

Abstract
Natural hybridization events provide unique windows into the barriers that keep species apart as well as the consequences of their breakdown. Here we characterize hybrid populations formed between the northern swordtail fish Xiphophorus cortezi and X. birchmanni from collection sites on two rivers. We use simulations and new genetic reference panels to develop sensitive and accurate local ancestry calling in this novel system. Strikingly, we find that hybrid populations on both rivers consist of two genetically distinct subpopulations: a cluster of pure X. birchmanni individuals and one of phenotypically intermediate hybrids that derive ~85–90% of their genome from X. cortezi. Simulations suggest that initial hybridization occurred ~150 generations ago at both sites, with little evidence for contemporary gene flow between subpopulations. This population structure is consistent with strong assortative mating between individuals of similar ancestry. The patterns of population structure uncovered here mirror those seen in hybridization between X. birchmanni and its sister species, X. malinche, indicating an important role for assortative mating in the evolution of hybrid populations. Future comparisons will provide a window into the shared mechanisms driving the outcomes of hybridization not only among independent hybridization events between the same species but also across distinct species pairs.
Keywords: hybridization, assortative mating, local ancestry inference, evolutionary genetics
Introduction
It has long been recognized that hybrids provide unique insights into the barriers between species and the consequences of their breakdown (Barton & Hewitt, 1985). While artificial hybrids, particularly in Drosophila, formed the foundation of early research into the genetic barriers that differentiate species (Coyne & Orr, 1997; Dobzhansky, 1936; Orr & Coyne, 1989), in recent years there has been a renaissance in the study of natural hybrid populations (Brandvain, Kenney, Flagel, Coop, & Sweigart, 2014; Calfee, Agra, Palacio, Ramírez, & Coop, 2020; El Ayari, Trigui El Menif, Hamer, Cahill, & Bierne, 2019; Hopkins, Guerrero, Rausher, & Kirkpatrick, 2014; Powell et al., 2020; Stukenbrock, Christiansen, Hansen, Dutheil, & Schierup, 2012; Turner & Harr, 2014). These natural experiments provide the unique opportunity to study hybridization in its ecological and evolutionary contexts, which are fundamental to fully characterizing the consequences of hybridization (Barton & Hewitt, 1985).
More recently, the increasing accessibility of dense genomic data has allowed granular studies of genome evolution in hybrid zones, revealing variation in ancestry among individuals and populations as well as selection on ancestry at particular loci (Taylor, Larson, & Harrison, 2015; Teeter et al., 2008; Torre, Ingvarsson, & Aitken, 2015). This increased resolution has allowed researchers to begin to compare distinct hybridization events, a first step towards tackling the important question of whether shared evolutionary mechanisms lead to predictable outcomes of hybridization at both the population and genomic level.
Though the complexity of natural hybrid zones provides an opportunity to study the interactions of different genetic, ecological, and evolutionary forces, it also creates challenges in disentangling them. For example, it can be difficult to determine whether patterns observed in individual hybrid zones are driven by intrinsic interactions between the genomes of hybridizing species, are dependent upon behavioral, demographic, or ecological context, or are stochastic (Ross & Harrison, 2002). Thus, the study of multiple hybrid zones provides the best of both worlds, with natural replication testing for shared mechanisms driving evolution after hybridization, and variation in environment or demographic history between populations creating informal tests for the relevance of these factors (Harrison & Larson, 2016; Janoušek et al., 2012). Each new case described offers a unique window into how eco-evolutionary history drives the outcomes of hybridization.
Due in part to their natural replication in multiple river systems, hybrid populations formed between swordtail fish Xiphophorus birchmanni and X. malinche have become an emerging model for the study of hybridization (Rosenthal et al., 2003). Research in this system has revealed that hybridization between X. birchmanni and X. malinche began recently (in the last ~100 generations) in several populations (Schumer et al., 2014), likely due to disrupted sensory communication as a result of human-mediated habitat disturbance (Fisher, Wong, & Rosenthal, 2006). Moreover, our work has indicated that differences in the strength of assortative mating by ancestry explain differences in population structure between X. birchmanni × X. malinche hybrid populations in distinct rivers (Schumer et al., 2017).
Here, we describe a previously unexplored hybridization event between X. birchmanni and its more distant relative, X. cortezi (Kallman & Kazianis, 2006; X. birchmanni - X. malinche sequence divergence 0.4% per basepair, X. birchmanni - X. cortezi 0.6% per basepair; Fig. 1A). We characterize the history of hybridization in two geographically separated tributaries of the Río Santa Cruz drainage in northern Hidalgo, Mexico (Fig 1B). Using sensitive and accurate local ancestry calling, we infer demographic history of each population and evaluate the potential role of assortative mating in maintaining ancestry structure. Like X. birchmanni and X. malinche, X. birchmanni and X. cortezi have overlapping ranges but largely are separated along an elevational gradient. X. malinche and X. cortezi are allopatric across their entire ranges. X. malinche occurs at the highest elevations of all three species. In streams where X. birchmanni and X. cortezi co-occur, X. cortezi is found at lower elevations. Moreover, both pairs of hybridization events include a sworded (X. malinche; X. cortezi) and swordless species (X. birchmanni), among other differences in sexual signals (Cui, Delclos, Schumer, & Rosenthal, 2017; Culumber & Rosenthal, 2013; Fernandez & Morris, 2008; Rosenthal et al., 2003). These analyses allow us to characterize another case of hybridization that is independent from the X. birchmanni × X. malinche hybridization event, yet broadly parallel in the recent onset and ongoing nature of hybridization, providing a valuable opportunity to study the population-level outcomes of hybridization in closely related species pairs. As such, these new hybrid populations provide a powerful window into the barriers between species and the consequences of their breakdown.
Figure 1.
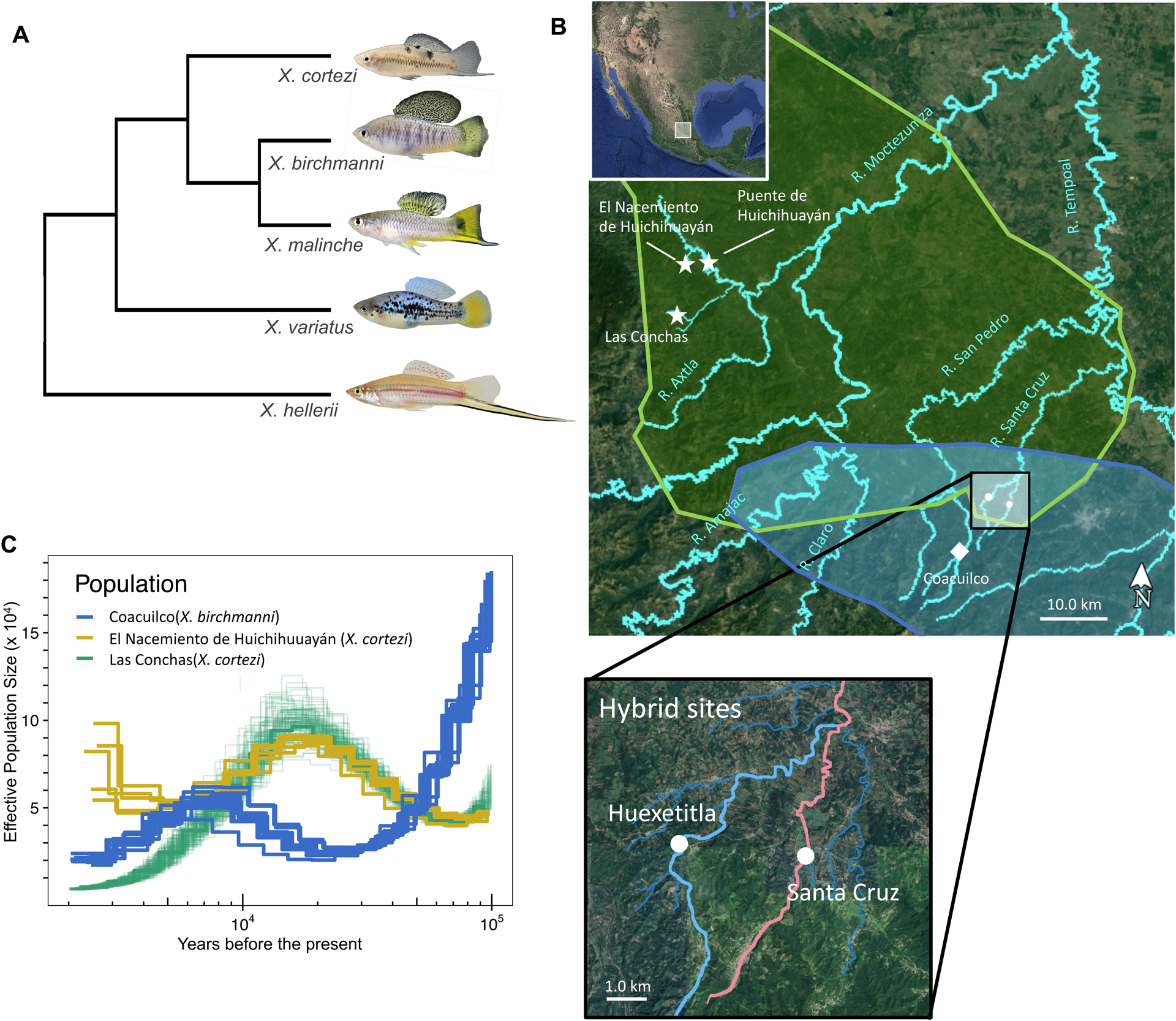
Phylogenetic relationships between species, location of sampling sites, and demographic history of parental species. A) Phylogenetic relationships between X. birchmanni, X. malinche, and X. cortezi (simplified from Cui et al., 2013). An outgroup, X. hellerii is also included, as is X. variatus, a distantly related species to the northern swordtail clade that co-occurs with X. birchmanni and X. cortezi. Representative individuals from parental species populations are pictured. B) Map of parental ranges and population collections. Polygons represent the known ranges of X. cortezi (green) and X. birchmanni (blue). Pure populations of X. cortezi (stars), X. birchmanni (square) and hybrid populations (circles) are labeled. Sampling sites for X. birchmanni × X. cortezi hybrid populations (Huextetitla n=76, Santa Cruz n=95) with the tributaries they occur in are highlighted in the lower inset. Major rivers are labeled in turquoise. C) Demographic history of pure X. birchmanni (blue lines) and X. cortezi (green and yellow lines) populations inferred by PSMC, assuming 2 generations a year and a per-base pair mutation rate of 3.5 × 10−9.
