

Abstract
Objective: To explore the molecular mechanisms underlying meniscus degeneration. Methods: We performed anterior cruciate ligament resection in the Hainan Wuzhishan pig to establish a meniscus degeneration model. We applied gene chip technology to detect differentially expressed genes (DEG) in the degenerative meniscus tissues. We applied Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, core gene network, and relevant MicroRNA analyses to identify regulatory networks relevant to meniscus degeneration. We detected 893 differentially expressed genes, mainly involved in hormone production, apoptosis, and inflammation. Results: We found that MUC13, inflammatory mediator regulation of TRP channels, MDFI, and miR-335-5p may play a key role in the degenerative meniscus tissue. Conclusion: We found that meniscus degeneration involves several molecular mechanisms and provide molecular targets for future research into the disease.
Keywords: Differentially expressed genes, gene ontology, Kyoto encyclopedia of genes and genomes, MicroRNA, Wuzhishan pig, molecular targets
Introduction
Meniscal degeneration plays an important role in knee pain triggered by various factors. With the increase of age, the meniscus will degenerate in micro and macro aspects, leading to pain and knee joint dysfunction [1-3]. Meniscus degeneration can lead to osteoarthritis (OA) [4]. Protecting the meniscus and delaying the degeneration of the meniscus are critical to prevent meniscus injury in young and middle-aged patients, and relieves the symptoms of elderly patients with knee arthritis. The molecular mechanism underlying the meniscus degeneration remains largely unclear.
Yubo Sun et al. [5] reported that many genes involved in meniscus degeneration by participating in the biological processes of immune response, inflammatory response, biomineral formation, and cell proliferation. These were expressed at significantly higher levels in OA meniscal cells than in normal meniscal cells. Jun Zhao et al. [6] identified lncRNA and mRNA biomarkers in the degenerative meniscus of patients with OA. The results revealed 208 differentially expressed RNAs, including 32 lncRNAs and 176 mRNAs. These were primarily involved in collagen fibril organization, extracellular matrix organization, endothelial cell migration, Staphylococcus aureus infection, complement and coagulation cascades, and rheumatoid arthritis. Trauma, chronic inflammation and apoptosis may involve unilaterally or multilaterally in the process of meniscus degeneration. Countless genes may play a role in meniscus degeneration [7,8], which makes the research on the mechanism of meniscus degeneration challenging.
The Gene-Cloud of Biotechnology Information (GCBI) laboratory performs gene chip data analysis online: (https://www.gcbi.com.cn/gclab/html/getLab/10025, 2021-1-20) [9]. In GCBI, complex biological information analysis can be completed just by dragging the ‘move’ module and clicking ‘operation’ [10]. Many studies [11-15] have used GCBI to discover new genes and molecular mechanisms in diseases and to understand the underlying molecular mechanisms. Studies on meniscus degeneration by GCBI are scarce. In order to identify more candidate genes involved in meniscal degeneration by GCBI, we compared the expression profiles of degenerative meniscus with those of normal controls.
Materials and methods
Animals
A total of 14 healthy, adult male Hainan Wuzhishan pigs (Institute of Animal Husbandry and Veterinary Medicine, Hainan Academy of Agricultural Sciences, China) were randomly divided into a normal meniscus group and a meniscus degeneration group. Anterior cruciate ligament resection was performed in the meniscus degeneration group. The skin was cut in the normal meniscus group. The weight of the pigs was 15-18 kg, and the age was 6-7 months. Normal light/dark circulation, diet, and group feeding were provided. The environmental temperature was maintained at 20-28°C. All pigs were sacrificed 24 weeks after the operation. All operations were performed under pentobarbital sodium anaesthesia (20 mg/kg BW, Merck Serono KGaA, Darmstadt, Germany). The Ethics Committee of Hainan Provincial People’s Hospital of China approved the experiment (Approval No: Med-Eth-Re [2018] 01). All methods were performed in accordance with the relevant guidelines and regulations of the Ethics Committee.
RNA extraction
The total RNA was extracted from meniscus tissues using E.Z.N. (Omega Bio-Tek Inc., Norcross, GA, USA) after DNase digestion, according to the manufacturer’s instructions. The quantity and quality of the RNA were measured using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Massachusetts, USA). RNA integrity was assessed by standard denatured agarose gel electrophoresis.
Microarray
The Pig Gene Expression 4x44K Microarray V2 (Agilent Technologies, Santa Clara, CA, USA) was used to compare mRNA expression profiles in normal and degenerative meniscus tissue. Microarray analysis was performed using the GCBI analysis platform [9].
Morphological observation and HE staining
The medial meniscus was taken out to observe the smoothness, gloss, and color of the surface. After cleaning, it was fixed with 10% formalin. After 72 hours, it was decalcified, dehydrated, embedded in paraffin, and then sectioned. After HE staining, the morphological and structural changes of the meniscus were observed under a microscope.
