

Abstract
The appearance of emerging variants of SARS‐CoV‐2 carrying mutations into the spike protein has recently raised concern with respect to tracking their transmission and mitigating the impact in the evolving pandemic across countries. AY.4.2, a recently detected Delta variant sublineage, is considered a new variant under investigation (VUI) as it carries specific genetic signatures present in the spike protein, called Y145H and A222V. Here, using genomic epidemiology, we provide the first preliminary insight regarding the circulation of this emerging VUI in Italy.
Keywords: AY.4.2 variant, genomic surveillance, Italy, SARS‐CoV‐2
Highlights
Genomic epidemiology suggests that multiple independent introductions have occurred trough time likely‐mediated by European countries.
Virus migration generally followed patterns of national and international human mobility, illustrating how the easing of restriction measures might facilitate the spread of those emerging variants worldwide.
Our data reveal how crucial appear to be the implementation of a widespread genomic monitoring to detect variants previously not yet identified across the country.
1. INTRODUCTION
Over the past year, the evolution of new, increasingly infectious variants of SARS‐CoV‐2 has fueled surges in the number of COVID‐19 cases and deaths around the world. 1 , 2 , 3 The Delta variant (B.1.167.2 lineage), first detected in India in late 2020, and recently designed as a new SARS‐CoV‐2 variant of concern (VOC), becoming during those past months, the dominant circulating strain globally. 4 , 5 As of August 2021, the Delta variant has been already subdivided in the Pango lineage designation system into sublineages from AY.1 to AY.95. It is said that, as of August 2021, the AY.4 is likely among the predominant forms circulating around the world. 6 Moreover, the recent identification in England, of a new subtype of Delta, called AY.4.2, recently defined as a “variant under investigation” (VUI), has raised concerns that this may ratchet up infection rates globally even further. 7 This new subvariant of the virus is distinguished by two mutations in its spike protein, called Y145H and A222V and it has been suggested that it might be 10%–15% more transmissible than the original strain. 7 However, neither mutation is in the receptor‐binding domain, which is the part of the spike that binds to a particular receptor on human cells. Thus, suggesting that these mutations are unlikely to cause major increases in transmissibility and/or in the immune escape. In an effort to quickly detect and characterize newly circulating SARS‐CoV‐2 variants in Italy, the University of Campus Biomedico in Rome has enhanced epidemiological and genomic investigation. Here, we provide the first preliminary insight regarding the circulation of this emerging VUI in Italy.
2. MATERIALS AND METHODS
Viral RNA from nasopharyngeal swab samples, obtained for routine purposes, were submitted to extraction and multiplex real‐time polymerase chain reaction (PCR) using the Allplex™ 2019‐nCoV Assay (Seegene). The RT‐PCR–positive samples were then submitted to viral genomic amplification and posterior sequencing using the Illumina MiSeq system in conformity with the manufacturer's instructions. Consensus sequences were generated by de novo assembling using the iVar with the default setting. 8 Lineage assignment was performed using the Pangolin lineage classification software tool. 9 All samples belonging to the emerging AY.4.2 sublineage (n = 10) were then compared to a diverse pool of genome sequences (n = 13 434) sampled worldwide collected up to October 28, 2021. All sequences were aligned using the ViralMSA tool, 10 , 11 and IQ‐TREE2 12 was used for phylogenetic analysis using the maximum likelihood approach. TreeTime 13 was then used to transform the preliminary ML tree topology into a dated tree using a constant mean rate of 8.0 × 10‐4 nucleotide substitutions per site per year, after the exclusion of outlier sequences. The mutation pattern of the VUI was analyzed using the NextClade online tool. 14
3. RESULTS
The sequenced samples obtained in this study were collected from 60% females and 40% males (Table S1), presenting mild symptoms, with a median age of 36 years (range: 11–69 years of age). All tested samples contained sufficient viral genetic material (≥2 ng/µl) for library preparation. For positive samples, PCR cycle threshold (Ct) values were on average 22.65 (range: 17.30–29.10). Sequences had a median genome coverage of 97% (range: 94.0–99.9). To accurately establish evolutionary relationships among the newly generated sequence and other isolates of SARS‐CoV‐2, we then subjected a combined data set to phylogenetic inference.