Materials and Methods
Sample collection
Fish were collected from wild populations in the states of Hidalgo and San Luis Potosí, Mexico using baited minnow traps. Putative X. cortezi × X. birchmanni hybrids were sampled from two distinct collection sites (hereafter sites; Fig. 1B), Huextetitla (21°9’43.82”N 98°33’27.19”W, n=87) and Santa Cruz (21°9’27.63”N 98°31’13.79”W, n=95). These sites occur in separate tributaries of the Río Santa Cruz in northern Hidalgo. Pure X. cortezi were collected in January 2020 from the Río Huichihuayán, a fully allopatric population with respect to X. birchmanni (Puente de Huichihuayán, 21°26’9.95”N 98°56’0.00”W, n=30). One X. cortezi individual from Las Conchas (21°23’33.30”N 98°59’23.33”W) and seven from El Nacimiento de Huichihuayán (21°27’34.10”N 98°58’36.70”W) that were previously sequenced to high coverage were included in genomic analyses (Powell et al., 2020; Schumer et al., 2018). Likewise, pure X. birchmanni from Coacuilco (21° 5’51.16”N 98°35’20.10”W, n=55) were collected previously for studies of hybridization with X. malinche (Schumer et al., 2018).
After collection, fish were anesthetized in a buffered solution of MS-222 and water diluted to 100 mg/mL (Stanford APLAC protocol #33071). Once anesthetized, fish were photographed against a grid background with dorsal and caudal fins spread using a Nikon d90 DSLR digital camera mounted to a copy stand and equipped with a macro lens. A small fin clip was taken from each individual and preserved in 95% ethanol for later DNA extraction. Females used for embryo comparisons were euthanized by MS-222 overdose in the field before being preserved in 95% ethanol.
Phenotyping and PCA analysis
Standard length, body depth, peduncle depth, caudal fin length, dorsal fin width, dorsal fin height were measured from photographs of adult fish using ImageJ (Schneider, Rasband, & Eliceiri, 2012). Additionally, length of the sword, a sexually selected male ornament that is present in X. cortezi and absent in X. birchmanni, was measured for adult males. Principal Component Analysis (PCA) was performed on adult males (X. birchmanni Coacuilco, n=23; X. cortezi Puente de Huichihuayán, n=10; Huextetitla, n=26; Santa Cruz, n=38) and adult females (X. birchmanni Coacuilco, n=25; X. cortezi Puente de Huichihuayán, n=16; Huextetitla, n=24; Santa Cruz, n=38) separately to compare phenotypic variance in the hybrid populations to phenotypic variance in the parental species using the princomp function in R v.3.6.3. Hybrid individuals were assigned to an ancestry group based on their genome-wide ancestry proportions (see Inferring Local Ancestry Section below).
Discriminant function analysis was performed using the R package MASS (Venables & Ripley, 2002) to assess how well morphological phenotypes from the samples described above could predict ancestry in males. A linear discriminant analysis model was trained using 75% of individuals categorized based on their genome-wide ancestry and collection site as pure X. birchmanni from an allopatric population, pure X. cortezi from an allopatric population, X. birchmanni from a hybrid population, or X. cortezi-like hybrid, and was then used to predict the ancestry and source population of the remaining 25% of individuals. Training on different subsets of the data ranging from 30–90% did not qualitatively change results.
DNA extraction and library preparation
DNA was extracted from fin tissue using the Agencourt DNAdvance kit (Beckman Coulter, Brea, California) as specified by the manufacturer but using half the recommended reaction volume. Extracted DNA was quantified using the TECAN Infinite M1000 microplate reader (Tecan Trading AG, Switzerland) at the High Throughput Biosciences Center at Stanford University, Stanford, CA.
Tagmentation-based whole genome libraries for low coverage sequencing were prepared from DNA extracted from fin clips collected from fish caught at the Huextetitla and Santa Cruz populations. Briefly, DNA was diluted to approximately 2.5 ng/μL and enzymatically sheared using the Illumina Tagment DNA TDE1 Enzyme and Buffer Kits (Illumina, San Diego, CA) at 55°C for 5 minutes. Sheared DNA samples were amplified in PCR reactions with dual indexed custom primers for 12 cycles. Amplified PCR reactions were pooled and purified using 18% SPRI magnetic beads.
Genomic libraries for high coverage sequencing of an individual collected in Huextetitla and one collected from Santa Cruz were prepared following Quail et al. (Quail, Swerdlow, & Turner, 2009). Briefly, approximately 500 ng of DNA was sheared to ~400 basepairs using a QSonica sonicator (QSonica Sonicators, Newton, Connecticut). To repair the sheared ends, DNA was mixed with dNTPs and T4 DNA polymerase, Klenow DNA polymerase and T4 PNK and incubated at room temperature for 30 minutes (NEB, Ipswich, MA) and then purified with the Qiagen QIAquick PCR purification kit (Qiagen, Valencia, CA). A-tails were added by mixing the purified end-repaired DNA with dATPs and Klenow exonuclease and incubating at 37° C for 30 minutes (NEB, Ipswich, MA) and then purified using the Qiagen QIAquick PCR purification kit (Qiagen, Valencia, CA). Adapter ligation reaction was performed followed by purification with the Qiagen QIAquick PCR purification kit (Qiagen, Valencia, CA). Adapter ligated DNA was amplified using indexed primers in individual Phusion PCR reactions for 12 cycles and then purified using 18% SPRI beads.
Libraries were quantified with a Qubit fluorometer (Thermo Scientific, Wilmington, DE). Library size distribution and quality were assessed using Agilent 4200 Tapestation (Agilent, Santa Clara, CA). Libraries were sequenced on an Illumina HiSeq 4000 at Admera Health Services, South Plainfield, NJ.
10X chromium library
To generate a draft genome assembly for X. cortezi, we made a 10X Chromium library using the Genomic Services Lab at the HudsonAlpha Institute for Biotechnology. High molecular weight DNA was extracted from fin tissue using the Genome Reagent Kit from 10X genomics. DNA was diluted to working concentrations of 0.4 ng/μL (quantified with a Qubit fluorometer). This is the recommended concentration given the Xiphophorus genome size of ~700 Mb. These working solutions were used as input to the library preparation protocol to begin the emulsion phase. The emulsion phase was broken as directed by the protocol, and bead purification was performed in 96-well plates. Final libraries were quantified using a Qubit fluorometer and library size was evaluated on a Bioanalyzer.
Admixtools analysis to evaluate evidence for hybridization between X. birchmanni and X. cortezi
To evaluate initial evidence for admixture, we sequenced one individual from Huextetitla and one Santa Cruz individual who appeared phenotypically intermediate between X. birchmanni and X. cortezi to ~30X coverage, as described above. We mapped reads from this individual to the X. birchmanni reference genome using bwa (Li & Durbin, 2009), marked and removed duplicates with Picard Tools and realigned insertion-deletion differences (indels) with GATK v3.4 (McKenna et al., 2010). We performed variant calling with GATK’s HaplotypeCaller in GVCF mode (McKenna et al., 2010). Because we lack an appropriate variant set for variant recalibration, we did not perform this step and instead implemented hard-calls based on several filters (DP, QD, MQ, FS, SOR, ReadPosRankSum, and MQRankSum) as described elsewhere (Schumer et al., 2018). In addition, we masked 5 bp windows surrounding indels and any site with greater than 2X or less than 0.5X the average genome-wide coverage. Based on past work quantifying Mendelian errors in swordtail pedigrees after applying these filters, we believe that this approach has high accuracy (Schumer et al., 2018).
We repeated these steps for previously sequenced X. malinche, X. birchmanni, and X. cortezi individuals to generate variant calls from an appropriate set of species for D-statistic analysis (Patterson et al., 2012). We used custom scripts available on our lab github to convert these files to admixtools format (https://github.com/Schumerlab/Lab_shared_scripts; https://openwetware.org/wiki/Schumer_lab:_Commonly_used_workflows#g.vcf_files_to_Admixtools_input). This resulted in 1,001,493 informative sites for analysis with admixtools. We used the qpDstat function from admixtools and a jack-knife bootstrap window size of 5 Mb to determine the most likely four-population tree, and calculate the D-statistic based on that tree. We also explored evidence of admixture with another Xiphophorus species that is sympatric with X. birchmanni and X. cortezi but deeply diverged from both species and found no evidence for hybridization with this species (Supporting Information 1–2).
Generation of a reference guided X. cortezi assembly
An initial draft assembly for X. cortezi was generated from the 10X Chromium library described above using the supernova software (v2.0.1; Weisenfeld, Kumar, Shah, Church, & Jaffe, 2017). The maximum reads used parameter was set to 280 million and the output style was specified as pseudohap, otherwise recommended parameters for the Xiphophorus genome size were used. This resulted in a draft assembly of 7,610 scaffolds (2,182 longer than 10 kb) with an N50 of 1.04 Mb and a total of 686 Mb assembled. The expected genome size of Xiphophorus is approximately 700 Mb.
Chromosome-scale synteny is conserved as 24 chromosomes across Xiphophorus species (Amores et al., 2014; Powell et al., 2020; Schartl et al., 2013). Thus, we decided to leverage the chromosome structure in other Xiphophorus assemblies to create chromosome-level scaffolds for X. cortezi. First, we created a multi-way whole genome alignment for swordtail species including X. birchmanni, X. variatus, and X. malinche (Powell et al., 2020), X. cortezi and X. xiphidium (this study), X. couchianus (RefSeq assembly GCF_001444195.1), and X. maculatus (RefSeq assembly GCF_002775205.1). Using the phylogenetic relationships from Cui et al. (R. Cui et al., 2013) as our guide tree, we ran progressive Cactus (Armstrong et al., 2019) to build the alignment. Parameters for the alignment are automatically determined by progressive Cactus based on branch lengths of the guide tree. Using this alignment and the same guide tree described previously, we arranged the scaffolds into 24 putative chromosomes using Ragout (Komolgorov et al. 2018), keeping the naming scheme consistent with that of the X. birchmanni genome (Fig. S1). Chromosome aligned scaffolds (n=28) were combined with unplaced scaffolds (n=4,777) to create the final assembly. Configuration files and associated scripts, as well as a Docker environment, are provided on github at https://github.com/Schumerlab/Xbir_xcor_hybridzone.