Strategy
The flow chart of the analysis is shown in Figure 1. Raw counts were used for testing differentially expressed genes (DEG) using the two-sample Welch’s t-test (unequal variances). DEG was defined as genes with at least two-fold up- or down-regulation and an FDR controlled at 5%. DEG analysis adopted a literature method (SAM, Significance Analysis of Microarrays) to screen genes with significant differences under pre-set groups [16]. The two-sample Welch’s t-test (unequal variances) was performed to identify mRNAs that showed significant (P<0.05) fold change (i.e., >1.5) between groups and presented a false discovery rate (FDR) <0.05. The identified DEGs were then sorted by P-value. The Gene Ontology (GO) system was used to classify the DEGs according to their biological functions. GO system is a database established by the Gene Ontology Consortium. It is a cross-species, comprehensive, and descriptive database. GO analysis annotates the gene function of the different genes between the two groups based on GO system to get all the functions involved by the genes. Fisher’s exact test and multiple comparison test were used to calculate the significance level (P-value) of each function, to screen out the significant functions embodied by the difference genes. P<0.05 was set to distinguish statistically significant enrichment results. Kyoto Encyclopedia of genes and genomes (KEGG) pathway analysis was performed to identify the affected pathways. KEGG is a database for systematic analysis of the relationship between genes (and their coding products), gene function, and genome information. It helps researchers to study genes and expression information as a whole network. Based on KEGG database, Fisher’s exact test was used to analyze the significance of the pathways involved in the target genes, to screen out the significant pathways embodied by the differential genes. P<0.05 was set as the significant pathway. DEGs were analysed by core gene network analysis and related miRNA analysis. The core gene network analysis displays the interaction between the input genes, which makes it easier for researchers to understand the core function of the input genes. The core network was based on the relationship between these genes and genes that have a database and literature basis in PubMed, Mesh, and KEGG databases. Related miRNA analysis showed at least two miRNAs that interact with the input gene. Related miRNAs were based on the relationship between genes and miRNAs that were documented in the PubMed and miRbase, to construct a network of related miRNAs. All data mentioned above were analysed by the GCBI analysis platform.
Figure 1.
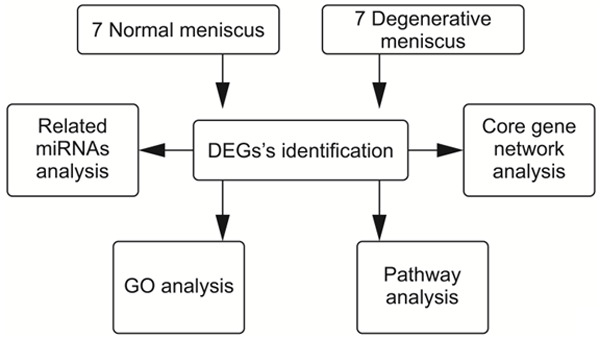
Flow diagram of the study design.
Real-time RT-PCR
Seven genes were selected for validation. The transcriptional level of the selected housekeeping gene, GAPDH, was quantified by the ViiA 7 Real-time PCR System (Applied Biosystems, Foster City, CA, USA). Primers were designed using Primer 5.0 (Premier Biosoft International, Palo Alto, CA, USA), and were based on the cDNA sequences in the NCBI sequence database (https://www.ncbi.nlm.nih.gov/refseq/, 2021-02-10). The main parameters are shown in Table 1. The Gene Amp PCR System 9700 (Applied Biosystems, Foster City, CA, USA) was used to generate the first chain cDNA from the separated RNA. According to the manufacturer’s instructions, cDNA was amplified by performing the initial step of Taq DNA polymerase activation at 95°C for 10 minutes, followed by 40 denaturation cycles at 95°C for 10 seconds, and annealed at 60°C for 60 seconds. Each reaction was repeated three times. The Ct value of each gene was obtained in triplicate. The 2-ΔΔCt method [16] was used to normalize the expression level of the selected gene to that of GAPDH. Each real-time RT-PCR experiment was repeated in triplicate. The data of PT-PCR was analysed using SPSS 19.0 software (IBM, Armonk, NY, USA). Student’s t-tests were used for comparisons between the two groups, and P<0.05 indicated statistical difference.
Table 1.
Primer sequences for GAPDH, CYP2C33, GCNT7, and NCDN
| GAPDH | F: 5’ TCTCTGCTCCTCCCCGTT 3’ |
| R: 5’ CGGCCAAATCCGTTCACT 3’ | |
| CYP2C33 | F: 5’ CATACATCGCCTTACTTCCTTCTAAC 3’ |
| R: 5’ CTGAAGACAGTAGCGGCAATACA 3’ | |
| GCNT7 | F: 5’ GGCTTACACTGGCTTTAGGAG 3’ |
| R: 5’ AGTTTGGCTTGTCTTGGATTTA 3’ | |
| NCDN | F: 5’ AGACCTGCTGTCACATCTTCCTC 3’ |
| R: 5’ AGCGACGCCATCAGAGTGTT 3’ |
Results
Degeneration model in the Hainan Wuzhishan pig
A Wuzhishan pig in Hainan province of China is the smallest, lightest, and most endangered Chinese miniature pig. Its anatomy, physiological characteristics, and disease mechanism are very similar to humans. In our study, Wuzhishan pigs provided the additional advantages of body size, facilitated surgical procedures, and generated sufficient joint tissue for molecular analysis. In the normal meniscus group, the meniscus was smooth and complete, with a white and shiny surface, without any signs of degeneration. In the degenerative meniscus group, the surface of the meniscus was rough, the color was dim, the elasticity was poor, and there were some small defects and erosion (Figure 2). In the normal meniscus group, HE staining of the meniscus showed normal meniscus structure. In the degenerative meniscus group, HE staining of the meniscus showed disorder, uneven staining and sparse arrangement of collagen fibers, reduction and swelling of chondrocytes, reduction or disappearance of cartilage lacunae, increased local fibers and hyaline changes, which were consistent with meniscal degeneration (Figure 3).
Figure 2.
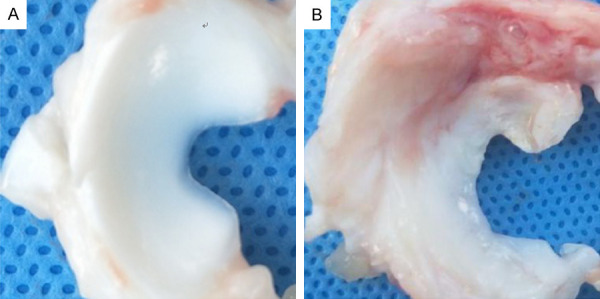
Meniscus in normal group and degeneration group. A: In the normal meniscus group, the meniscus was crescent shaped, with complete and smooth surface, no tear, white color and good elasticity. The medial part of the meniscus was thin, and the lateral part was thick. B: In the degenerative meniscus group, the color of the meniscus was light yellow, the elasticity was worse than that of the normal meniscus, the surrounding synovial membrane was congested with edema, the inner part was thinner with uneven wear, and the free edge was incomplete and cracked.
