout layers, respectively.
Encoders except for the original U-Net encoder, VGG16, or VGG19 were pretrained with ImageNet pretrained weights provided by Keras. We pretrained U-Net encoder on the ImageNet classification task by the same training process used for VGG16 and VGG19 ImageNet pretraining(Simonyan and Zisserman, 2015). When pretrained U-Net encoder with three fully connected layers for classification was evaluated against the ImageNet validation set, its top-1 accuracy and top-5 accuracy were 0.56 and 0.8, respectively. For every convolutional layer in the decoder or convolutional layer in the encoder that is non-pretrained, the kernel weights were initialized with Glorot uniform(Glorot and Bengio, 2010), and the bias weights were initialized with zeros. The pretrained models were fine-tuned without freezing any weights.
The convolutional filter size is 3x3, and the zero-padding in each convolutional layer of U-Net and VGG models yields the feature map with the same spatial size after convolution. The size of the input patch is 128x128, and the size of the max-pooling and up-sampling filter is both 2x2. The same size of max-pooling and up-sampling filters make the max-pooled feature map and the up-sampled feature map at the same hierarchical level to have the same spatial size. The last layer of the network crops the 128x128 output by 30 pixels on all sides to get the segmented image of size 68x68. Cropping is necessary to eliminate the boundary effects. Without cropping, the segmented image is hazy along the image boundary, lowering the segmentation accuracy.
In the prediction step, every frame of the movie in the test set is segmented by the trained model. The image is not cropped into 128x128 pixel patches, but its width and height are padded with its reflection by about 30 pixels. Then, the models take the padded input images, segment their binary masks, and remove the padded regions from the binary masks to avoid boundary effects.
Neural network training settings
For a fair comparison of models, each model’s hyperparameters were configured the same as follows: Adam(Kingma and Ba, 2015) optimizer with learning rate=10-5, batch size=64, input size=128, output size=68, early stopping patience=3. The binary cross-entropy was used as a loss function for training. To avoid overfitting, we used the early stopping, so training stopped when the validation loss did not decrease during the three consecutive epochs. For the phase contrast and SDC datasets, early stopping patience was 3, and the maximum epoch was 100. For the TIRF dataset, early stopping patience was 10, and the maximum epoch was 300. We used default parameters in the Keras for other parameters. The neural network training was performed using TensorFlow(Abadi et al., 2016) 2.3 on RTX Titan GPU with CUDA 10.1 for multiple-microscopy-type phase contrast models and multiple-microscopy-type models and TensorFlow 1.15 on GTX 1080Ti GPU with CUDA 10.0 for multiple-microscopy-type SDC models and multiple-microscopy-type TIRF models.
The number of training frames
The number of training frames per movie in leave-one-movie-out cross validation (FC) and the total number of training frames (FT) used to train the model in the results section are described here. For phase contrast and SDC datasets, FT is four times the Fc because each dataset has five movies in total and training frames are obtained from four movies in leave-one-movie-out cross validation. For TIRF dataset, FT is five times the Fc because TIRF dataset has six movies in total and training frames are obtained from five movies in leave-one-movie-out cross validation. For multiple-microscopy-type models, FT is fifteen times the Fc because there are sixteen movies in total and training frames are obtained from fifteen movies in leave-one-movie-out cross validation. All training frames are randomly selected at each validation step.
-
•
FC = 34, FT = 136 (Figure 3C)
- •
-
•
FC = 10, FT = 50 (Figures 5D, 5F, and 5H)
-
•
FC = 2, FT = 8 (Figure 4)
-
•
FC = 2, FT = 30 (Figure 6A)
-
•
FC = 2, and FT = 30 for multiple-microscopy-type models, FT = 8 for single-microscopy-type model trained on phase contrast or SDC datasets or FT = 10 for single-microscopy-type model trained on TIRF dataset (Figures 6B–6G, 7, and S2–6).