PSMC demographic inference
We inferred the demographic history of all X. cortezi individuals sequenced to high coverage. We used data from the 10X Chromium library generated for the X. cortezi genome assembly, as well as previously sequenced X. cortezi individuals from El Nacemiento de Huichihuayán and X. birchmanni individuals from Coacuilco (Powell et al., 2020; Schumer et al., 2018). Briefly, raw reads were mapped to the X. birchmanni reference assembly (Powell et al., 2020), after which GATK v3.4 (McKenna et al., 2010) was used to call variant sites as described above. These variants were then quality filtered as described above and used to create pseudo-reference genomes for each individual, which were input to PSMC (Li & Durbin, 2011). PSMC output was converted to effective population size assuming a mutation rate of 3.5×10−9 per basepair per generation and a generation time of 0.5 years, as described elsewhere (Schumer et al., 2018). We note that although other methods such as MSMC allow for simultaneous inference of demographic history in multiple individuals, they also require phasing, which can introduce errors, especially in cases where haplotype reference panels are not available (Schiffels & Wang, 2020).
Inferring local ancestry
We used a series of approaches to develop ancestry informative sites that distinguished X. birchmanni and X. cortezi. We first used a panel of 25 high coverage X. birchmanni individuals from the Coacuilco population, 7 X. cortezi individuals from El Nacimiento de Huichihuayán, and the reference individual from Las Conchas from other studies (Powell et al., 2020; Schumer et al., 2018) to identify candidate ancestry informative sites. With this candidate set and low coverage whole-genome sequence data that we collected for X. cortezi collected from Puente de Huichihuyán in this study (n=30) and data for X. birchmanni collected from Coacuilco in a previous study (Schumer et al., 2018), we evaluated population level counts for X. cortezi and X. birchmanni alleles at these ancestry informative sites (Powell et al., 2020). Any candidate ancestry informative site where the major allele in either parental population was at less than 90% frequency was excluded, yielding a set of 1.1 million ancestry informative sites genome-wide (~1.5 per kb). We describe our approach for identifying ancestry informative sites and determining parameters for local ancestry inference in more detail in Supporting Information 3–4; we have also explored these issues in previous work (Powell et al., 2020; Schumer, Powell, & Corbett-Detig, 2020).
With this set of ancestry informative sites, we used a hidden Markov model (HMM) approach to infer local ancestry with our previously developed local ancestry inference tool, ancestryinfer (Schumer et al., 2020), and evaluated performance on a set of parental individuals that were not used in previous steps (Fig. S2). We also performed simulations to evaluate expected performance under a range of demographic scenarios. Together these results suggest that we expect to have high accuracy in calling local ancestry in X. birchmanni × X. cortezi hybrids (Supporting Information 3–4; Fig. 2B; Fig. S3).
Figure 2.
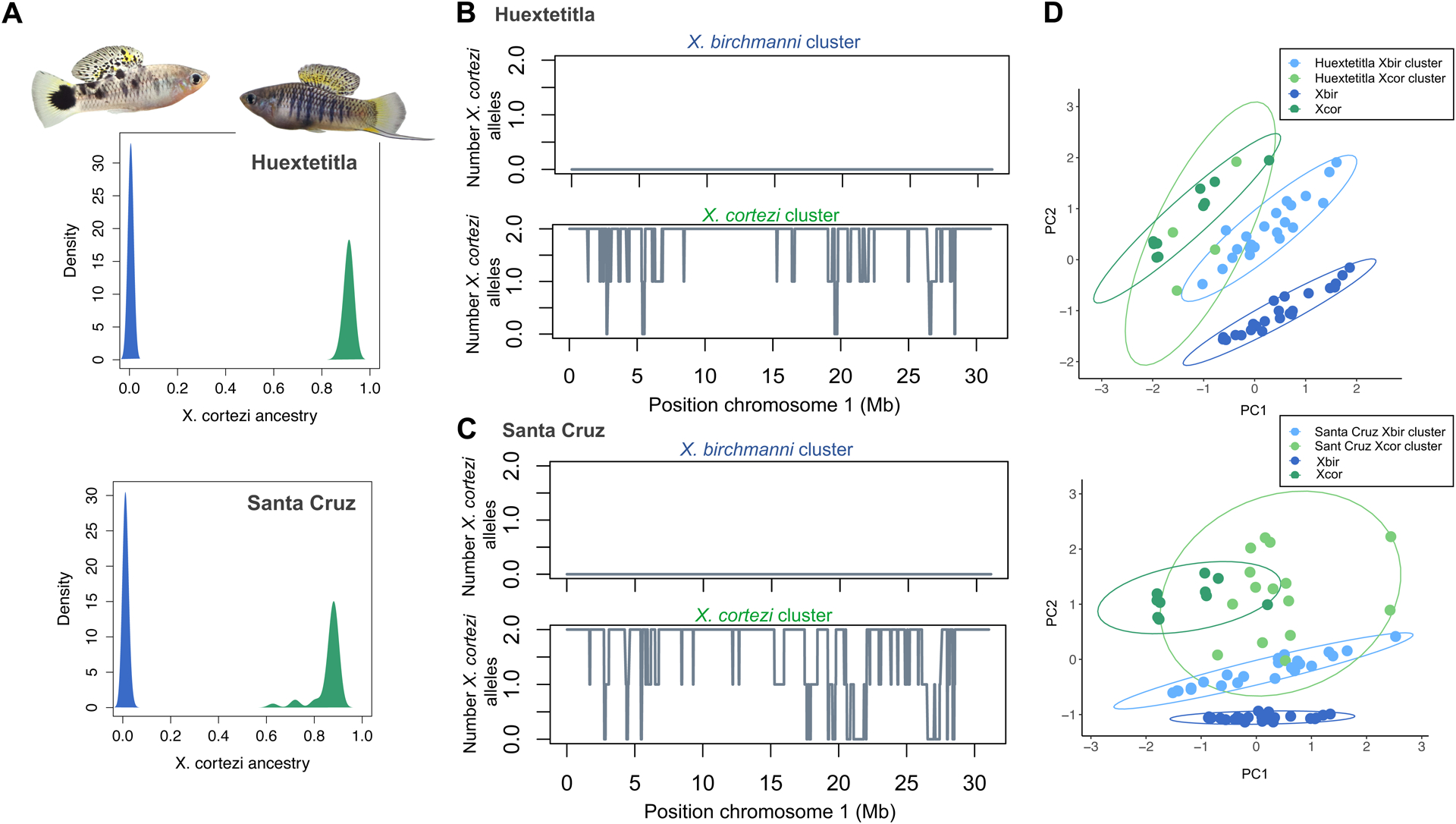
Ancestry structure of X. birchmanni × X. cortezi populations and example local ancestry inference. A) Genome-wide ancestry in the Huextetitla (top) and Santa Cruz (bottom) populations. Plotted here is the proportion of the genome derived from X. cortezi in all sampled individuals in both populations. Individuals plotted in green were assigned to the X. cortezi ancestry cluster and in blue were assigned to the X. birchmanni ancestry cluster. Representative individuals from each ancestry cluster from the Huextetitla population are shown. B) Local ancestry on chromosome 1 for one X. birchmanni cluster and one X. cortezi cluster individual for the Huextetitla population. C) Local ancestry on chromosome 1 for one X. birchmanni cluster and one X. cortezi cluster individual for the Santa Cruz population. D) PCA plots of phenotypic data from Huextetitla population males (top) and Santa Cruz population males (bottom) compared with parental species male phenotypic data. Xbir – X. birchmanni, Xcor – X. cortezi. Each point represents one individual and ellipses represent the 95% confidence interval. Loadings for each phenotype can be found in Table S1.
Confident in the accuracy of our local ancestry inference approach, we next applied these methods to individuals collected from putative hybrid populations. Based on the results of an initial analysis with uniform ancestry priors, we identified the presence of two distinct ancestry clusters at both collection sites (Supporting Information 3–4). We thus re-ran the HMM for each genetic cluster using cluster-specific ancestry priors (X. birchmanni cluster: 1% X. cortezi; X. cortezi hybrid cluster: 15% X. birchmanni) and generated a merged dataset for the two populations.
Leveraging local ancestry and high-coverage sequencing of individual hybrids
For the two hybrid individuals that were deep sequenced (described above), we were able to perform both local ancestry inference and population genomic analyses. Specifically, using local ancestry inference we inferred tracts of the genome that were homozygous for the X. cortezi parental species in each individual. We extracted these tracts and analyzed nucleotide diversity (π) within homozygous X. cortezi tracts, as well as pairwise sequence divergence (Dxy) in tracts that were homozygous X. cortezi in both samples. We also evaluated pairwise divergence sequence between X. cortezi ancestry tracts in hybrids and pure X. cortezi populations.
Due to skewed admixture proportions, there were too few homozygous X. birchmanni tracts to perform the reciprocal analysis. However, we also sequenced one individual from the X. birchmanni subpopulation from the Huextetitla and Santa Cruz populations to ~10X genome-wide coverage (11X for Huextetitla individual and 12X for Santa Cruz individual). We used these sequences to evaluate divergence between sympatric X. birchmanni in the hybrid populations and the X. birchmanni reference population collected from Coacuilco. Because we did not have sufficient coverage to confidently call heterozygous sites, for sites where mapped reads supported both the reference and alternative allele we used binomial sampling weighted by the counts for the reference and alternative alleles to randomly sample one read at each variant site. We calculated Dxy relative to the X. birchmanni reference genome, which was originally generated from the Coacuilco population (Powell et al., 2020).
Approximate Bayesian computation for inferring hybrid population history
We used a variety of approaches to investigate the time since admixture in the Santa Cruz and Huextetitla hybrid populations, described in detail in Supporting Information 5. However, many approaches assume a single pulse of admixture, which may not be realistic for the Santa Cruz and Huextetitla hybrid populations where hybrids and pure X. birchmanni coexist (see Results).
To investigate this, we used an approximate Bayesian computation (ABC) approach to infer admixture scenarios that were consistent with observed data in the Santa Cruz and Huextetitla populations, focusing on the hybrid X. cortezi ancestry cluster (Fig. S4). We performed simulations in SLiM (Haller & Messer, 2019). Because our empirical data relies on local ancestry inference, we wanted our simulated data to mimic this structure. As a result, we used the tree sequence recording functions in SLiM to track ancestry tracts in each simulated individual (Haller, Galloway, Kelleher, Messer, & Ralph, 2018), and used this information to summarize overall ancestry on the individual and population level. Since our analyses are based on ancestry information in hybrids rather than other population genetic summaries, we simply initialized two parental populations at the start of each simulation and did not simulate parental population history. All simulations were performed without selection on hybrids. Although biologically unrealistic, the possible parameter space in simulations with selection is too large to explore in the ABC framework implemented here.