Figure 3.
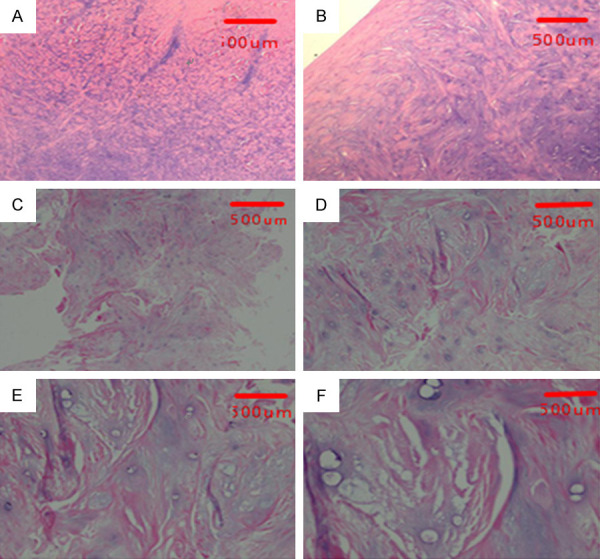
H-E staining of meniscus of normal group and degeneration group. A, B: The HE staining of normal meniscus tissue. The chondrocyte nucleus was large and round, the cell distribution was regular, the collagen fibers were abundant, and the fiber bundles were thick and neat. C-F: The HE staining of degenerative meniscus. The arrangement of collagen fibers was disordered, the staining was uneven, and the arrangement was sparse; the chondrocytes were arranged disorderly and reduced, and the visible cells were swollen, the cartilage lacuna was reduced or disappeared, and the local area was fibrotic and hyalinized. A: ×40, B: ×100, C: ×40, D: ×100, E: ×200, F: ×400.
893 DEGs of degenerative meniscus tissue
There were 893 DEGs in the two groups, including 537 upregulated genes and 356 downregulated genes. The results are shown in the volcano plot and the dendrogram (Figure 4). The top 10 most significant genes were CYP2C33, GCNT7, NCDN, EXD3, MUC13, PPP1R3D, NPHP3, UPB81, CD81, and PRPH (Table 2).
Figure 4.
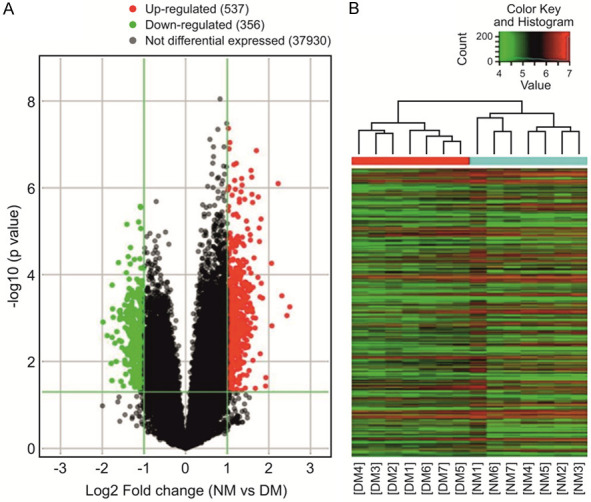
Volcano plot and heatmap of DEGs. A: Volcano plot. Red and green represent differentially expressed genes (DEGs); the downregulated DEGs are shown in green, and the upregulated DEGs are shown in red. B: The dendrogram. The ordinate represents the grouping information of the sample. Red indicates high relative expression and green indicates low relative expression. NM: normal meniscus; DM: degenerative meniscus.
Table 2.
Top 10 most significant genes based on the comparative expression profiles of the degenerative and control menisci in a pig degeneration model
| Probe Name | P-value | FDR | Fold Change | Regulation | Gene Symbol | Rank |
|---|---|---|---|---|---|---|
| A_72_P526917 | 1.2593E-07 | 0.000537464 | 2.0593695 | up | CYP2C33 | 1 |
| A_72_P362758 | 1.38326E-07 | 0.000537464 | 3.2506906 | up | GCNT7 | 2 |
| A_72_P050406 | 6.10533E-07 | 0.001031403 | 2.5418055 | up | NCDN | 3 |
| A_72_P258107 | 8.6355E-07 | 0.001198329 | 2.5599119 | up | EXD3 | 4 |
| A_72_P165116 | 9.46523E-07 | 0.001225904 | 2.046065 | up | MUC13 | 5 |
| A_72_P442485 | 1.08108E-06 | 0.001272895 | 2.2891991 | up | PPP1R3D | 6 |
| A_72_P222892 | 1.13988E-06 | 0.001302652 | 2.0178437 | up | NPHP3 | 7 |
| A_72_P234107 | 1.23101E-06 | 0.001328635 | 2.5152315 | up | UPB1 | 8 |
| A_72_P496803 | 1.4119E-06 | 0.001443671 | 3.0181273 | up | CD81 | 9 |
| A_72_P352838 | 1.81842E-06 | 0.001613149 | 2.4487246 | up | PRPH | 10 |
FDR: false discovery rate.
GO analysis of 55 biological processes
A total of 55 biological processes enriched by the DEGs were obtained. The top 10 biological processes were cell response to hormone stimulus, response to hormones, sex determination, muscle fibre development, mesenchyme morphogenesis, cell response to endogenous stimulus, C21 steroid hormone metallic process, neuropeptide signalling pathway, negative regulation of the reactive oxygen species metallic process, and regulation of the nitric oxide biosynthetic process (Table 3).