Up to October 28, about 60 Italian AY.4.2 strains were collected and sequenced and even if the number of detected sub‐lineage is still small‐scale, it appears to be already widespread within Italian regions (about 50% of them, Figure 1A). Our time‐stamped phylogeny revealed that the Italian isolates are scattered throughout the tree and cluster together with viral strains isolated mainly in other European countries, suggesting that multiple independent introductions have occurred through time (Figure 1B). Together, our results reinforce that virus migration generally followed patterns of national and international human mobility, illustrating how the easing of restriction measures facilitated the spread of those emerging variants within the country and around the world. Additionally, using NextClade, the mutation pattern of the newly generated strains (Figure 1C) was determined, and no newly acquired mutations were identified.
Figure 1.
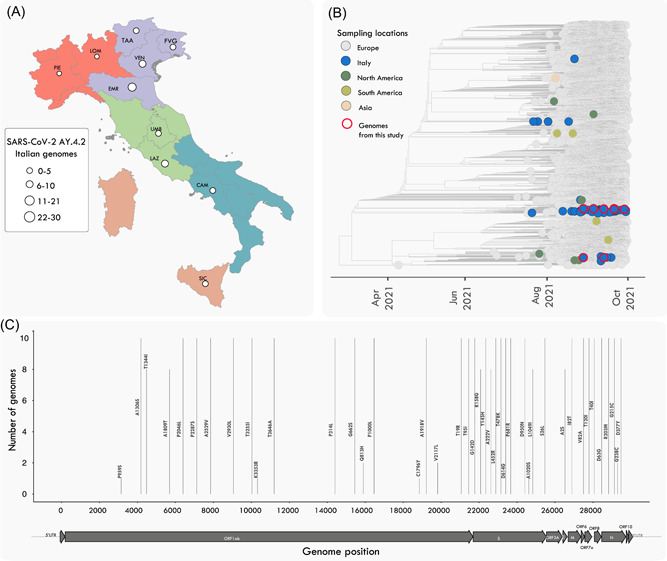
Genomic epidemiology of the SARS‐CoV‐2 AY.4.2 variant in Italy. (A) Map of Italy with the number of AY.4.2 available genomes on GISAID as of October 28, 2021. The size of the circles is proportional to the number of genomes by region. Italian regions are colored according to five macroregions (NUTS, nomenclature of territorial units for statistics): Northeast, Northwest, Central, South, and Insular. (B) Maximum likelihood (ML) phylogenetic tree including the newly (n = 10) AY.4.2 isolates obtained in this study plus n = 13 434 representative SARS‐CoV‐2 genomes collected up to October 28, 2021. The genomes are colored according to their location (continent) and the color legend is on the left of the tree. (C) Variant maps of the AY.4.2 lineage‐defining‐mutations mapped against the SARS‐CoV‐2 genome structure. Most common mutations are highlighted
4. DISCUSSION
Here we provide the first preliminary overview regarding the circulation of the AY.4.2 VUI in Italy, highlighting how crucial appear to be the implementation of widespread genomic monitoring to detect variants previously not yet identified across the country. This emerging lineage carries two additional mutations in the spike protein (Y145H and A222V), that may have functional importance, in addition to several mutations seen in the Delta variant related to increased risk of disease severity, risk of vaccine escape and higher transmissibility. In Italy, the VUI detected is still limited but already widespread across the Italian region and a boost in real‐time genomic monitoring will be crucial to mitigate the possible impact of any potential emerging variants that might face the fight against the pandemic. Moreover, multiple introductions in Italy were revealed probably from European countries highlighting how critical is to identify this pattern and to track human mobility to promptly react with epidemiological measures. Furthermore, as recently reported, no impairment of neutralization sensitivity of the Delta variant and its sublineages have been already observed, except for the Delta Plus that showed resistance for monoclonal antibody used for COVID‐19 patient treatment. 15
In conclusion, considering the recent concern regarding the rapid rise of this novel variant mainly across Europe and the constellation of mutation into the S protein, our findings reinforce how crucial is the implementation of nonpharmaceutical measures together with the need for active monitoring, in containing and preventing the spread of any potential emerging viral strains with possible implications for public health policies and immunization strategies.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ETHICS STATEMENT
This study was approved by the University of Campus Biomedico Ethics Review Committee (Approval number 8.1(21).21 OSS). Individual participant consent was not required for genomic surveillance–this requirement was waived by the Research Ethics Committees.