We initialized simulations with two parental populations (generation 1) and formed a hybrid population between them (generation 2). We drew parameters for each simulation from uniform prior distributions. Guided by results of initial simulations (see Results), we drew from a prior for the time since initial admixture of 10–200 generations, admixture proportion of 0.7–1 X. cortezi, and hybrid population size ranging from 50–3,000 diploid individuals (Fig. S4). We also implemented migration into the hybrid population from sympatric X. birchmanni individuals. Based on the number of early generation hybrids between ancestry clusters observed in the empirical data (see Results), we knew migration rates were low. We thus drew a per-generation migration rate from sympatric X. birchmanni individuals of 0–2%. All scripts to implement these simulations are available on github (https://github.com/Schumerlab/Xbir_xcor_hybridzone).
While we see evidence in our empirical data to support mating between sympatric X. birchmanni individuals and individuals in the X. cortezi hybrid cluster (see Results), we did not sample any pure X. cortezi individuals at Santa Cruz or Huextetitla. Moreover, when we attempted to sample downstream of Huextetitla and Santa Cruz during our field collections, where we expected to find more X. cortezi-like individuals, we found that swordtails had been extirpated from these sites (see Supporting Information 9). We thus did not simulate migration from pure X. cortezi source populations.
To identify the subset of simulations most closely matching patterns in our data, we performed rejection sampling at a 5% threshold based on summary statistics from our data and from simulations. As summary statistics we used average genome-wide ancestry, population-level variance in genome-wide ancestry, and the average length of minor parent ancestry tracts. We performed simulations until 500 parameter sets had been accepted. After an initial set of 1 million simulations resulted in only tens of accepted parameter sets when fitting summary statistics for the Huextetitla population, we restricted parameter space guided by those accepted simulations to evaluate a narrower range of initial admixture proportions (0.85–1) and migration rates (0–0.5%), and a broader range of generations since initial admixture (10–500). Otherwise simulations for Huextetitla were performed as described above.
Evaluating evidence for assortative mating in the Santa Cruz hybrid population
Evidence of bimodal ancestry structure in both hybrid populations (see Results) is suggestive of ancestry assortative mating, strong selection on hybrids, habitat partitioning, or some combination of these factors. To investigate this, we collected 87 females from the Santa Cruz hybrid population in March of 2020, euthanized them, and dissected and developmentally staged their offspring (Supporting Information 6). Forty-six females had developing embryos, with an average of 18 and standard deviation of 10 embryos per female; past work has suggested that a brood typically contains ~3 sires (Paczolt et al., 2015; Schumer et al., 2017). For each brood, we randomly selected two offspring for sequencing from each developmental stage present (to account for possible developmental differences associated with mating type). This resulted in a total of 159 sequenced embryos across mothers (mean 4.4, standard deviation 6.8 per mother), which were used in low-coverage library preparation and sequencing as described above.
To evaluate evidence for assortative mating by ancestry, we took advantage of expectations about maternal-offspring ancestry differences as a function of different types of mating events. Given the extreme differences in ancestry observed across the two genetic clusters in the Santa Cruz hybrid population (Fig. 2A), the difference between a mother and her offspring in ancestry allows us to infer the ancestry of the father. Specifically, if a female mates with a male from her own genetic cluster, she and her offspring will have very similar genome-wide ancestry, with the difference between them falling close to zero. If a female instead mates with a male from the other subpopulation, she and her offspring are expected to differ by ~40% in their genome-wide ancestry, given a difference of more than 80% in ancestry between the two clusters (Fig. 2A). This allowed us to quantify the evidence for assortative mating by ancestry by comparing observed mating events to simulations with varying strengths of assortative mating (Supporting Information 7). We had originally planned to analyze evidence for differential development as a function of mating type, but found too few mating events between ancestry clusters for this analysis to be conducted (Supporting Information 6).
Another biological mechanism that could cause patterns that might be confused with assortative mating are strong sperm-egg incompatibilities that prevent fertilization and the formation of embryos. To evaluate this, we set up crosses between pure parental species. Since livebearers store sperm we set up crosses between virgin X. birchmanni females and X. cortezi males, as well as crosses between virgin X. cortezi females and X. birchmanni males in the lab. We dissected 8 females ~30 days after they were introduced to males and evaluated development stage of all embryos (Haynes, 1995), as well as whether unfertilized eggs were present in the broods.
Analysis of videos from the Santa Cruz hybrid population
As a first step towards evaluating whether there is evidence of habitat partitioning in this structured hybrid population, we took underwater videos at the Santa Cruz site. Because males of the two clusters can be reliably distinguished based on their morphological characteristics, we scored videos to evaluate whether males of both clusters were inhabiting the same space.
Underwater video footage was recorded at the Santa Cruz locality to determine whether there is spatial and temporal overlap between X. birchmanni and X. cortezi-cluster males at this site. Videos were taken consecutively in 50 second to 23 minute sections (20 videos, total of 267 minutes) in July 2020. Cameras were set up in shallow pools isolated by riffles up and down-stream. The frame of view spanned ~1.5 meters. Males of the two clusters are visually distinguishable by the presence or absence of a sword (see Results), so the number of sworded and unsworded adult males observed was recorded for each video. Each time an adult male swordtail entered the ~1.5 meter frame of view was considered an independent observation and we observed 52 instances of male swordtails entering the frame of view. The presence of sworded and unsworded adult males in the same video was considered evidence for spatial and temporal overlap between the two genetic clusters. Females of the two genetic clusters are not visually distinguishable and thus were not evaluated.
Results
Demographic history of X. cortezi and split from X. birchmanni
We used the X. cortezi data obtained from 10X sequencing (Powell et al., 2020), along with pre-existing sequence data for single individuals of X. birchmanni (Coacuilco locality) and X. cortezi (El Nacimiento de Huichihuayán locality, San Luis Potosí; Powell et al., 2020; Schumer et al., 2018), to compare the demographic histories of X. cortezi and X. birchmanni (see Supporting Information 8). PSMC analysis of each individual points to distinct demographic histories of X. birchmanni and X. cortezi populations (Fig. 1C; assuming two generations per year and a mutation rate of 3.5 × 10−9). Interestingly, our results also suggest divergent demographic trends between two X. cortezi populations allopatric to the hybrid zones (Las Conchas and El Nacimiento de Huichihuayán; Fig. 1B). Declines in effective population size over the last 20,000 years inferred from the individual sampled from Las Conchas may reflect the demographic effects of colonization of this small tributary (Fig. 1C).
Despite differences in the timing of population size fluctuations, the long-term effective population size across species and sampling sites, estimated based on the harmonic mean (Supporting Information 8), was quite similar between the X. cortezi population at El Nacimiento de Huichihuayán and X. birchmanni. Specifically, we estimated that the long-term effective population size for X. cortezi ranged from 47,000–56,000 across populations compared to 48,000–53,000 in X. birchmanni, consistent with the observation that levels of genetic diversity (π) are similar between these X. cortezi and X. birchmanni populations (0.1% and 0.12% per basepair respectively). Assuming a long-term effective population size of 50,000 for both species, we estimate that X. cortezi and X. birchmanni diverged from each other approximately 250,000 years ago (Supporting Information 8).
Santa Cruz and Huextetitla populations are composed of pure X. birchmanni and X. birchmanni × X. cortezi hybrids
Initial analysis of a high-coverage individual sampled from Huextetitla and Santa Cruz indicated that these individuals were hybrids between X. birchmanni and X. cortezi (Huextetitla D= −0.49, Z= −30; Santa Cruz D=−0.55, Z=28; see also Supporting Information 1; Fig. S5, Fig. S6; Powell et al., 2020), motivating us to develop the local ancestry inference approaches as described in the Methods and Supporting Information 2–4. After inferring local ancestry based on the 1.1 million ancestry informative sites developed for X. birchmanni × X. cortezi hybrids, we summarized genome-wide ancestry for each individual sampled at the Huextetitla and Santa Cruz locations. To do so, we converted posterior probabilities at each ancestry informative site to hard-calls, requiring that the posterior probability for a given ancestry state exceed 0.9.
This analysis uncovered two genetically distinct subpopulations present in both the Huextetitla and Santa Cruz locations. One cluster consisted of pure X. birchmanni individuals (Huextetitla - n=64; Santa Cruz - n=59), coexisting with the second cluster of X. birchmanni × X. cortezi hybrids, with mean X. cortezi ancestry of 91±1% (n=12) and 86±6% (n=36) at Huextetitla and Santa Cruz respectively (Fig. 2A; see Fig. 2B and 2C for chromosome 1 local ancestry plots from representative individuals assigned to each ancestry cluster at Huextetitla and Santa Cruz). These results suggest the presence of strong barriers to gene flow between the two ancestry clusters at both locations, which we explore in more detail below. Notably, given the geography of these river systems (Fig. 1B), the two hybrid populations are likely independent in their recent demographic histories.
Wild X. birchmanni and X. birchmanni × X. cortezi hybrids can be distinguished by their phenotypic differences
Due to morphological differences between species (Fig. 1A) and striking differences in ancestry between the two genetic clusters at both the Huextetitla and Santa Cruz sampling sites (Fig. 2A), we predicted that males of the two clusters could be distinguished phenotypically. As is the case with many swordtail species, females of the two species are not visually distinguishable (Fig. S7). Using traits that differentiated males in PCA analysis (Fig. 2D; Table S1), we tested how well male genotypes could be predicted based on these phenotypes using discriminant function analysis. We found that a linear discriminant function analysis model fit to 75% of individuals (allopatric parental species, sympatric X. birchmanni, and hybrids from both sites) accurately predicted the ancestry cluster of 90.9% of individuals not used to fit the model (N = 22). We note that we did not have sufficient individuals to perform training separately on the two sampling sites. Training on different subsets of the data ranging from 30–90% did not qualitatively change results (accuracy range: 89–94%).
ABC simulations indicate that hybrid populations formed recently
Because the X. birchmanni and hybrid X. cortezi ancestry clusters are sympatric, we realized that typical approaches to estimate the time of admixture between the two species would likely underestimate the time of initial admixture. We also see genomic evidence for low levels of ongoing gene flow (see Evidence for ongoing admixture and assortative mating below). As a result, we used an ABC approach with simulation implemented in SLiM, allowing for ongoing migration to infer population histories consistent with our data. We focused these simulations on the hybrid cortezi ancestry cluster, as the X. birchmanni cluster shows little evidence of admixture.
From both the Santa Cruz and Huextetitla populations, we inferred well-resolved posterior distributions for the time since initial admixture and migration rates from the X. birchmanni cluster into the hybrid X. cortezi cluster (Fig. 3A). For both Santa Cruz and Huextetitla, we did not recover a well-resolved posterior distribution for population size, but posterior distributions are skewed away from very small population sizes for both sets of simulations (<500 individuals; Fig. S8). Although our simulations allow us to infer initial admixture proportions in the Huextetitla population (Fig. 3C), we were surprised that we were unable to recover a well resolved posterior distribution for initial admixture proportion for the Santa Cruz hybrid population. However, this appears to be driven by a strong correlation in the posterior distributions between admixture proportion, time of initial admixture, and migration rate parameters inferred for Santa Cruz. Joint posteriors for these parameters for the Santa Cruz population are shown in Fig. 3D and Fig. S9.