Cross validations
To rigorously test our deep learning model's generalizability and reproducibility, we evaluated every model by leave-one-movie-out cross validation. It is similar to the leave-one-subject-out cross validation(Koul et al., 2018) but with the subject replaced by the live cell movie. We set aside one movie as a test set, and the rest of the movies are used for training and validation. Frames in the same live cell movie have little difference in image features, but there is a distinctive visual difference even among the live cell movies taken by the same microscopy. Therefore, we consider frames from the same live cell movie to be independent and identically distributed (i.i.d) and frames from different live cell movies to be out of distribution (o.o.d). The leave-one-movie-out cross-validation ensures that our model is assessed on the o.o.d test set to prevent shortcut learning(Geirhos et al., 2020). For instance, given five live cell movies called A, B, C, D, and E, movie E is set aside as a test set, and movies A, B, C, and D become training/validation sets. In the subsequent validation, movie D is set aside as a test set, and movies A, B, C, and E become training/validation sets. This process is repeated until every movie is set aside as the test set once. Then, the test performance measures are averaged.
Our segmentation pipeline trained on the same dataset can yield different segmentation results due to random selection of frames, random cropping, and random train/validation set splits. In order to reduce the variations caused by them, the leave-one-movie-out cross-validation is repeated five times for single-microscopy-type phase contrast models in Figures 3 and S1A–S1C. Frames and patches were randomly selected and randomly split into training and validation set in each repetition.
Evaluation metrics
Precision, recall, and F1 score between ground truth edges and segmented edges are calculated by the edge correspondence algorithm in the Berkeley Segmentation Benchmark(Arbelaez et al., 2010; Martin et al., 2004), with the search radii (Phase Contrast: 3 pixels; SDC: 5 pixels, TIRF: 5 pixels). The package performs bipartite matching of two edge images by iteratively matching edge pixels in one image with edge pixels in another image. For instance, an edge pixel in the first image is counted as a match if there is an edge pixel in the second image within the search radii of the target pixel.
Before evaluation, both ground truth masks and segmentation by the models are image processed. The image processing steps include thresholding the grayscale images into binary images with a threshold value of 0.5, given intensity values ranging from 0 to 1, filling small holes, and extracting edge by the canny edge detector. Since images are binarized before evaluation, the intensity value of each pixel in the image is either 0 or 1. The match between ground truth pixels and segmented pixels of intensity 1 are true positives (tp). The segmented pixels of intensity 1 that do not match with ground truth pixels are false positives (fp). And the ground truth pixels of intensity 1 that do not match with segmented pixels are false negatives (fn). True negatives, which are the match between ground truth pixel of intensity 0 and segmented pixel of intensity 0, are ignored. After counting tp, fp, and fn in an image, the metrics are calculated as follows.
Every segmented frame is evaluated by precision, recall and F1. The evaluated frames are bootstrapped with 1000 replicates to calculate bootstrap mean and 95% confidence interval. The image processing and evaluation are performed in MATLAB (2019b).
Profiling of cellular morphodynamics
The steps taken to perform quantitative profiling of cellular morphodynamics(Machacek and Danuser, 2006) on segmented movies are describedin Figure 1C. The live cell movie is cropped as illustrated by the dashed white rectangle, and the velocity of the cell along its boundary is estimated based on the difference of segmented area in the previous and present frames. Then, the estimated velocity is grouped into rectangular blocks called “window” to get a smoother estimate of the velocity. At the local sampling step, the outermost band of windows along the boundary of the cell is sampled to draw a protrusion activity map showing the velocity at each window number and frame number. Inner bands inside the cell are ignored. The size of each window is 6 pixels, or 1.95μm for phase contrast dataset, 7 pixels, or 504nm for SDC, and 8 pixels, or 520nm for TIRF datasets.