Table 3.
The top 10 enriched GO terms for the DEGs by P-value in ascending order
| Term | Count | P-value | Regulation | Rank |
|---|---|---|---|---|
| cellular response to hormone stimulus | 7 | 0.000311929 | Up | 1 |
| response to hormone | 7 | 0.001152029 | Up | 2 |
| sex determination | 2 | 0.001281445 | Down | 3 |
| muscle fibre development | 2 | 0.002753211 | Down | 4 |
| mesenchyme morphogenesis | 2 | 0.002753211 | Down | 5 |
| cellular response to endogenous stimulus | 8 | 0.003566943 | Up | 6 |
| C21-steroid hormone metabolic process | 2 | 0.003588081 | Up | 7 |
| neuropeptide signalling pathway | 3 | 0.003940886 | Up | 8 |
| negative regulation of reactive oxygen species metabolic process | 2 | 0.004359628 | Up | 9 |
| regulation of nitric oxide biosynthetic process | 2 | 0.007086781 | Up | 10 |
Modulation of 36 pathway
A total of 36 pathways were modulated by the alteration of the gene expression. The top 10 pathways are Type II diabetes mellitus, thyroid hormone synthesis, taste transduction, prolactin signalling pathway, longevity regulating pathway, ovarian steroidogenesis, neuroactive ligand-receptor interaction, inflammatory mediator regulation of TRP channels, pantothenate and CoA biosynthesis, and cocaine addiction (Table 4).
Table 4.
Enriched pathways for the differentially expressed genes (DEGs) identified by their comparative expression profiles in the degenerative and control menisci in a pig degeneration model
| Definition | Fisher P-value | Regulation | Rank |
|---|---|---|---|
| Type II diabetes mellitus | 0.000759595 | Up | 1 |
| Thyroid hormone synthesis | 0.002669959 | Down | 2 |
| Taste transduction | 0.002858715 | Up | 3 |
| Prolactin signalling pathway | 0.003352503 | Up | 4 |
| Longevity regulating pathway | 0.007875137 | Up | 5 |
| Ovarian steroidogenesis | 0.009215694 | Up | 6 |
| Neuroactive ligand-receptor interaction | 0.01021098 | Up | 7 |
| Inflammatory mediator regulation of TRP channels | 0.01098087 | Up | 8 |
| Pantothenate and CoA biosynthesis | 0.01205545 | Up | 9 |
| Cocaine addiction | 0.01468413 | Down | 10 |
Core gene network and 101 relevant miRNA analysis
Core network analysis yielded 40 core genes. MDFI had the largest number of connections, indicating that it was in the most core gene (Figure 5). The corresponding genes and data sources of MDFI are detailed in Table 5. Relevant miRNA analysis yielded 101 related miRNAs; of these, miR-335-5p was the most connected related miRNA (Figure 6). The correspondence and data source of miR-335-5p are detailed in Table 6.
Figure 5.
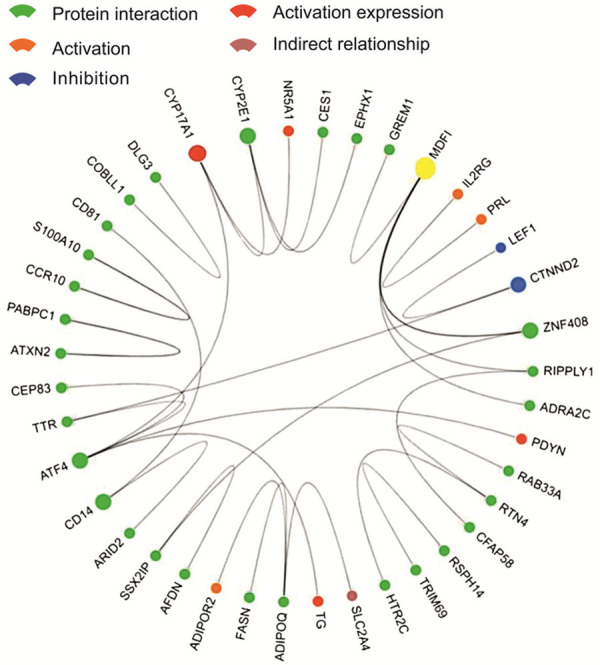
Interaction between core genes. Dots indicate core genes; lines between dots indicate relationships, and the colors of dots indicate specific relationships (see the icon in the upper left corner); larger dots indicate more relationships; yellow dots indicate genes with the most relationships (MDFI).
Table 5.
The Correspondence and data source of MDFI
| Gene 1 | Protein 1 | Gene 2 | Protein 2 | Relationship | Experiment | Source |
|---|---|---|---|---|---|---|
| MDFI | Q99750 | GREM1 | O60565 | protein interaction | two hybrid array | PMID: unassigned 1304 |
| MDFI | Q99750 | ZNF408 | Q9H9D4 | protein interaction | two hybrid array | PMID: 19060904 |
| MDFI | Q99750 | ZNF408 | Q9H9D4 | protein interaction | two hybrid array | PMID: unassigned 1304 |
| MDFI | Q99750 | ZNF408 | Q9H9D4 | protein interaction | two hybrid pooling approach | PMID: 16189514 |
| MDFI | Q99750 | RIPPLY1 | Q0D2K3 | protein interaction | two hybrid array | PMID: unassigned 1304 |
| MDFI | Q99750 | ADRA2C | P18825 | protein interaction | two hybrid array | PMID: unassigned 1304 |
Figure 6.