Figure 3.
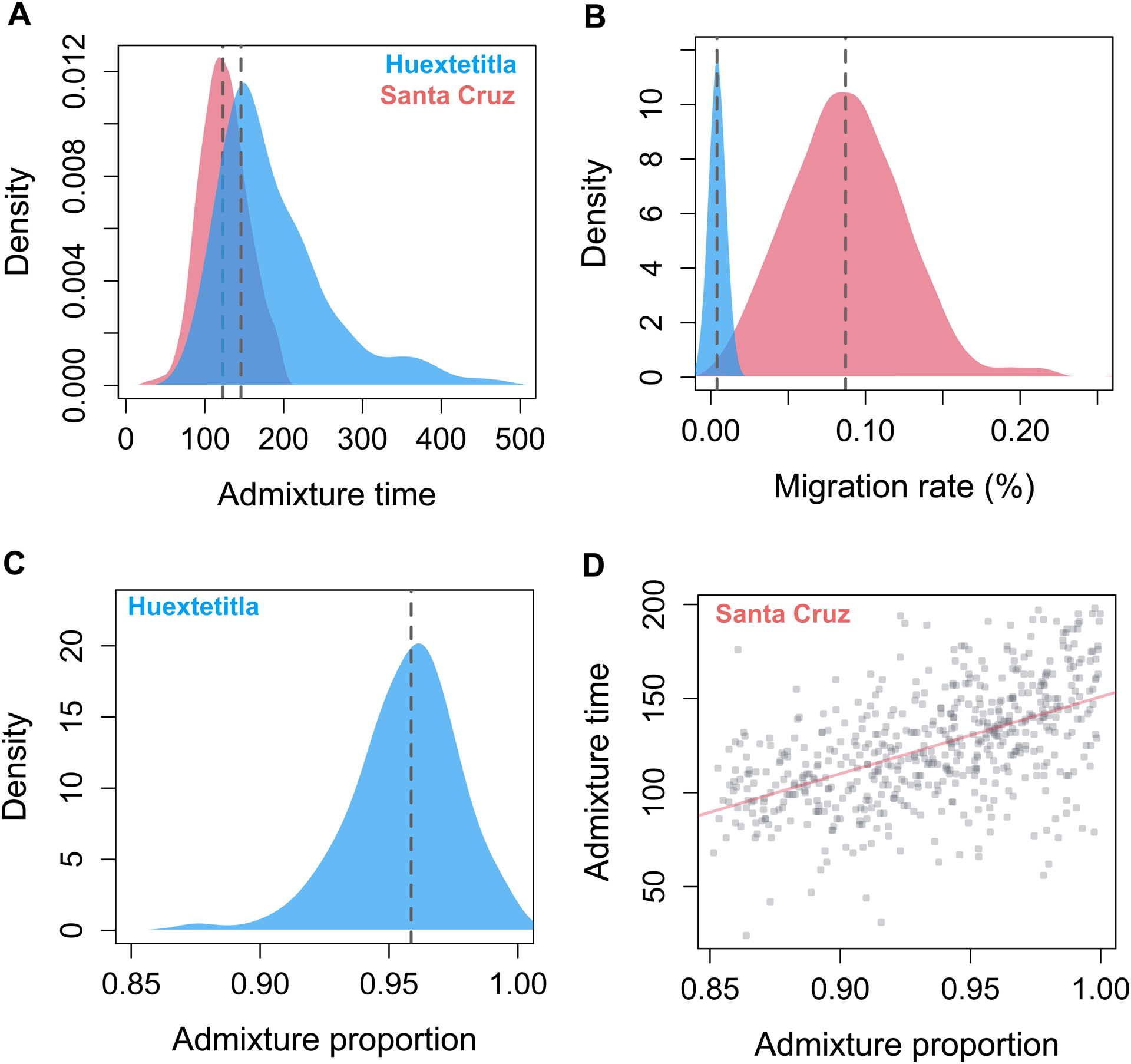
Posterior distributions from Approximate Bayesian Computation simulations inferring demographic history of the X. cortezi ancestry cluster in Huextetitla (blue) and Santa Cruz (pink). A) Posterior distributions for admixture time indicate that both populations formed relatively recently, in the last ~150 generations. B) Posterior distributions of per-generation migration rate reflect substantial differences between populations which can also be observed in variation in admixture proportions (Fig. 2A). C) For the Huextetitla population, where cross-cluster migration rates are much lower, we recovered a well-resolved posterior distribution of initial admixture proportion. D) For the Santa Cruz population, accepted initial admixture proportions span a wide range of parameters and co-vary with both the time since initial admixture (shown here) as well as the cross-cluster migration rate (Fig. S9). For C and D, an admixture proportion of 1 indicates pure X. cortezi ancestry genome-wide.
Posterior distributions for admixture time suggest that the hybrid X. cortezi populations at Santa Cruz and Huextetitla formed recently, within the last ~140 and ~167 generations respectively (95% confidence intervals – Santa Cruz: 101–183 generations; Huextetitla: 92–384 generations). These estimates are older than estimates from LD decay methods (Supporting Information 5), which put the time of initial admixture ~40 generations ago. This discrepancy is not entirely surprising because LD decay methods tend to underestimate the time since initial admixture in cases where there is ongoing hybridization, and ABC simulations suggest moderate levels of ongoing gene flow from sympatric X. birchmanni individuals into the cortezi hybrid ancestry cluster at Santa Cruz (maximum a posteriori or MAP estimate of m=0.1%, 95% confidence intervals: 0.02–0.15%). Ongoing migration appears to be much more limited at the Huextetitla collection site (MAP estimate of m=0.001%, 95% confidence intervals: 0.0002–0.01%; Fig. 3B).
Notably, the low inferred migration rates despite the populations existing in sympatry suggests some substantial barrier to gene flow – whether it be due to genetic incompatibilities, ecological factors, or assortative mating. We explore these possible barriers in more detail below.
Population genetic signals suggest independent recent demographic history of Huextetitla and Santa Cruz populations
Despite similarities in overall population structure and recent demographic history, given river distance between the Huextetitla and Santa Cruz populations, data on elevation changes, and available flooding records, we consider it unlikely that frequent migration occurs between the two hybrid populations (Fig. 1B; Supporting Information 9). As a second line of evidence, we evaluated evidence for differences in source populations and the recent demographic history of these two populations using population genomic summaries of high coverage individuals. We found little evidence of nucleotide divergence between the homozygous X. cortezi ancestry tracts present in both Huextetitla and Santa Cruz (Dxy Santa Cruz-Huextetitla = 0.05%, Dxy Santa Cruz-Santa Cruz = 0.07% per basepair), indicating that the X. cortezi source populations for the two hybrid sites are closely related. However, we do detect substantial differences in π across the Huextetitla and Santa Cruz populations (πSanta Cruz = 0.06 ± 0.003, πHuextetitla = 0.02 ± 0.002; Fig. S10). This suggests either substantial differences in the genetic diversity of the X. cortezi source populations or differences in the parental diversity contributed to the hybridization events in Huextetitla and Santa Cruz. Both possibilities indicate that these two hybrid populations are largely independent, at least in their recent demographic history. However, high concordance in local ancestry between the X. cortezi cluster in Huextetitla and Santa Cruz could indicate past connections between these populations (Supporting Information 9; Fig. S11).
In developing our local ancestry inference approaches, we used parental reference populations that were geographically isolated from the hybrid populations to ensure that reference panels did not contain X. birchmanni × X. cortezi hybrids. However, this raises the possibility that reference panels may be substantially diverged from the parental individuals that generated the focal hybrid populations. We evaluated this by calculating Dxy between homozygous X. cortezi ancestry tracts in hybrids and the X. cortezi source populations (using the data described above) and between pure X. birchmanni individual collected from the hybrid populations versus the reference X. birchmanni population at Coacuilco. This analysis indicated moderate genetic drift between the reference and admixing populations: Dxy of Huextetitla hybrid tracts versus X. cortezi – 0.0016, Huextetitla X. birchmanni versus Coacuilco X. birchmanni – 0.0014, Santa Cruz hybrid tracts versus X. cortezi – 0.0018, Santa Cruz X. birchmanni versus Coacuilco X. birchmanni – 0.0016. In all cases, divergence is higher than within population expectations for the pure parental populations (Dxy of 0.0008 – 0.0012), but ~4-fold lower than Dxy between X. birchmanni and X. cortezi. Nonetheless, we incorporated genetic drift into simulations evaluating the accuracy of local ancestry inference and are able to conclude that it will likely have a minor effect on accuracy given the degree of overall divergence between X. birchmanni and X. cortezi (Supporting Information 3–4).
Evidence for ongoing admixture and assortative mating
Out of 49 pregnant females collected from the Santa Cruz hybrid population, we successfully sequenced the mother and at least one offspring for 46 mother-offspring pairs. Thirty of these mothers belonged to the hybrid X. cortezi genotype cluster and 16 were pure X. birchmanni. Based on observed ancestry in embryos, none of the offspring collected were the product of a first generation cross-cluster mating event, however we infer that two females from the X. cortezi genotype cluster had mated with males of intermediate ancestry (males with approximately 25% and 55% X. birchmanni ancestry respectively). The proportion of sampled individuals with intermediate ancestry did not differ between the embryonic and adult populations at Santa Cruz (4.3±3% of sampled embryos and 3.2±2% of sampled adults with ancestry between 5–75% X. cortezi; Fig. 2A and Fig S11). Notably, all these individuals are inferred to have a X. cortezi mother, which could hint at weaker assortative mating by ancestry among females of the cortezi cluster (Supporting Information 10; Fig. S12). We found no evidence of differences in number of embryos, variation in embryo stage, or developmental abnormalities between females of the two clusters (all P values > 0.7; Supporting Information 6).
Analysis of the maternal-offspring ancestry patterns indicate clear deviations from expectations under random mating (Fig. 4; Supporting Information 7). We used simulations to quantify the strength of ancestry-assortative mating consistent with our data (Fig. 4B, Supporting Information 7). These simulations indicated that our data is consistent with a strength of ancestry assortative mating of approximately 98% (Fig. 4C). Strong assortative mating by ancestry is thus one likely factor maintaining the two distinct subpopulations at Santa Cruz.
Figure 4.