The signal in the protrusion map is found by the cubic smoothing spline interpolation of the protrusion maps with the smoothing parameter (p=0.7). The error/noise is calculated by subtracting the original protrusion map with the spline filtered protrusion map. To ignore the regions of error/noise and protrusion of small magnitudes, thresholding operation set magnitude lower than 3μm/min to zero for the phase contrast movie and lower than 2μm/min to zero for the SDC and the TIRF movies. Then, small connected regions of error/noise or protrusion signal containing less than 10 pixels are removed (Figures 7B, 7F, and S6) to highlight the large error from U-NetS or protrusion signal from MARS-Net that facilitates further analysis. For the SDC dataset (Figure 7D), the histogram of all error magnitude without thresholding was plotted against its log frequency as a line graph.
Class activation map
The technique called SEG-GRAD-CAM (Vinogradova et al., 2020) visualizes the feature maps that are positively associated with the increase in the intensity of output pixels. Unlike GRAD-CAM(Selvaraju et al., 2017), which is designed for classifiers that output a vector, SEG-GRAD-CAM can explain the decision of the segmentation model that outputs a 2-dimensional matrix. Our region-of-interest is the cell boundary, so we visualized activation of feature maps with respect to the edge extracted from the ground truth mask.
Let Ak be the kth feature map in the filter. Among convolutional layers in the VGG19D-U-Net, we are interested in the last layer of each block in the encoder. The total number of feature maps from the first to last blocks are as follows: 64, 128, 256, and 512. The output of the model, y, only has one channel, and its value ranges from 0 to 1. i and j are indexes of the pixels that correspond to the cell boundary C in the output image y, and u and v are indexes of the spatial location in Ak. N is the total number of pixels in Ak. Then, the importance of the feature map at each spatial location can be computed as follows.
For every pixel of y, the gradients with respect to all pixels in the feature map are calculated by backpropagation and global average pooled across the spatial dimensions of Ak. The weight matrix ak, which has the same spatial dimension as Ak, dot products with Ak to get the weighted sum of the feature maps.
The weighted sum of the feature maps or heatmap is spatially scaled up by bilinear interpolation to match the input image size. Scaling up is necessary because the heatmaps from different convolutional feature maps have different spatial sizes. Scaled-up heatmaps are overlaid with their corresponding ground truth edge and can be compared with each other. Finally, ReLU is applied to ignore the negative influence of the feature maps on the prediction of a cell boundary.
Quantification and statistical analysis
All quantification and statistical analyses are performed in MATLAB (2019b). All values in line graphs and bar graphs are shown as bootstrap mean ± 95% confidence intervals of the bootstrap mean. Precision, Recall, or F1 values from evaluating each frame are aggregated and bootstrapped with 1000 replicates to calculate bootstrap mean and confidence interval. Precision, recall, and F1 score between ground truth edges and segmented edges are calculated by the edge correspondence algorithm in the Berkeley Segmentation Benchmark (Arbelaez et al., 2010; Martin et al., 2004), with the search radii (Phase Contrast: 3 pixels; SDC: 5 pixels, TIRF: 5 pixels). For Figure 7, quantitative profiling of cellular morphodynamics on a segmented live cell movie is performed using the Windowing software in MATLAB (Machacek and Danuser, 2006).
For Figures 3A, 5A, 5D, and 6A, loss values are presented as the average of each loss from the cross validated models. For Figures 3B, 3D–3F, 5B, 5E, and 6B, values are presented as bootstrap mean ± 95% confidence intervals of the bootstrap mean. A two-sided Wilcoxon signed-rank test is used to test statistical significance between the model performances because the distribution of values is not normally distributed. The statistical test is considered significant if the p-value is less than 0.05. For Figure 3C, the accuracy of each model is presented as the mean of F1 scores without bootstrapping. Given n represents the number of frames evaluated, n=202 for Figures 3B–3F, n=1000 for Figures 5B and 5C, n=132 for Figures 5E and 5F, and n=335 for Figures 6B and 6C. For Figures 5C, 5F, and 6C, F1 score of all evaluated frames are plotted as small dots on the violin plots without bootstrapping, and their median as a white circle and interquartile ranges as black boxes are presented. For Figures 5C and 5F, a two-sided Wilcoxon signed-rank test is used to test statistical significance between the F1 scores. The statistical test is considered significant if the p-value is less than 0.05. For Figure 7, the velocity of cellular morphodynamics is in a micrometer per minute scale, measured at every 5 second interval. The maximum and minimum velocities are ±10. Any velocity greater than +10 is set to +10, and any velocity lower than -10 is set to -10. Additional statistical details of experiments can be found in figure legends, results, and method details.