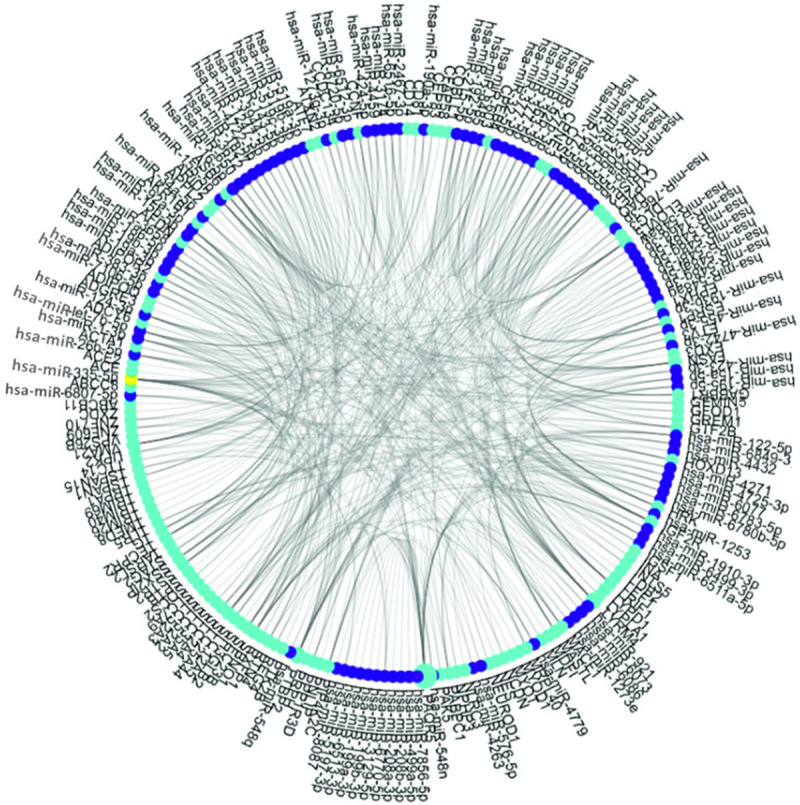
The relationship network between DEGs and miRNAs. Royal blue dots indicate DEGs; purple dots indicate miRNAs; larger dots indicate more relationships; yellow dots indicate miRNAs with the most relationships (miR-335-5p). DEGs, differentially expressed genes. miRNAs, microRNA.
Table 6.
The Correspondence and data source of miR-335-5p
| MiRNA name | Target gene name | Experimental verification method | PMID |
|---|---|---|---|
| miR-335-5p | ABCC8 | Microarray | 18185580 |
| miR-335-5p | ACE | Microarray | 18185580 |
| miR-335-5p | ADCY6 | Microarray | 18185580 |
| miR-335-5p | ADGRE3 | Microarray | 18185580 |
| miR-335-5p | AFDN | Microarray | 18185580 |
| miR-335-5p | CD14 | Microarray | 18185580 |
| miR-335-5p | CFAP58 | Microarray | 18185580 |
| miR-335-5p | CPEB4 | Microarray | 18185580 |
| miR-335-5p | CYP2E1 | Microarray | 18185580 |
| miR-335-5p | DLG3 | Microarray | 18185580 |
| miR-335-5p | ESRP1 | Microarray | 18185580 |
| miR-335-5p | EXD3 | Microarray | 18185580 |
| miR-335-5p | GABBR1 | Microarray | 18185580 |
| miR-335-5p | MACF1 | Microarray | 18185580 |
| miR-335-5p | MDFI | Microarray | 18185580 |
| miR-335-5p | MLN | Microarray | 18185580 |
| miR-335-5p | MYOD1 | Microarray | 18185580 |
| miR-335-5p | MYOT | Microarray | 18185580 |
| miR-335-5p | NCDN | HITS-CLIP | 27418678 |
| miR-335-5p | PNLIP | Microarray | 18185580 |
| miR-335-5p | PPP1R3D | Microarray | 18185580 |
| miR-335-5p | PTK2B | Microarray | 18185580 |
| miR-335-5p | SLC2A4 | Microarray | 18185580 |
| miR-335-5p | SLC35A5 | HITS-CLIP | 23313552 |
| miR-335-5p | SLC7A9 | Microarray | 18185580 |
| miR-335-5p | SPTB | Microarray | 18185580 |
| miR-335-5p | TRIM40 | Microarray | 18185580 |
| miR-335-5p | TRIM69 | Microarray | 18185580 |
| miR-335-5p | TSPAN15 | Microarray | 18185580 |
| miR-335-5p | ZNF609 | Microarray | 18185580 |
Strong agreement between the microarray and RT-PCR results
To verify the reliability of the microarray results, the expression levels of the 10 genes (CYP2C33, GCNT7, NCDN, EXD3, MUC13, PPP1R3D, NPHP3, UPB81, CD81, and PRPH) were detected by RT-PCR and the results were consistent with the microarray data. The 10 genes were chosen because they showed maximum significance in the microarray results. The differential mRNA expression of GCNT7, NCDN, EXD3, MUC13, PPP1R3D, NPHP3, and CD81 between the degenerative meniscus group and the normal meniscus group was statistically significant (P<0.05) (Figure 7). The results proved the strong agreement between the microarray and RT-PCR results.
Figure 7.
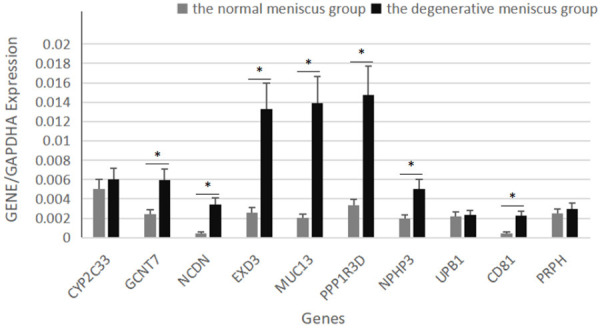
Verification of 10 selected genes by real-time RT-PCR for their comparative expression profiles in the degenerative and control menisci in a pig degeneration model. Error bars indicate the mean ± standard errors of the mean (*P<0.05).