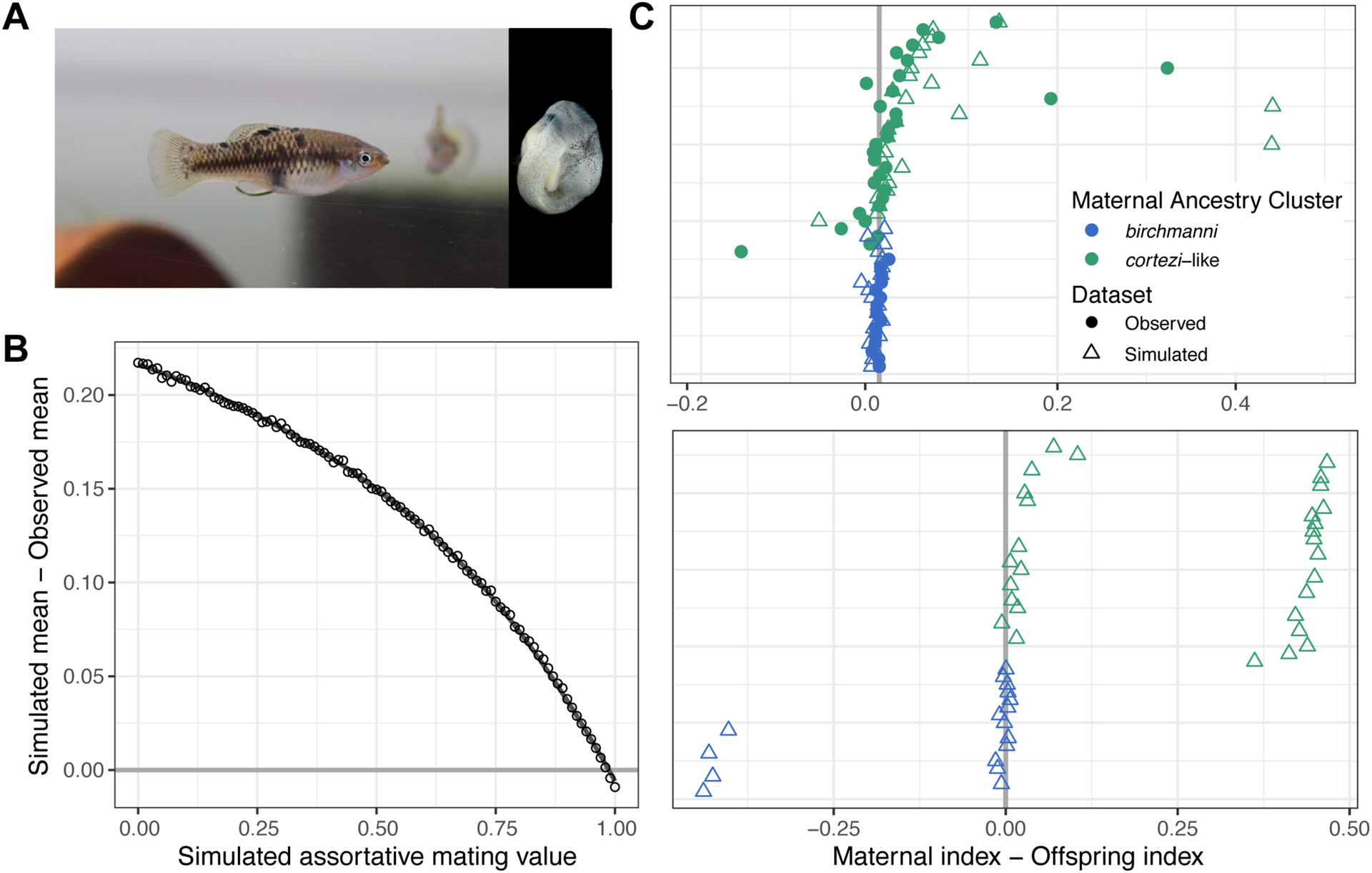
Results of assortative mating simulations in the Santa Cruz population. A) Photos of a pregnant X. cortezi-cluster female and a representative embryo. B) Results of simulations ranging from 0–100% assortative mating in increments of 1% comparing the simulated versus observed difference in maternal and offspring ancestry index. Simulations of 98% assortative mating minimized the difference between the observed and simulated datasets. C) Top – In the observed data (circles; see Fig. S13) and simulated data of 98% assortative mating (triangles), few offspring have dramatically different ancestry from their mothers. Bottom – In contrast, many such individuals are observed in simulations of random mating. Points close to the zero line represent females that mated with males from their own ancestry cluster. Individuals are colored based on their maternal ancestry cluster and are placed on the y-axis based on increasing X. cortezi ancestry.
Because strong sperm-egg incompatibilities or near complete mortality of hybrid offspring in the earliest stages of embryonic development could also explain the patterns we observe, we took advantage of crosses in the lab to determine that embryos could be formed in both cross directions. We dissected four females of each cross direction and found developing embryos in four out of four X. cortezi females (3 broods developmental stage 4; one brood developmental stage 9; n=12–26) and three out of four X. birchmanni females (3 broods developmental stage 4, one female not pregnant; n=14–18) using staging following Haynes 1995. There was no evidence for unfertilized eggs within broods (Supporting Information 6). This suggests that neither sperm-egg incompatibilities nor developmental incompatibilities are sufficient to explain the patterns of ancestry observed in wild-caught mothers and embryos. It should be noted these crosses do not rule out hybrid incompatibilities affecting later embryonic developmental stages, and indeed selection on cross-cluster hybrids (via incompatibilities or other mechanisms) could be an important factor driving the observed patterns of assortative mating.
We also note that our current results do not allow us to distinguish between assortative mating mediated via mate preferences and post-mating mechanisms such as near-perfect sperm precedence for males of similar ancestry. While this seems unlikely given what is known about the strength of sperm precedence in related species (Evans & Magurran, 2001; Gasparini, Simmons, Beveridge, & Evans, 2010; Paczolt et al., 2015), we cannot completely rule out this possibility.
Evidence of sympatry of the X. birchmanni and cortezi populations
Ancestry assortative mating could be driven by processes such as mate discrimination or by spatial isolation that prevents individuals from different genotype clusters from encountering each other. We suspected that the latter scenario was not the case at these collection sites as we repeatedly collected both male and gravid female X. birchmanni and X. cortezi cluster hybrid individuals from the same minnow traps (over three collections at Santa Cruz and one collection at Huextetitla). This suggests that these individuals are sympatric in the wild and have the opportunity to mate with each other.
As a first step towards investigating this further, we took underwater videos during the summer of 2020 at Santa Cruz and scored the videos for interactions between males of the two ancestry clusters, which can be distinguished with high accuracy based on their sword phenotypes (see above). We found both sworded and unsworded males in 4 of the 12 videos in which male swordtails were observed (12 to 15 minutes each, total of 158 minutes), showing that individuals of both hybrid clusters inhabit the same areas at the same time. There were 8 videos (50 seconds to 23 minutes each, total of 109 minutes) in which no male swordtails were observed. Raw video footage and scored data are available on Dryad (https://doi.org/10.5061/dryad.pzgmsbcmn).
Ancestry on chromosome 13 – a major QTL for the development of the sword
Several sexually dimorphic traits distinguish the pure forms of the two species, including the sword ornament (Fig. 1A) and the large dorsal fin (found in X. birchmanni; Rosenthal et al., 2003). In addition, both species show preferences for species-specific olfactory signals, as in other swordtail species studied to date (Crapon de Caprona & Ryan, 1990; Fisher, Mascuch, & Rosenthal, 2009; Fisher et al., 2006; McLennan & Ryan, 1997, 1999). For most of these sexually selected traits the genetic basis is unknown. However, regions of the genome associated with the sword in F2 crosses between X. birchmanni and X. malinche have recently been mapped (Powell et al., 2021), identifying a major effect QTL on chromosome 13 that explains ~10% of the heritable variation in sword length. This QTL also contributes to variation in sword length in other species in the genus (Schartl et al., 2021). We examined local ancestry in hybrids from the Santa Cruz and Huextetitla populations within this region, focusing on the most promising candidate gene identified – the regulator of fin and limb development sp8. Intriguingly, X. birchmanni ancestry is relatively high surrounding sp8 in both populations (Santa Cruz - upper 90% of genome-wide distribution; Huextetitla – upper 95% of genome-wide distribution; Fig. S14). The coincidence of high X. birchmanni ancestry in this region in both populations is unexpected by chance (p<0.005 by permutation). This result hints that the sword ornament is not a strong barrier to gene flow between the X. birchmanni and X. cortezi clusters, and alleles associated with reduced sword length may be introgressing more than expected by chance. Indeed, males from the X. cortezi hybrid cluster in both populations have shorter swords than pure X. cortezi on average (X. cortezi normalized sword length: 0.33 ± 0.08; Santa Cruz X. cortezi cluster: 0.17 ± 0.1; Huextetitla X. cortezi cluster: 0.20 ± 0.1).
Discussion
As hybridization is documented across ever-growing swaths of the tree of life, research can now address the extent to which the evolutionary outcomes of hybridization are predictable across pairs of species, from the genetic to the population level. Here, we develop sensitive local ancestry calling and infer the history of hybridization and ancestry structure in two newly characterized hybrid populations between non-sister Xiphophorus species, X. birchmanni and X. cortezi (Fig. 2). This species pair is more distantly related than the well-studied sister species X. birchmanni and X. malinche (Fig. 1A), and opens a new array of questions within an emerging model system for studying the consequences of hybridization.
Given that the X. birchmanni and X. cortezi hybrid populations at Santa Cruz and Huextetitla are geographically separated, the similarities in overall ancestry structure and inferred demographic parameters between the populations are striking. Local ancestry patterns are also markedly concordant across the two hybrid populations (Fig. S11). This could indicate past connections between Santa Cruz and Huextetitla, but the two populations appear to have had different genetic contributions from the X. cortezi source population (Fig. S10), suggesting that they are largely independent in their recent demographic history. Disentangling these factors to determine the degree to which ancestry concordance across populations is driven by shared sources of selection versus shared demographic history will be an exciting direction for future work.
Notably, like the X. birchmanni × X. malinche system, our demographic inference in Huextetitla and Santa Cruz suggests that both of these hybrid populations formed recently (in the last ~150 generations; Fig. 3A), providing a window into evolution in the first tens of generations after hybridization. Similar timing of initial admixture in the two populations could reflect shared histories of disturbance due to their geographical proximity to growing human populations. Indeed, disturbance appears to be a key factor in the recent formation of X. birchmanni × X. malinche hybrid zones (Fisher et al., 2006).