Acknowledgments
We thank Microsoft for providing us with Azure cloud computing resources (Microsoft Azure Research Award), and Boston Scientific for providing us with the gift for deep learning research. This work was supported by NIH, United States (Grant Numbers: R15GM122012 and R35GM133725).
Author contributions
C.W. and X.Z. initiated the project. J.J. and C.W. designed the pipeline. J.J. wrote the final version of the manuscript and supplement. J.J., C.W., X.Z., B.L., and Y.Y. wrote the code for the segmentation model. J.J. built the labeling tool. H.C. performed the live cell imaging experiments. J.J., C.W., H.C., X.P., M.R., and Y.C. prepared the training sets. J.J. visualized feature maps using SEG-GRAD-CAM and profiled cellular morphodynamics from the segmented movies. K.L. coordinated the study and wrote the final version of the manuscript and supplement. All authors discussed the results of the study.
Declaration of interests
The authors declare no competing interests.
Published: October 27, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100105.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at Github (https://github.com/kleelab-bch/MARS-Net) and Zenodo (https://doi.org/10.5281/zenodo.5541392) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abadi M.i., Barham P., Chen J., Chen Z., Davis A., Dean J., Devin M., Ghemawat S., Irving G., Isard M., et al. Tensorflow: A System for Large-Scale Machine Learning. USENIX conference on Operating Systems Design and Implementation. 2016:265–283. [Google Scholar]
- Ahmed I., Ahmad M., Khan F.A., Asif M. Comparison of deep-learning-based segmentation models: using top view person images. IEEE Access. 2020;8:136361–136373. [Google Scholar]
- Al-Kofahi Y., Zaltsman A., Graves R., Marshall W., Rusu M. A deep learning-based algorithm for 2-D cell segmentation in microscopy images. BMC Bioinformatics. 2018;19:365. doi: 10.1186/s12859-018-2375-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M.A.S., Misko O., Salumaa S.-O., Papkov M., Palo K., Fishman D., Parts L. Evaluating very deep convolutional neural networks for nucleus segmentation from brightfield cell microscopy images. SLAS Discov. 2021 doi: 10.1177/24725552211023214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambühl M.E., Brepsant C., Meister J.J., Verkhovsky A.B., Sbalzarini I.F. High-resolution cell outline segmentation and tracking from phase-contrast microscopy images. J. Microsc. 2012;245:161–170. doi: 10.1111/j.1365-2818.2011.03558.x. [DOI] [PubMed] [Google Scholar]
- Arbelaez P., Maire M., Fowlkes C., Malik J. Contour detection and hierarchical image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2010;33:898–916. doi: 10.1109/TPAMI.2010.161. [DOI] [PubMed] [Google Scholar]
- Badrinarayanan V., Kendall A., Cipolla R. SegNet: a deep convolutional encoder-decoder architecture for image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2017;39:2481–2495. doi: 10.1109/TPAMI.2016.2644615. [DOI] [PubMed] [Google Scholar]
- Baheti B., Innani S., Gajre S., Talbar S. CVPR Workshops; 2020. Eff-UNet: A Novel Architecture for Semantic Segmentation in Unstructured Environment. [Google Scholar]
- Bensch R., Ronneberger O. 2015 IEEE 12th International Symposium on Biomedical Imaging. ISBI; 2015. Cell segmentation and tracking in phase contrast images using graph cut with asymmetric boundary costs; pp. 1220–1223. [Google Scholar]
- Bertasius G., Shi J., Torresani L. Deepedge: a multi-scale bifurcated deep network for top-down contour detection. Proc. IEEE Conf. Comput. Vis. Pattern Recogn. 2015:4380–4389. [Google Scholar]
