
Table 2.
Summary of the applications of in vitro 2D and 3D cell culture methods for MM research and their main advantages and disadvantages.
| Method | Application to MM research | Advantages | Disadvantages |
|---|---|---|---|
2D cell culture 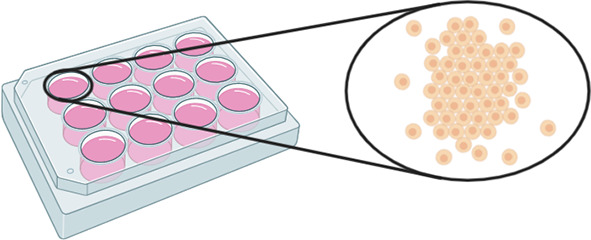
|
*Large scale drug testing *Identification and/or validation of novel biomarkers. *Investigating the role of genes in MM progression. |
*Cost-effective *Easy handling. *Easy to maintain. *High throughput capacity. |
*Drug sensitivity data generated from this method does not always reflect that of the in vivo/clinical counterpart. * Lack of 3D structure; limited cell-cell interactions; unnatural substrate. *Lack of cellular heterogeneity/complexity compared to the original tumour. * Gene expression less similar to in vivo tumours. |
3D cell culture (includes spheroids, TFS and organ-on-a-chip) 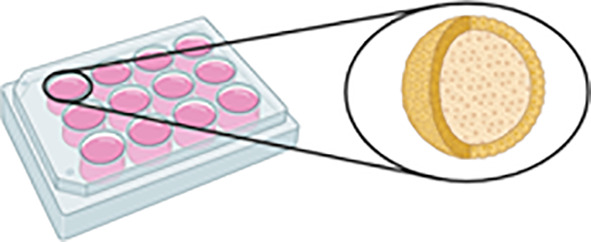
|
*Studying therapeutic efficacy of novel drugs. *Identification and/or validation of novel biomarkers. *Studying cell-to-cell and cell-to-extracellular matrix signaling. |
*More representative of the in vivo tumour structure/complexity. *Gene expression more similar to in vivo tumours. * Drug response better reflects in vivo/clinical drug response. *Increased cell-to-cell and cell-to-extracellular matrix signalling. |
*TFS and organ-on-a-chip require access to fresh surgical MM tumour samples = low throughput capacity. *Complex handling. *Less cost-effective. |
Whole genome sequencing 
|
*Studying all types of MM-specific genetic variation across the entire genome. | *Detects coding, non-coding and structural variants across the entire genome. | *High associated cost. *Large volume of data to process and store. *Numerous variants of unknown significance can be detected. I.e. limited knowledge to fully understand / appreciate the significance of detected unknown variants. |
Transcriptome sequencing 
|
*Studying all types of aberrant MM-specific mRNA / transcript variation. | *Rapid, precise, quantitative measurement of gene expression. *High sensitivity enables detection of low-abundance transcripts. *DNA sequences can be unambiguously mapped to unique regions of the genome instead of relying on existing genome sequence data. *Useful for the discovery of single-nucleotide polymorphisms and rare mutations. *More affordable compared to whole genome sequencing. |
*Transcript quantitation can be affected by biases introduced during cDNA library construction and sequence alignment. *Accurate sequence annotation and data interpretation can be computationally challenging in the absence of pre-existing reference genome(s). |
Targeted sequencing 
|
*Studying unique MM-specific alterations at the sites of specific regions of the genome (i.e. exosomes) or subset of genes. | *Significantly less time-consuming and more cost-effective than whole genome sequencing. *Specific areas of the genome can be sequenced at a greater depth than whole genome sequencing. *Reduced volume of data to process and store than whole genome sequencing. |
*Only focuses on limited regions of the genome, meaning it does not take into account any other genetic variants outside of the focus/target gene panel. |
Droplet digital PCR 
|
*Studying unique MM-specific gene copy number variations, DNA mutations or deletions. *Detection and validation of MM-specific biomarkers. |
*Provides an absolute and independent quantification of DNA without the need for a standard curve. *Generated data is more accurate and reproducible than conventional qPCR. *Capable of detecting very low concentrated target molecules from variably contaminated samples. |
*Equipment and reaction running costs are more expensive than conventional qPCR. *Requires advanced skill and handling compared to conventional qPCR. |