The existence of distinct ancestry clusters in both X. birchmanni × X. cortezi populations suggests substantial reproductive barriers. While the simplest explanation for this pattern would be some form of spatial isolation by ancestry, several observations argue against this hypothesis. Namely, we collected reproductively active males and females of both ancestry clusters in the same minnow traps over multiple collections, and we identify males of both clusters in underwater videos capturing small geographic areas. Likewise, successful embryonic development in lab crosses rules out the possibility of sperm-egg incompatibilities or hybrid incompatibilities that arrest early stage embryonic development. Instead, the sequencing of wild-caught mothers and their offspring from the Santa Cruz hybrid populations provides clear evidence for nearly complete assortative mating by ancestry cluster. We thus conclude that assortative mating is an important force contributing to the observed ancestry structure in the Santa Cruz hybrid population. It is interesting to note, then, that there is substantial introgression of X. birchmanni ancestry into the X. cortezi ancestry cluster at a QTL underlying the sword ornament, suggesting that this trait is not a strong barrier to gene flow in these populations (Powell et al. 2021). While this may seem surprising at first, several traits are important for mate preferences in swordtails and existing data indicates that olfactory preferences may play an especially large role in assortative mating (Crapon de Caprona & Ryan, 1990; Fisher, Mascuch, & Rosenthal, 2009; Fisher et al., 2006; McLennan & Ryan, 1997, 1999).
Although we find evidence of strong ancestry assortative mating in both the X. birchmanni and hybrid X. cortezi ancestry clusters, the results of ABC simulations are consistent with low levels of ongoing gene flow between clusters (Fig. 3B). Intriguingly, our data show that all individuals originating from cross-cluster mating events had mothers from the cortezi ancestry cluster. This hints that mating barriers may be weaker between X. cortezi females and X. birchmanni males than in the alternative direction, consistent with higher levels of X. birchmanni ancestry in the cortezi ancestry cluster (Fig. 2A).
Perhaps the most striking finding of this study is the similarity of ancestry structure across diverse hybrid zones. The bimodal population structure we observe is repeated not only between the Santa Cruz and Huextetitla populations of pure X. birchmanni and X. birchmanni × X. cortezi hybrids but also echoes our previous findings in a X. birchmanni × X. malinche hybrid population on the Río Calnali (“Aguazarca”). This population also exhibits bimodal structure, with a cluster of birchmanni-skewed hybrids deriving ~75% of their genome from X. birchmanni and introgressed X. malinche individuals deriving ~5% of their genome from X. birchmanni (Culumber, Ochoa, & Rosenthal, 2014; Schumer et al., 2017). Consistent with our findings here, ancestry assortative mating helps maintain isolation between ancestry clusters in this X. birchmanni × X. malinche hybrid population (Culumber et al., 2014; Schumer et al., 2017).
Together, these results support an important role of premating mechanisms of assortative mating in maintaining population structure after hybridization. Although many hybridizing populations show evidence of bimodal population structure (Jiggins & Mallet, 2000), similar to the structure we describe in X. birchmanni × X. cortezi populations, it is rare to be able to distinguish between the many possible causes of such structure. Our findings highlight the need to better understand mate preferences in hybrids, which is often made difficult by the context dependence of mate choice (Callander, Backwell, & Jennions, 2012; Rongfeng Cui et al., 2017; Filice & Long, 2017; Jennions & Petrie, 1997; Willis, Rosenthal, & Ryan, 2012) and the fact that hybrids can have distinct preferences from parental species (Mavarez et al., 2006; Melo, Salazar, Jiggins, & Linares, 2009), or suffer from conflicting sensory signals that make them less choosy (Rosenthal, 2013). Another important challenge is understanding how mechanisms of assortative mating in hybrid populations interact with or are driven by selection on hybrids (Irwin, 2020), which is likely to be an important factor in X. birchmanni × X. cortezi hybrids. We predict that hybrid incompatibilities likely exist between X. cortezi × X. birchmanni given findings in related species (Schumer et al., 2014; Powell et al., 2020; Schumer et al., 2018), and strong selection against hybrids could be a major factor driving assortative mating.
Overall similarities in hybrid population structure between X. birchmanni × X. cortezi hybrid populations at Huextetitla and Santa Cruz and X. birchmanni × X. malinche hybrid populations at Aguazarca may belie important biological differences. For example, in the Huextetitla and Santa Cruz hybrid populations, the X. birchmanni subpopulation experiences less gene flow, whereas the opposite is true in the Aguazarca population. Among many possible explanations for such a pattern, differences in responses to pairs of sensory cues between species could play an important role (Crapon de Caprona & Ryan, 1990; Cui et al., 2017; McLennan & Ryan, 1999), suggesting another rich area for future study.
The web of hybridization between X. cortezi, X. birchmanni, and X. malinche provides a novel opportunity to investigate the consequences of hybridization across scales. Greater sample sizes across both X. birchmanni × X. cortezi populations will allow us to test shared drivers of local ancestry across systems (Schumer et al., 2018), identify hybrid incompatibilities, and ask whether observed patterns are a function of phylogenetic history or other biological variables. Such comparative approaches, made possible by the work described here, will ultimately allow us to evaluate the degree to which outcomes of hybridization are predictable across independent hybridization events.
Supplementary Material
Acknowledgements
We thank Gil Rosenthal, Andrea Sweigart, Vaclav Alexei Sotola, Matthew Farnitano, Kira Delmore, Yaniv Brandvain, members of the Schumer lab, and two anonymous reviewers for helpful discussion and/or feedback on earlier versions of this work. We also thank Baruc Zago-Mazzocco for field work support. We are grateful to the Mexican federal government for permission to collect samples. We thank Stanford University and the Stanford Research Computing Center for providing computational support for this project. This work was supported by NSF GRFP 2019273798 to B. Moran, NRSA F32 GM135998 to B. Kim, a Cornell University Provost Diversity Fellowship to S. M. Aguillon, a CEHG fellowship and NSF PRFB (2010950) to Q. Langdon, and a Hanna H. Gray fellowship and NIH 1R35GM133774 to M. Schumer.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Data availability
Code generated from this project is available on github (https://github.com/Schumerlab/Xbir_xcor_hybridzone). All newly collected DNA sequence data generated for this project are available through the NCBI sequence read archive (PRJNA746607). Genome assembly files, video and morphometric data are available on Dryad (https://doi.org/10.5061/dryad.pzgmsbcmn).
References
- Amores A, Catchen J, Nanda I, Warren W, Walter R, Schartl M, & Postlethwait JH (2014). A RAD-Tag Genetic Map for the Platyfish (Xiphophorus maculatus) Reveals Mechanisms of Karyotype Evolution Among Teleost Fish. Genetics, 197(2), 625–641. doi: 10.1534/genetics.114.164293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong J, Hickey G, Diekhans M, Deran A, Fang Q, Xie D, … Paten B (2019). Progressive alignment with Cactus: A multiple-genome aligner for the thousand-genome era. BioRxiv, 730531. doi: 10.1101/730531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH, & Hewitt GM (1985). Analysis of Hybrid Zones. Annual Review of Ecology and Systematics, 16(1), 113–148. [Google Scholar]
- Brandvain Y, Kenney AM, Flagel L, Coop G, & Sweigart AL (2014). Speciation and Introgression between Mimulus nasutus and Mimulus guttatus. PLOS Genetics, 10(6), e1004410. doi: 10.1371/journal.pgen.1004410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfee E, Agra MN, Palacio MA, Ramírez SR, & Coop G (2020). Selection and hybridization shaped the rapid spread of African honey bee ancestry in the Americas. PLOS Genetics, 16(10), e1009038. doi: 10.1371/journal.pgen.1009038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callander S, Backwell PRY, & Jennions MD (2012). Context-dependent male mate choice: The effects of competitor presence and competitor size. Behavioral Ecology, 23(2), 355–360. doi: 10.1093/beheco/arr192 [DOI] [Google Scholar]
- Coyne JA, & Orr HA (1997). “ Patterns of speciation in Drosophila” revisited. Evolution, 51(1), 295–303. [DOI] [PubMed] [Google Scholar]
- Crapon de Caprona MD, & Ryan MJ (1990). Conspecific mate recognition in swordtails, Xiphophorus nigrensis and X. pygmaeus (Poeciliidae): Olfactory and visual cues. Animal Behaviour, 39(2). doi: 10.1016/s0003-3472(05)80873-5 [DOI] [Google Scholar]
- Cui R, Schumer M, Kruesi K, Walter R, Andolfatto P, & Rosenthal G (2013). Phylogenomics reveals extensive reticulate evolution in Xiphophorus fishes. Evolution, 67(8), 2166–2179. [DOI] [PubMed] [Google Scholar]
- Cui Rongfeng, Delclos PJ, Schumer M, & Rosenthal GG (2017). Early social learning triggers neurogenomic expression changes in a swordtail fish. Proceedings. Biological Sciences, 284(1854). doi: 10.1098/rspb.2017.0701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culumber Z, & Rosenthal G (2013). Population-level mating patterns and fluctuating asymmetry in swordtail hybrids. Die Naturwissenschaften, 100. doi: 10.1007/s00114-013-1072-z [DOI] [PubMed] [Google Scholar]
- Culumber ZW, Ochoa OM, & Rosenthal GG (2014). Assortative Mating and the Maintenance of Population Structure in a Natural Hybrid Zone. The American Naturalist, 184(2), 225–232. doi: 10.1086/677033 [DOI] [PubMed] [Google Scholar]
- Dobzhansky T (1936). Studies on hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics, 21(2), 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ayari T, Trigui El Menif N, Hamer B, Cahill AE, & Bierne N (2019). The hidden side of a major marine biogeographic boundary: A wide mosaic hybrid zone at the Atlantic–Mediterranean divide reveals the complex interaction between natural and genetic barriers in mussels. Heredity, 122(6), 770–784. doi: 10.1038/s41437-018-0174-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JP, & Magurran AE (2001). Patterns of sperm precedence and predictors of paternity in the Trinidadian guppy. Proceedings of the Royal Society B: Biological Sciences, 268(1468), 719–724. doi: 10.1098/rspb.2000.1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AA, & Morris MR (2008). Mate choice for more melanin as a mechanism to maintain a functional oncogene. Proceedings of the National Academy of Sciences, 105(36), 13503–13507. doi: 10.1073/pnas.0803851105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filice DCS, & Long TAF (2017). Phenotypic plasticity in female mate choice behavior is mediated by an interaction of direct and indirect genetic effects in Drosophila melanogaster. Ecology and Evolution, 7(10), 3542–3551. doi: 10.1002/ece3.2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher HS, Mascuch SJ, & Rosenthal GG (2009). Multivariate male traits misalign with multivariate female preferences in the swordtail fish, Xiphophorus birchmanni. Anim Behav, 78(2), 265–269. doi: 10.1016/j.anbehav.2009.02.029 [DOI] [Google Scholar]