- Bertram C.A., Veta M., Marzahl C., Stathonikos N., Maier A., Klopfleisch R., Aubreville M. Springer International Publishing; 2020. Are Pathologist-Defined Labels Reproducible? Comparison of the TUPAC16 Mitotic Figure Dataset with an Alternative Set of Labels. [Google Scholar]
- Buggenthin F., Buettner F., Hoppe P.S., Endele M., Kroiss M., Strasser M., Schwarzfischer M., Loeffler D., Kokkaliaris K.D., Hilsenbeck O., et al. Prospective identification of hematopoietic lineage choice by deep learning. Nat. Methods. 2017;14:403–406. doi: 10.1038/nmeth.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo J.C., Goodman A., Karhohs K.W., Cimini B.A., Ackerman J., Haghighi M., Heng C., Becker T., Doan M., McQuin C., et al. Nucleus segmentation across imaging experiments: the 2018 data science bowl. Nat. Methods. 2019;16:1247–1253. doi: 10.1038/s41592-019-0612-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canny J. A computational approach to edge detection. IEEE Trans. pattern Anal. Mach. Intell. 1986:679–698. [PubMed] [Google Scholar]
- Chai X., Ba Q., Yang G. 2018 25th IEEE International Conference on Image Processing. ICIP; 2018. Characterizing robustness and sensitivity of convolutional neural networks in segmentation of fluorescence microscopy images; pp. 3838–3842. [Google Scholar]
- Chamier L.V., Laine R.F., Jukkala J., Spahn C., Krentzel D., Nehme E., Lerche M., Hernández-Pérez S., Mattila P.K., Karinou E., et al. ZeroCostDL4Mic: An Open Platform to Use Deep-Learning in Microscopy. Cold Spring Harb. Lab. 2020 doi: 10.1101/2020.03.20.000133. [DOI] [Google Scholar]
- Chan T.F., Vese L.A. Active contours without edges. IEEE Trans. Image Process. 2001;10:266–277. doi: 10.1109/83.902291. [DOI] [PubMed] [Google Scholar]
- Chen C., Zuo Y., Ye W., Li X., Ong S.P. Learning properties of ordered and disordered materials from multi-fidelity data. Nat. Comput. Sci. 2021;1:46–53. doi: 10.1038/s43588-020-00002-x. [DOI] [PubMed] [Google Scholar]
- Choi J.Y., Yoo T.K., Seo J.G., Kwak J., Um T.T., Rim T.H. Multi-categorical deep learning neural network to classify retinal images: a pilot study employing small database. PLoS One. 2017;12:e0187336. doi: 10.1371/journal.pone.0187336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conze P.-H., Naciye Ali Emilie, Yannick M., Rousseau F.c. arXiv; 2020. Abdominal Multi-Organ Segmentation with Cascaded Convolutional and Adversarial Deep Networks. [DOI] [PubMed] [Google Scholar]
- Deng J., Dong W., Socher R., Li L.-J., Kai L., Li F.-F. CVPR, IEEE Computer Society Conference on Computer Vision and Pattern Recognition. IEEE; 2009. ImageNet: a large-scale hierarchical image database. [Google Scholar]
- Donahue J., Jia Y., Vinyals O., Hoffman J., Zhang N., Tzeng E., Darrell T. International conference on machine learning; 2014. Decaf: A Deep Convolutional Activation Feature for Generic Visual Recognition; pp. 647–655. [Google Scholar]
- Falk T., Mai D., Bensch R., Cicek O., Abdulkadir A., Marrakchi Y., Bohm A., Deubner J., Jackel Z., Seiwald K., et al. U-Net: deep learning for cell counting, detection, and morphometry. Nat. Methods. 2019;16:67–70. doi: 10.1038/s41592-018-0261-2. [DOI] [PubMed] [Google Scholar]
- Geirhos R., Jacobsen J.-H., Michaelis C., Zemel R., Brendel W., Bethge M., Wichmann F.A. Shortcut learning in deep neural networks. Nat. Mach. Intell. 2020;2:665–673. [Google Scholar]
- Ghiasi G., Lin T.-Y., Quoc . Advances in Neural Information Processing Systems. Vol. 31. Curran Associates, Inc.; 2018. DropBlock: A Regularization Method for Convolutional Networks. [Google Scholar]