Discussion
In this study, an Agilent-026440 Sus scrofa (Pig) Oligo Microarray v2 chip was used to detect DEGs in the degenerative meniscus tissue. There were 893 DEGs between the meniscus degeneration group and the normal group, of which 537 genes were upregulated, and 356 genes were downregulated. We found 10 genes with the most significant expression. They were CYP2C33, GCNT7, NCDN, EXD3, MUC13, PPP1R3D, NPHP3, UPB1, CD81, and PRPH. The transmembrane mucin encoded by MUC13 has a variety of physiological functions, mainly to lubricate and protect the mucosal epithelium. Under pathological conditions, MUC13 expression is abnormal and participates in the occurrence and development of inflammation and tumours [18,19]. Our study found that MUC13 was overexpressed in degenerative meniscus tissues, suggesting that it may play an important role in this process, presumably by exerting anti-inflammatory effects. IL-8 is an important inflammatory factor. MUC13 promotes NF-кB activity through NF-κB-dependent pathways and increases IL-8 production. OA synovial fluid contains high levels of IL-6 and IL-8 [20]. The increased expression of IL-8 has a chemotactic effect on inflammatory cells and stimulates the secretion of IL-6, promoting joint inflammation [20]. MUC13 may play a role in degenerative meniscus tissue through its anti-inflammatory effect.
According to the GO analysis, 55 biological processes were significantly expressed. After comprehensive analysis, the top 10 biological processes were cell response to hormone stimulus, response to hormones, sex determination, muscle fibre development, mesenchyme morphogenesis, cell response to endogenous stimulus, C21 steroid hormone metallic process, neuropeptide signalling pathway, negative regulation of reactive oxygen species, metallic process, and regulation of nitric oxide biosynthetic process. Studies [21-23] have reported that the inhibitory effect of sex hormones seems to be related to cystic degeneration of meniscus tissue. Growth hormones and parathyroid hormones have effects on the meniscus and chondrocytes. NO is the main cause of human articular chondrocyte apoptosis [24-27]. These studies are consistent with our findings. The regulatory biosynthesis of nitric oxide is one of the most enriched biological processes in GO analysis. Studies have shown that NO induces chondrocyte apoptosis. NO is the main cause of human articular chondrocyte apoptosis [24,28-31]. Our study shows that No is involved in the biological process of meniscus degeneration, indicating that NO induced apoptosis of meniscus cells also occurs, which could eventually lead to the degeneration of meniscus tissue. The consistency between our findings and previous studies strongly supports our hypothesis that degenerated meniscus cells are different from normal meniscus cells.
Inflammatory mediator regulation of TRP channels was one of the 10 lowest P-values in the pathway analysis. With a variety of cellular signal receptors, it plays a very important role in the inflammatory response [32,33]. In this study, we found that the inflammatory mediators of TRP channel were up-regulated in the degenerative meniscus, indicating that the degeneration of meniscus is related to TRP channel and inflammatory response.
MDFI and miR-335-5p identified in this study, both regulate the WNT signalling pathway [34,35], which is a core signal pathway of OA synovitis [36,37]. The result showed that synovitis and meniscal degeneration may have similar mechanisms of action. Synovial and meniscal tissues are also involved in the pathogenesis of OA. Tornero-Esteban’s [38] study showed a correlation between miR-335-5p expression and OA. The possible mechanism is that miRNA-335-5p alleviates inflammation in OA chondrocytes through activation of autophagy [39]. Our study found a strong correlation between miR-335-5p and meniscus degeneration. MiR-335-5p may play an important role in meniscus degeneration, indicating that miR-335-5p may become a new target for clinical prevention and treatment of meniscus degeneration. The specific mechanism of mediating meniscus degeneration will be the focus of follow-up research.
There are some shortcomings in this study. The ‘knee’ joint in pigs is different from humans in biomechanics. The animal model research cannot be completely equivalent to the study of meniscus degeneration in human patients. This model is similar to traumatic meniscus degeneration, such as human meniscus and cruciate ligament injury, rather than primary meniscus degeneration. Considering the various challenges (such as the ethical issues) involved with the study of human specimens, the difficulty of obtaining a sufficient sample size, the obvious heterogeneity of specimens, and the difficulty of obtaining specimens from special body parts, this animal model is an ideal research tool.
In summary, our study presents a comprehensive bioinformatics analysis of meniscus degeneration. The results may help us to improve our understanding of the molecular mechanisms underlying meniscus degeneration. MDFI and miR-335-5p may represent targets for the diagnosis and treatment of meniscus degeneration.
Acknowledgements
This work was supported by Key R&D plan of Hainan Province, China (grant number ZDYF2019180), and Scientific Research Project of the Health Industry in Hainan Province, China (grant number 20A200488).
Disclosure of conflict of interest
None.
