

In the article “Connecting TNF-α Signaling Pathways to iNOS Expression in a Mouse Model of Alzheimer's Disease: Relevance for the Behavioral and Synaptic Deficits Induced by Amyloid β Protein,” by Rodrigo Medeiros, Rui D. S. Prediger, Giselle F. Passos, Pablo Pandolfo, Filipe S. Duarte, Jeferson L. Franco, Alcir L. Dafre, Gabriella Di Giunta, Cláudia P. Figueiredo, Reinaldo N. Takahashi, Maria M. Campos, and João B. Calixto, which appeared on pages 5394–5404 of the May 16, 2007 issue, panels were inadvertently duplicated in Figure 7, A and B, and Supplementary Figure 1. Two Western blot panels in Figure 7, A and B, that represent cytoplasmic p65 NF-κB levels “p65 (C)” were duplicated and have been removed. The cytoplasmic p65 NF-κB from Figure 7, C and D, were also removed for overall consistency. The study's conclusions are not altered by these changes since the activation p65 NF-κB is demonstrated by its levels in the nuclear (N) compartment. The corrected figures and legends are shown below. The authors regret this oversight.
Figure 7.

Aβ1-40-induced NF-κB pathway activation. A, B, Nuclear [(N)] samples of hippocampus (A) and prefrontal cortex (B) from Swiss mice were probed with a p65 NF-κB antibody at the time points indicated. Data indicate that Aβ1-40 induced a translocation of p65 NF-κB into the nucleus. Pretreatment with the specific antibody against mouse TNF-α (AbTNF-α; 10 ηg, i.c.v., per mouse) prevented Aβ1-40 induction of p65 NF-κB translocation in hippocampus and prefrontal cortex. Immunoblot for lamin A/C was used as a nuclear loading control. Treatment with the reverse peptide Aβ40-1 (6 h) had no significant effect on the p65 NF-κB translocation. As a positive control, LPS (2.5 μg, i.c.v., 1 h) treatment resulted in p65 NF-κB migration into the nucleus. C, D, Aβ1-40-induced p65 NF-κB translocation was prevented by pretreatment with NF-κB inhibitor PDTC (100 mg/kg, i.p.), but not by pretreatment with JNK inhibitor SP600125 (25 mg/kg, i.p.), in hippocampus (C) and prefrontal cortex (D). These data indicate that JNK/c-Jun and NF-κB pathways are independently activated by Aβ1-40 intracerebroventricular injection. N, Naive, untreated mice.
Figure S1.
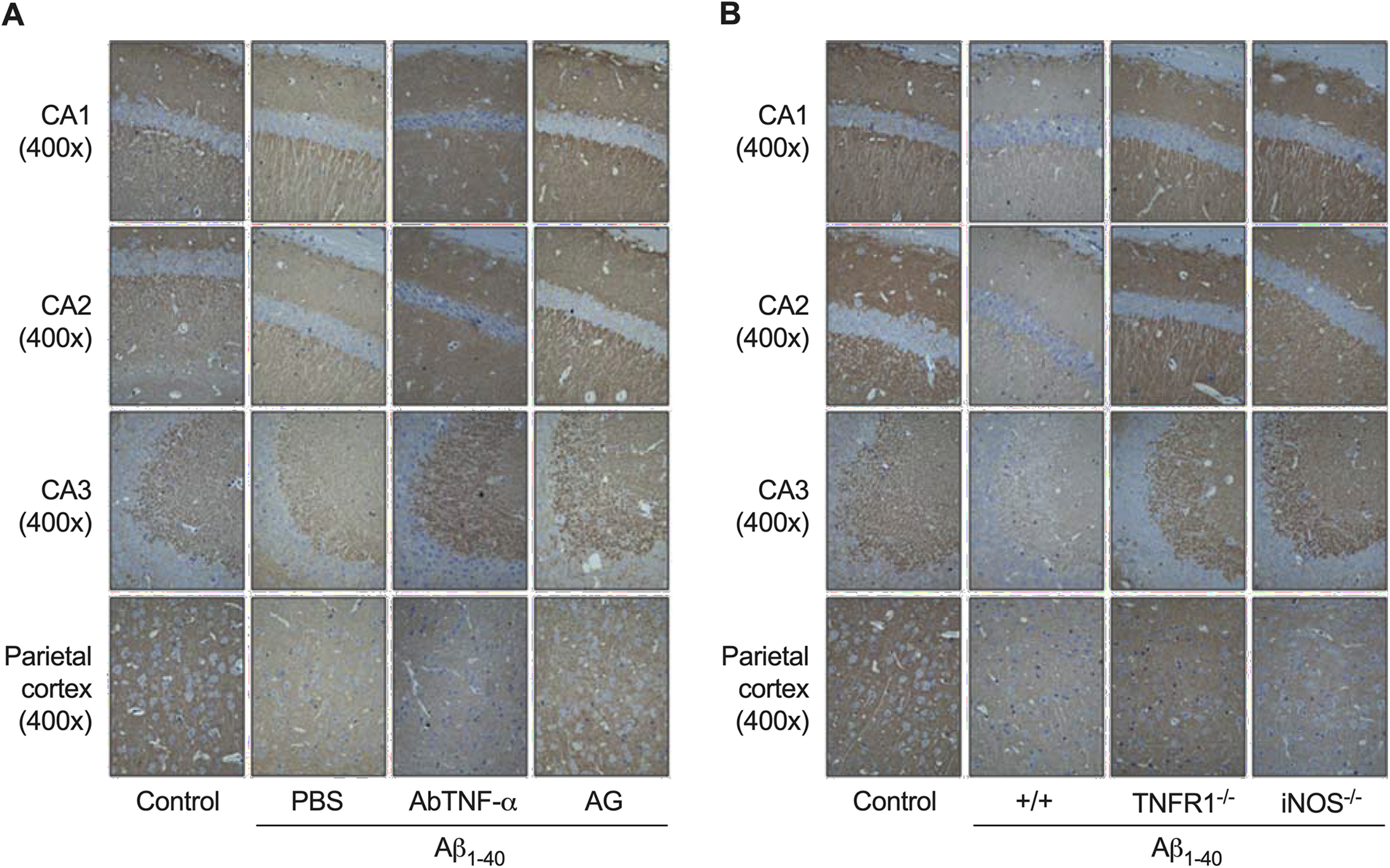
Supplementary Figure 1. Representative figures of synaptophysin immunostaining in the CA1, CA2, and CA3 subregions of the hippocampus, and parietal cortex. Immunohistochemistry analysis for the presynaptic protein synaptophysin was carried out on day 8 after aggregated Aβ1-40 (400 pmol/mouse) intracerebroventricular injection. Synaptophysin immunoreactivity was used as a measure of synaptic density. A, Pretreatment with the specific antibody against mouse TNF-α (AbTNF-α; 10 ng/mouse, i.c.v.) or with the preferential iNOS inhibitor aminoguanidine (AG; 100 mg/kg, i.p., once a day) prevented the Aβ1-40-induced synaptic disruption in Swiss mice. B, TNFR1 knock-out (TNF–/–) and iNOS knock-out (iNOS–/–) mice were significantly more resistant than wild-type C57BL/6 mice to the Aβ1-40-induced synaptic disruption. Magnification: 40× or 400×.