- Glorot X., Bengio Y. Understanding the difficulty of training deep feedforward neural networks. Proc. Thirteenth Int. Conf. Artif. Intell. Stat. JMLR Workshop Conf. Proc. 2010;9:249–256. [Google Scholar]
- Gómez-De-Mariscal E., García-López-De-Haro C., Donati L., Unser M., Muñoz-Barrutia A., Sage D. Cold Spring Harbor Laboratory; 2019. DeepImageJ: A User-Friendly Plugin to Run Deep Learning Models in ImageJ. [DOI] [PubMed] [Google Scholar]
- Gonzalez G., Fusco L., Benmansour F., Fua P., Pertz O., Smith K. Automated Quantification of Morphodynamics for High-Throughput Live Cell Time-Lapse Datasets. IEEE 10th International Symposium on Biomedical Imaging; 2013. pp. 664–667. [Google Scholar]
- Guo C., Szemenyei M., Pei Y., Yi Y., Zhou W. 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering. BIBE; 2019. SD-unet: a structured dropout U-net for retinal vessel segmentation; pp. 439–444. [Google Scholar]
- He K., Zhang X., Ren S., Sun J. Deep residual learning for image recognition. Proc. IEEE Conf. Comput. Vis. Pattern Recogn. 2016:770–778. [Google Scholar]
- He K., Zhang X., Ren S., Sun J. arXiv; 2016. Identity Mappings in Deep Residual Networks. [Google Scholar]
- Hermans T.M., Pilans D., Huda S., Fuller P., Kandere-Grzybowska K., Grzybowski B.A. Motility efficiency and spatiotemporal synchronization in non-metastatic vs. metastatic breast cancer cells. Integr. Biol. (Camb) 2013;5:1464–1473. doi: 10.1039/c3ib40144h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglovikov V., Seferbekov S.S., Buslaev A., Shvets A. CVPR Workshops; 2018. TernausNetV2: Fully Convolutional Network for Instance Segmentation; pp. 233–237. [Google Scholar]
- Isola P., Zoran D., Krishnan D., Adelson E.H. European Conference on Computer Vision; 2014. Crisp Boundary Detection Using Pointwise Mutual Information; pp. 799–814. [Google Scholar]
- Jha D., Michael Johansen D., Halvorsen P., Hvard . arXiv; 2020. DoubleU-Net: A Deep Convolutional Neural Network for Medical Image Segmentation. [Google Scholar]
- Kim S.J., Wang C., Zhao B., Im H., Min J., Choi H.J., Tadros J., Choi N.R., Castro C.M., Weissleder R., et al. Deep transfer learning-based hologram classification for molecular diagnostics. Sci. Rep. 2018;8:17003. doi: 10.1038/s41598-018-35274-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma D.P., Ba J. ICLR; 2015. ADAM: A METHOD FOR STOCHASTIC OPTIMIZATION. [Google Scholar]
- Koul A., Becchio C., Cavallo A. Cross-validation approaches for replicability in psychology. Front. Psychol. 2018;9:1117. doi: 10.3389/fpsyg.2018.01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhevsky A., Sutskever I., Hinton G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012:1097–1105. [Google Scholar]
- LeCun Y., Bengio Y., Hinton G. Deep learning. Nature. 2015;521:436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- Lee K., Elliott H.L., Oak Y., Zee C.T., Groisman A., Tytell J.D., Danuser G. Functional hierarchy of redundant actin assembly factors revealed by fine-grained registration of intrinsic image fluctuations. Cell Syst. 2015;1:37–50. doi: 10.1016/j.cels.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithner A., Eichner A., Muller J., Reversat A., Brown M., Schwarz J., Merrin J., de Gorter D.J., Schur F., Bayerl J., et al. Diversified actin protrusions promote environmental exploration but are dispensable for locomotion of leukocytes. Nat. Cell Biol. 2016;18:1253–1259. doi: 10.1038/ncb3426. [DOI] [PubMed] [Google Scholar]