References
- 1.Howell R, Kumar NS, Patel N, Tom J. Degenerative meniscus: pathogenesis, diagnosis, and treatment options. World J Orthop. 2014;5:597–602. doi: 10.5312/wjo.v5.i5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuan X, Arkonac DE, Chao PG, Vunjak-Novakovic G. Electrical stimulation enhances cell migration and integrative repair in the meniscus. Sci Rep. 2014;4:3674. doi: 10.1038/srep03674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goetz JE, Coleman MC, Fredericks DC, Petersen E, Martin JA, McKinley TO, Tochigi Y. Time-dependent loss of mitochondrial function precedes progressive histologic cartilage degeneration in a rabbit meniscal destabilization model. J Orthop Res. 2017;35:590–599. doi: 10.1002/jor.23327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.López-Franco M, López-Franco O, Murciano-Antón MA, Cañamero-Vaquero M, Fernández-Aceñero MJ, Herrero-Beaumont G, Gómez-Barrena E. Meniscal degeneration in human knee osteoarthritis: in situ hybridization and immunohistochemistry study. Arch Orthop Trauma Surg. 2016;136:175–183. doi: 10.1007/s00402-015-2378-4. [DOI] [PubMed] [Google Scholar]
- 5.Sun Y, Mauerhan DR, Honeycutt PR, Kneisl JS, Norton JH, Hanley EN Jr, Gruber HE. Analysis of meniscal degeneration and meniscal gene expression. BMC Musculoskelet Disord. 2010;11:19. doi: 10.1186/1471-2474-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Y, Mauerhan DR. Meniscal calcification, pathogenesis and implications. Curr Opin Rheumatol. 2012;24:152–157. doi: 10.1097/BOR.0b013e32834e90c1. [DOI] [PubMed] [Google Scholar]
- 7.Aigner T, Fundelm K, Saas J, Gebhard PM, Haag J, Weiss T, Zien A, Obermayr F, Zimmer R, Bartnik E. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006;54:3533–44. doi: 10.1002/art.22174. [DOI] [PubMed] [Google Scholar]
- 8.Brophy RH, Sandell LJ, Cheverud JM, Rai MF. Gene expression in human meniscal tears has limited association with early degenerative changes in knee articular cartilage. Connect Tissue Res. 2016;58:295–304. doi: 10.1080/03008207.2016.1211114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Gene-Cloud of Biotechnology Information (GCBI) https://www.gcbi.com.cn/gclab/html/getLab/10025, 2021-1-20.
- 10.Huang H, Zheng J, Shen N, Wang G, Zhou G, Fang Y, Lin J, Zhao J. Identification of pathways and genes associated with synovitis in osteoarthritis using analyses. Sci Rep. 2018;8:10050. doi: 10.1038/s41598-018-28280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng A, Tu Z, Yin B. The effect of HMGB1 on the clinicopathological and prognostic features of non-small cell lung cancer. Oncotarget. 2016;7:20507–20519. doi: 10.18632/oncotarget.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu Y, Qin B, Hu H, Zhang J, Wang Y, Wang Q, Wang S. Integrative microRNA-gene expression network analysis in genetic hypercalciuric stone-forming rat kidney. PeerJ. 2016;4:e1884. doi: 10.7717/peerj.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Xu S, Jin P, Yang X, Li X, Wan D, Zhang T, Long S, Wei X, Chen G, Meng L, Liu D, Fang Y, Chen P, Ma D, Gao Q. MARCKS contributes to stromal cancer-associated fibroblast activation and facilitates ovarian cancer metastasis. Oncotarget. 2016;7:37649–37663. doi: 10.18632/oncotarget.8726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao JR, Qin XJ, Jiang H, Wang T, Song JM, Xu SZ. Screening and functional analysis of differentially expressed genes in chronic glomerulonephritis by whole genome microarray. Gene. 2016;589:72–80. doi: 10.1016/j.gene.2016.05.031. [DOI] [PubMed] [Google Scholar]
- 15.Ling Q, Xie H, Li J, Liu J, Cao J, Yang F, Wang C, Hu Q, Xu X, Zheng S. Donor graft MicroRNAs: a newly identified player in the development of new-onset diabetes after liver transplantation. Am J Transplant. 2017;17:255–264. doi: 10.1111/ajt.13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takagaki N, Ohta A, Ohnishi K, Kawanabe A, Minakuchi Y, Toyoda A, Fujiwara Y, Kuhara A. Identification of differentially expressed gene transcripts in porcine endometrium during early stages of pregnancy. EMBO Rep. 2020;21:e48671. doi: 10.15252/embr.201948671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reis M, Vieira P, Morales C, Ramiro H. Fold change in regucalcin expression after chill-coma recovery (ChCR) obtained by qRT-PCR using the 2-ΔΔCt method. PLoS One. 2011;6:e25520. [Google Scholar]
- 18.Gupta B, Sikander M, Jaggi M. Overview of mucin (MUC13) in gastrointestinal cancers. J Gastroenterol Dis. 2016;1:18–19. [Google Scholar]
- 19.Sheng YH, Triyana S, Wang R, Das I, Gerloff K, Florin TH, Sutton P, McGuckin MA. MUC1 and MUC13 differentially regulate epithelial inflammation in response to inflammatory and infectious stimuli. Mucosal Immunol. 2013;6:557–568. doi: 10.1038/mi.2012.98. [DOI] [PubMed] [Google Scholar]
- 20.Zhang B, Ren J, Yan X, Huang X, Ji H, Peng Q, Zhang Z, Huang L. Investigation of the porcine MUC13 gene: isolation, expression, polymorphisms and strong association with susceptibility to enterotoxigenic Escherichia coli F4ab/ac. Anim Genet. 2008;39:258–266. doi: 10.1111/j.1365-2052.2008.01721.x. [DOI] [PubMed] [Google Scholar]
- 21.Rokkanen P, Paatsama S, Rissanen P. The influence of certain hormones on the meniscus of the femoro-tibial (stifle) joint: a histological and histo-quantitative study on young dogs. J Small Anim Pract. 1967;8:221–227. doi: 10.1111/j.1748-5827.1967.tb04546.x. [DOI] [PubMed] [Google Scholar]