- Fisher HS, Wong BBM, & Rosenthal GG (2006). Alteration of the chemical environment disrupts communication in a freshwater fish. Proc R Soc London Ser B, 273(1591), 1187–1193. doi: 10.1098/rspb.2005.3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini C, Simmons LW, Beveridge M, & Evans JP (2010). Sperm Swimming Velocity Predicts Competitive Fertilization Success in the Green Swordtail Xiphophorus helleri. Plos One, 5(8). e12146 doi: 10.1371/journal.pone.0012146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller BC, Galloway J, Kelleher J, Messer PW, & Ralph PL (2018). Tree-sequence recording in SLiM opens new horizons for forward-time simulation of whole genomes. BioRxiv, 407783. doi: 10.1101/407783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller BC, & Messer PW (2019). SLiM 3: Forward Genetic Simulations Beyond the Wright–Fisher Model. Molecular Biology and Evolution, 36(3), 632–637. doi: 10.1093/molbev/msy228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RG, & Larson EL (2016). Heterogeneous genome divergence, differential introgression, and the origin and structure of hybrid zones. Molecular Ecology, 25(11), 2454–2466. doi: 10.1111/mec.13582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes JL (1995). Standardized Classification of Poeciliid Development for Life-History Studies. Copeia, 1995(1), 147–154. doi: 10.2307/1446809 [DOI] [Google Scholar]
- Hopkins R, Guerrero RF, Rausher MD, & Kirkpatrick M (2014). Strong Reinforcing Selection in a Texas Wildflower. Current Biology, 24(17), 1995–1999. doi: 10.1016/j.cub.2014.07.027 [DOI] [PubMed] [Google Scholar]
- Irwin DE (2020). Assortative Mating in Hybrid Zones Is Remarkably Ineffective in Promoting Speciation. The American Naturalist, 195(6), E150–E167. doi: 10.1086/708529 [DOI] [PubMed] [Google Scholar]
- Janoušek V, Wang L, Luzynski K, Dufková P, Vyskočilová MM, Nachman MW, … Tucker PK (2012). Genome-wide architecture of reproductive isolation in a naturally occurring hybrid zone between Mus musculus musculus and M. m. Domesticus. Molecular Ecology, 21(12), 3032–3047. doi: 10.1111/j.1365-294X.2012.05583.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennions MD, & Petrie M (1997). Variation in mate choice and mating preferences: A review of causes and consequences. Biological Reviews of the Cambridge Philosophical Society, 72(2), 283–327. doi: 10.1017/s0006323196005014 [DOI] [PubMed] [Google Scholar]
- Jiggins CD, & Mallet J (2000). Bimodal hybrid zones and speciation. Trends in Ecology & Evolution, 15(6), 250–255. doi: 10.1016/s0169-5347(00)01873-5 [DOI] [PubMed] [Google Scholar]
- Kallman KD, & Kazianis S (2006). The genus Xiphophorus in Mexico and central america. Zebrafish, 3(3), 271–285. doi: 10.1089/zeb.2006.3.271 [DOI] [PubMed] [Google Scholar]
- Li H, & Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25(14). doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, & Durbin R (2011). Inference of human population history from individual whole-genome sequences. Nature, 475(7357), 493–496. doi: 10.1038/nature10231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavarez J, Salazar CA, Bermingham E, Salcedo C, Jiggins CD, & Linares M (2006). Speciation by hybridization in Heliconius butterflies. Nature, 441(7095), 868–871. doi: 10.1038/nature04738 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, … DePristo MA (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20(9), 1297–1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan DA, & Ryan MJ (1997). Responses to conspecific and heterospecific olfactory cues in the swordtail Xiphophorus cortezi. Animal Behaviour, 54. doi: 10.1006/anbe.1997.0504 [DOI] [PubMed] [Google Scholar]
- McLennan DA, & Ryan MJ (1999). Interspecific recognition and discrimination based upon olfactory cues in northern swordtails. Evolution, 53(3), 880–888. doi: 10.2307/2640728 [DOI] [PubMed] [Google Scholar]
- Melo MC, Salazar C, Jiggins CD, & Linares M (2009). Assortative Mating Preferences Among Hybrids Offers a Route to Hybrid Speciation. Evolution, 63(6), 1660–1665. doi: 10.1111/j.1558-5646.2009.00633.x [DOI] [PubMed] [Google Scholar]
- Orr HA, & Coyne JA (1989). The genetics of postzygotic isolation in the Drosophila virilis group. 121(3), 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paczolt KA, Passow CN, Delclos PJ, Kindsvater HK, Jones AG, & Rosenthal GG (2015). Multiple Mating and Reproductive Skew in Parental and Introgressed Females of the Live-Bearing Fish Xiphophorus Birchmanni. Journal of Heredity, 106(1), 57–66. doi: 10.1093/jhered/esu066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, … Reich D (2012). Ancient Admixture in Human History. Genetics, 192(3), 1065–1093. doi: 10.1534/genetics.112.145037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DL, García-Olazábal M, Keegan M, Reilly P, Du K, Díaz-Loyo AP, … Schumer M (2020). Natural hybridization reveals incompatible alleles that cause melanoma in swordtail fish. Science, 368(6492), 731–736. doi: 10.1126/science.aba5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DL, Payne C, Banerjee SM, Keegan M, Bashkirova E, Cui R, … Schumer M (2021). The Genetic Architecture of Variation in the Sexually Selected Sword Ornament and Its Evolution in Hybrid Populations. Current Biology. doi: 10.1016/j.cub.2020.12.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail MA, Swerdlow H, & Turner DJ (2009). Improved Protocols for the Illumina Genome Analyzer Sequencing System. Current Protocols in Human Genetics, 62(1), 18.2.1–18.2.27. doi: 10.1002/0471142905.hg1802s62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal GG (2013). Individual mating decisions and hybridization. Journal of Evolutionary Biology, 26(2), 252–255. doi: 10.1111/jeb.12004 [DOI] [PubMed] [Google Scholar]
- Rosenthal Gil G., de la Rosa Reyna XF, Kazianis S, Stephens MJ, Morizot DC, Ryan MJ, & Garcia de Leon FJ (2003). Dissolution of sexual signal complexes in a hybrid zone between the swordtails Xiphophorus birchmanni and Xiphophorus malinche (Poeciliidae). Copeia, 2003(2), 299–307. doi: 10.1643/0045-8511(2003)003[0299:dossci]2.0.co;2 [DOI] [Google Scholar]
- Ross CL, & Harrison RG (2002). A Fine-Scale Spatial Analysis of the Mosaic Hybrid Zone Between Gryllus Firmus and Gryllus Pennsylvanicus. Evolution, 56(11), 2296–2312. doi: 10.1111/j.0014-3820.2002.tb00153.x [DOI] [PubMed] [Google Scholar]
- Schartl M, Kneitz S, Ormanns J, Schmidt C, Anderson JL, Amores A, … Postlethwait JH (2020). The Developmental and Genetic Architecture of the Sexually Selected Male Ornament of Swordtails. Current Biology. doi: 10.1016/j.cub.2020.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartl M, Walter RB, Shen Y, Garcia T, Catchen J, Amores A, … Warren WC (2013). The genome of the platyfish, Xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nature Genetics, 45, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffels S, & Wang K (2020). MSMC and MSMC2: The Multiple Sequentially Markovian Coalescent. In Dutheil JY (Ed.), Statistical Population Genomics (pp. 147–166). New York, NY: Springer US. doi: 10.1007/978-1-0716-0199-0_7 [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, & Eliceiri KW (2012). NIH Image to ImageJ: 25 years of Image Analysis. Nature Methods, 9(7), 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Cui R, Powell DL, Dresner R, Rosenthal GG, & Andolfatto P (2014). High-resolution mapping reveals hundreds of genetic incompatibilities in hybridizing fish species. ELife, 3, e02535. doi: 10.7554/eLife.02535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Powell DL, & Corbett-Detig R (2020). Versatile simulations of admixture and accurate local ancestry inference with mixnmatch and ancestryinfer. Molecular Ecology Resources, 20(4), 1141–1151. doi: 10.1111/1755-0998.13175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Powell DL, Delclós PJ, Squire M, Cui R, Andolfatto P, & Rosenthal GG (2017). Assortative mating and persistent reproductive isolation in hybrids. Proceedings of the National Academy of Sciences, 114(41), 10936. doi: 10.1073/pnas.1711238114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, … Przeworski M (2018). Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science, 360(6389), 656. doi: 10.1126/science.aar3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock EH, Christiansen FB, Hansen TT, Dutheil JY, & Schierup MH (2012). Fusion of two divergent fungal individuals led to the recent emergence of a unique widespread pathogen species. Proceedings of the National Academy of Sciences, 109(27), 10954–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SA, Larson EL, & Harrison RG (2015). Hybrid zones: Windows on climate change. Trends in Ecology & Evolution, 30(7), 398–406. doi: 10.1016/j.tree.2015.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeter KC, Payseur BA, Harris LW, Bakewell MA, Thibodeau LM, O’Brien JE, … Tucker PK (2008). Genome-wide patterns of gene flow across a house mouse hybrid zone. Genome Research, 18(1), 67–76. doi: 10.1101/gr.6757907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre ADL, Ingvarsson PK, & Aitken SN (2015). Genetic architecture and genomic patterns of gene flow between hybridizing species of Picea. Heredity, 115(2), 153–164. doi: 10.1038/hdy.2015.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner LM, & Harr B (2014). Genome-wide mapping in a house mouse hybrid zone reveals hybrid sterility loci and Dobzhansky-Muller interactions. ELife, 3, e02504. doi: 10.7554/eLife.02504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, & Ripley BD (2002). Modern Applied Statistics with S (4th ed.). New York: Springer-Verlag. doi: 10.1007/978-0-387-21706-2 [DOI] [Google Scholar]
- Weisenfeld NI, Kumar V, Shah P, Church DM, & Jaffe DB (2017). Direct determination of diploid genome sequences. Genome Research. doi: 10.1101/gr.214874.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis PM, Rosenthal GG, & Ryan MJ (2012). An indirect cue of predation risk counteracts female preference for conspecifics in a naturally hybridizing fish Xiphophorus birchmanni. PloS One, 7(4), e34802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code generated from this project is available on github (https://github.com/Schumerlab/Xbir_xcor_hybridzone). All newly collected DNA sequence data generated for this project are available through the NCBI sequence read archive (PRJNA746607). Genome assembly files, video and morphometric data are available on Dryad (https://doi.org/10.5061/dryad.pzgmsbcmn).