- Li K., Kanade T. Nonnegative mixed-norm preconditioning for microscopy image segmentation. Int. Conf. Inf. Process. Med. Imaging. 2009:362–373. doi: 10.1007/978-3-642-02498-6_30. [DOI] [PubMed] [Google Scholar]
- Li X., Chen S., Hu X., Yang J. 2019 IEEE/CVF Conference on Computer Vision and Pattern Recognition. CVPR; 2019. Understanding the disharmony between dropout and batch normalization by variance shift; pp. 2677–2685. [Google Scholar]
- Long J., Shelhamer E., Darrell T. Fully convolutional networks for semantic segmentation. Proc. IEEE Conf. Comput. Vis. Pattern Recogn. 2015:3431–3440. doi: 10.1109/TPAMI.2016.2572683. [DOI] [PubMed] [Google Scholar]
- Ma X., Dagliyan O., Hahn K.M., Danuser G. Profiling cellular morphodynamics by spatiotemporal spectrum decomposition. PLoS Comput. Biol. 2018;14:e1006321. doi: 10.1371/journal.pcbi.1006321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M., Danuser G. Morphodynamic profiling of protrusion phenotypes. Biophys. J. 2006;90:1439–1452. doi: 10.1529/biophysj.105.070383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M., Hodgson L., Welch C., Elliott H., Pertz O., Nalbant P., Abell A., Johnson G.L., Hahn K.M., Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manak M.S., Varsanik J.S., Hogan B.J., Whitfield M.J., Su W.R., Joshi N., Steinke N., Min A., Berger D., Saphirstein R.J., et al. Live-cell phenotypic-biomarker microfluidic assay for the risk stratification of cancer patients via machine learning. Nat. Biomed. Eng. 2018;2:761–772. doi: 10.1038/s41551-018-0285-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.R., Fowlkes C.C., Malik J. Learning to detect natural image boundaries using local brightness, color, and texture cues. IEEE Trans. Pattern Anal. Mach. Intell. 2004:530–549. doi: 10.1109/TPAMI.2004.1273918. [DOI] [PubMed] [Google Scholar]
- McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., Doan M., Ding L., Rafelski S.M., Thirstrup D., et al. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 2018;16:e2005970. doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moen E., Bannon D., Kudo T., Graf W., Covert M., Van Valen D. Deep learning for cellular image analysis. Nat. Methods. 2019;16:1233–1246. doi: 10.1038/s41592-019-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oquab M., Bottou L., Laptev I., Sivic J. IEEE Conference on Computer Vision and Pattern Recognition (CVPR) IEEE; 2014. Learning and transferring mid-level image representations using convolutional neural networks; pp. 1717–1724. [Google Scholar]
- Otsu N. A threshold selection method from gray-level histograms. IEEE Transactions Systems, Man, Cybernetics. 1979;9:62–66. [Google Scholar]
- Pratt L.Y. Discriminability-based transfer between neural networks. Adv. Neural Inf. Process. Syst. 1993:204–211. [Google Scholar]
- Raza S.E.A., Cheung L., Shaban M., Graham S., Epstein D., Pelengaris S., Khan M., Rajpoot N.M. Micro-Net: a unified model for segmentation of various objects in microscopy images. Med. Image Anal. 2019;52 doi: 10.1016/j.media.2018.12.003. [DOI] [PubMed] [Google Scholar]
- Razavian A.S., Azizpour H., Sullivan J., Carlsson S. 2014 IEEE Conference on Computer Vision and Pattern Recognition Workshops. IEEE; 2014. CNN features off-the-shelf: an astounding baseline for recognition; pp. 512–519. [Google Scholar]
- Ronneberger O., Fischer P., Brox T. U-net: convolutional networks for biomedical image segmentation. Int. Conf. Med. Image Comput. Comput. Assist. Interven. 2015:234–241. [Google Scholar]