- 22.Noone TJ, Millis DL, Korvick DL, Athanasiou K, Cook JL, Kuroki K, Buonomo F. Influence of canine recombinant somatotropin hormone on biomechanical and biochemical properties of the medial meniscus in stifles with altered stability. Am J Vet Res. 2002;63:419–426. doi: 10.2460/ajvr.2002.63.419. [DOI] [PubMed] [Google Scholar]
- 23.Huang H, Skelly JD, Ayers DC, Song J. Age-dependent changes in the articular cartilage and subchondral bone of C57BL/6 mice after surgical destabilization of medial meniscus. Sci Rep. 2017;7:42294. doi: 10.1038/srep42294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanco FJ, Ochs RL, Schwarz H, Lotz M. Chondrocyte apoptosis induced by nitric oxide. Am J Pathol. 1995;146:75–85. [PMC free article] [PubMed] [Google Scholar]
- 25.Shen P, Nguyen M, Fuchs M, Reisener MJ, Löhning M. TLR1/2 signaling impairs mitochondrial oxidative phosphorylation in human chondrocytes via the induction of nitric oxide. Osteoarthritis Cartilage. 2020;28:S119. [Google Scholar]
- 26.Kobayashi K, Mishima H, Hashimoto S, Goomer RS, Amiel D. Chondrocyte apoptosis and regional differential expression of nitric oxide in the medial meniscus following partial meniscectomy. J Orthop Res. 2010;19:802–808. doi: 10.1016/S0736-0266(01)00023-7. [DOI] [PubMed] [Google Scholar]
- 27.Kao XB, Chen Q, Gao Y, Fan P, Chen JH, Wang ZL, Wang YQ, Chen YN, Yan YP. SP600125 blocks the proteolysis of cytoskeletal proteins in apoptosis induced by gas signaling molecule (NO) via decreasing the activation of caspase-3 in rabbit chondrocytes. Eur J Pharmacol. 2018;824:40–47. doi: 10.1016/j.ejphar.2018.01.032. [DOI] [PubMed] [Google Scholar]
- 28.Kim S, Hwang S, Shin DY, Kang S, Chun J. p38 kinase regulates nitric oxide-induced apoptosis of articular chondrocytes by accumulating p53 via NFκB-dependent transcription and stabilization by serine 15 phosphorylation. J Biol Chem. 2002;277:33501–33508. doi: 10.1074/jbc.M202862200. [DOI] [PubMed] [Google Scholar]
- 29.Relić B, Bentires-Alj M, Ribbens C, Franchimont N, Guerne PA, Benoît V, Merville MP, Bours V, Malaise MG. TNF-α protects human primary articular chondrocytes from nitric oxide-induced apoptosis via nuclear factor-κB. Lab Invest. 2002;82:1661–1672. doi: 10.1097/01.lab.0000041714.05322.c0. [DOI] [PubMed] [Google Scholar]
- 30.Surendran S, Kim SH, Jee BK, Ahn SH, Goinathan P, Han CW. Anti-apoptotic Bcl-2 gene transfection of human articular chondrocytes protects against nitric oxide-induced apoptosis. J Bone Joint Surg Br. 2006;88:1660–1665. doi: 10.1302/0301-620X.88B12.17717. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y, Ming J, Li Y, Deng M, Chen Q, Ma Y, Chen Z, Zhang Y, Liu S. Ligustilide attenuates nitric oxide-induced apoptosis in rat chondrocytes and cartilage degradation via inhibiting JNK and p38 MAPK pathways. J Cell Mol Med. 2019;2:3357–3368. doi: 10.1111/jcmm.14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramirez GA, Coletto LA, Sciorati C, Bozzolo EP, Manunta P, Rovere-Querini P, Manfredi AA. Ion channels and transporters in inflammation: special focus on TRP channels and TRPC6. Cells. 2018;7:70. doi: 10.3390/cells7070070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oehler B, Kistner K, Martin C, Schiller J, Mayer R, Mohammadi M, Sauer RS, Filipovic MR, Nieto FR, Kloka J, Pflücke D, Hill K, Schaefer M, Malcangio M, Reeh PW, Brack A, Blum R, Rittner HL. Inflammatory pain control by blocking oxidized phospholipid-mediated TRP channel activation. Sci Rep. 2017;7:5447. doi: 10.1038/s41598-017-05348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Tu Q, Bonewald LF, He X, Stein G, Lian J, Chen J. Effects of miR-335-5p in modulating osteogenic differentiation by specifically downregulating Wnt antagonist DKK1. J Bone Miner Res. 2011;26:1953–1963. doi: 10.1002/jbmr.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Q, Young TM, Mathews MB, Pe’ery T. Developmental regulators containing the I-mfa domain interact with T cyclins and Tat and modulate transcription. J Mol Biol. 2007;367:630–646. doi: 10.1016/j.jmb.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van den Bosch MH, Blom AB, Sloetjes AW, Koenders MI, van de Loo FA, van den Berg WB, van Lent PL, ven der Kraan PM. Induction of canonical Wnt signaling by synovial overexpression of selected Wnts leads to protease activity and early osteoarthritis-like cartilage damage. Am J Pathol. 2015;185:1970–1980. doi: 10.1016/j.ajpath.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 37.Kitanaka T, Nakano R, Kitanaka N, Kimura T, Okabayashi K, Narita T, Sugiya H. JNK activation is essential for activation of MEK/ERK signaling in IL-1β-induced COX-2 expression in synovial fibroblasts. Sci Rep. 2017;7:39914. doi: 10.1038/srep39914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tornero-Esteban P, Rodríguez-Rodríguez L, Abásolo L, Tomé M, López-Romero P, Herranz E, González MA, Marco F, Moro E, Fernández-Gutiérrez B, Lamas JR. Signature of microRNA expression during osteogenic differentiation of bone marrow MSCs reveals a putative role of miR-335-5p in osteoarthritis. BMC Musculoskelet Disord. 2015;16:182. doi: 10.1186/s12891-015-0652-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong G, Long H, Ma S, Shunhan Y, Li J, Yao J. MiRNA-335-5p relieves chondrocyte inflammation by activating autophagy in osteoarthritis. Life Sci. 2019;226:164–172. doi: 10.1016/j.lfs.2019.03.071. [DOI] [PubMed] [Google Scholar]