- Sadanandan S.K., Ranefall P., Le Guyader S., Wahlby C. Automated training of deep convolutional neural networks for cell segmentation. Sci. Rep. 2017;7:7860. doi: 10.1038/s41598-017-07599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U., Weigert M., Broaddus C., Myers G. Springer International Publishing; 2018. Cell Detection with Star-Convex Polygons; pp. 265–273. [Google Scholar]
- Selvaraju R.R., Cogswell M., Das A., Vedantam R., Parikh D., Batra D. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. IEEE; 2017. pp. 618–626. [Google Scholar]
- Shen W., Wang X., Wang Y., Bai X., Zhang Z. Deepcontour: a deep convolutional feature learned by positive-sharing loss for contour detection. Proc. IEEE Conf. Comput.r Vis. Pattern Recogn. 2015:3982–3991. [Google Scholar]
- Simonyan K., Zisserman A. International Conference on Learning Representations. International Conference on Learning Representations; 2015. Very deep convolutional networks for large-scale image recognition. [Google Scholar]
- Srivastavanitish N., Hinton G., Krizhevskykriz A., Sutskeverilya I., Salakhutdinov R. Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014;15:1929–1958. [Google Scholar]
- Stephens D.J., Allan V.J. Light microscopy techniques for live cell imaging. Science. 2003;300:82–86. doi: 10.1126/science.1082160. [DOI] [PubMed] [Google Scholar]
- Stringer C., Wang T., Michaelos M., Pachitariu M. Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods. 2021;18:100–106. doi: 10.1038/s41592-020-01018-x. [DOI] [PubMed] [Google Scholar]
- Tan M., Le Q.V. ICML; 2020. EfficientNet: Rethinking Model Scaling for Convolutional Neural Networks; p. 2019. [Google Scholar]
- Van Valen D.A., Kudo T., Lane K.M., Macklin D.N., Quach N.T., DeFelice M.M., Maayan I., Tanouchi Y., Ashley E.A., Covert M.W. Deep learning automates the quantitative analysis of individual cells in live-cell imaging experiments. PLoS Comput. Biol. 2016;12:e1005177. doi: 10.1371/journal.pcbi.1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicar T., Balvan J., Jaros J., Jug F., Kolar R., Masarik M., Gumulec J. Cell segmentation methods for label-free contrast microscopy: review and comprehensive comparison. BMC Bioinformatics. 2019;20 doi: 10.1186/s12859-019-2880-8. https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2880-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova K., Dibrov A., Myers G. Towards interpretable semantic segmentation via gradient-weighted class Activation mapping (student abstract) Proc. AAAI Conf. Artif. Intell. 2020;34:13943–13944. [Google Scholar]
- Wang C., Choi H.J., Kim S.J., Desai A., Lee N., Kim D., Bae Y., Lee K. Deconvolution of subcellular protrusion heterogeneity and the underlying actin regulator dynamics from live cell imaging. Nat. Commun. 2018;9:1688. doi: 10.1038/s41467-018-04030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Choi H.J., Woodbury L., Lee K. Deep learning-based subcellular phenotyping of protrusion dynamics reveals fine differential drug responses at subcellular and single-cell levels. bioRxiv. 2021 doi: 10.1101/2021.05.25.445699. [DOI] [Google Scholar]
- Yosinski J., Clune J., Bengio Y., Lipson H. How transferable are features in deep neural networks? Adv. Neural Inf. Process. Syst. 2014:3320–3328. [Google Scholar]
- Zeiler M.D., Fergus R. Springer International Publishing; 2014. Visualizing and Understanding Convolutional Networks; pp. 818–833. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at Github (https://github.com/kleelab-bch/MARS-Net) and Zenodo (https://doi.org/10.5281/zenodo.5541392) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.