AUTHOR CONTRIBUTIONS
Molecular screening and produced SRS‐CoV‐2 genomic data: Silvia Angeletti, Marta Fogolari, Lucia De Florio, Carla Lintas, Roberta Veralli, Maria Francesconi, Francesca Caccuri, Marina De Cesaris, Cecilia De Flora, Stefano Pascarella, and Elisabetta Riva. Collected samples and curated metadata: Silvia Angeletti, Marta Fogolari, Lucia De Florio, Carla Lintas, Roberta Veralli, Marta Fogolari, Francesca Caccuri, Marina De Cesaris, Cecilia De Flora, Stefano Pascarella, and Elisabetta Riva. Analysed the data: Silvia Angeletti, Marta Giovanetti, Eleonora Cella, and Massimo Ciccozzi. Helped with study design and data interpretation: Silvia Angeletti, Marta Giovanetti, Eleonora Cella, and Massimo Ciccozzi. Wrote the initial manuscript, which was reviewed by all authors: Silvia Angeletti, Marta Giovanetti, Eleonora Cella, and Massimo Ciccozzi.
Supporting information
Supplementary information.
ACKNOWLEDGMENTS
Marta Giovanetti is supported by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro—FAPERJ. This study was supported in part through the CRP‐ICGEB RESEARCH GRANT 2020 Project CRP/BRA20‐03. We also would like to thank all the authors who have kindly deposited and shared genome data on GISAID.
Angeletti S, Giovanetti M, Fogolari M, et al. SARS‐CoV‐2 AY.4.2 variant circulating in Italy: Genomic preliminary insight. J Med Virol. 2022;94:1689‐1692. 10.1002/jmv.27451
Silvia Angeletti and Marta Giovanetti equally contributed to this study.
DATA AVAILABILITY STATEMENT
Newly generated SARS‐CoV‐2 sequences have been deposited in GISAID under accession numbers EPI_ISL_4498838, EPI_ISL_4498842, EPI_ISL_4571292, EPI_ISL_4884907, EPI_ISL_4884919, EPI_ISL_4884920, EPI_ISL_5163108, EPI_ISL_5163112, EPI_ISL_5163673, and EPI_ISL_5421318.
REFERENCES
- 1. Tegally H, Wilkinson E, Giovanetti M, et al. Detection of a SARS‐CoV‐2 variant of concern in South Africa. Nature. 2021;592(7854):438‐443. 10.1038/s41586-021-03402-9 [DOI] [PubMed] [Google Scholar]
- 2. Davies NG, Abbott S, Barnard RC, et al. Estimated transmissibility and impact of SARS‐CoV‐2 lineage B.1.1.7 in England. Science. 2021;372(6538):eabg3055. 10.1126/science.abg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Faria NR, Mellan TA, Whittaker C, et al. Genomics and epidemiology of the P.1 SARS‐CoV‐2 lineage in Manaus, Brazil. Science. 2021;372(6544):815‐821. 10.1126/science.abh2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tegally H, Wilkinson E, Althaus CL, et al. Rapid replacement of the Beta variant by the Delta variant in South Africa. medRxiv. 2021. 10.1101/2021.09.23.21264018 [DOI] [Google Scholar]
- 5. Earnest R, Uddin R, Matluk N, et al. Comparative transmissibility of SARS‐CoV‐2 variants Delta and Alpha in New England, USA. medRxiv. 2021. 10.1101/2021.10.06.21264641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Public Health England . SARS‐CoV‐2 variants of concern and variants under investigation. Tech Brief. 2021;26:31. [Google Scholar]
- 7. Public Health England . SARS‐CoV‐2 variants of concern and variants under investigation. Tech Brief. 2021;16:71. [Google Scholar]
- 8. Grubaugh ND, Gangavarapu K, Quick J, et al. An amplicon‐based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20(1):8. 10.1186/s13059-018-1618-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rambaut A, Holmes EC, O'Toole Á, et al. A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403‐1407. 10.1038/s41564-020-0770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moshiri N. ViralMSA: massively scalable reference‐guided multiple sequence alignment of viral genomes. Bioinformatics. 2021;37(5):714‐716. 10.1093/bioinformatics/btaa743 [DOI] [PubMed] [Google Scholar]
- 11. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094‐3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minh BQ, Schmidt HA, Chernomor O, et al. IQ‐TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37(5):1530‐1534. 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sagulenko P, Puller V, Neher RA. TreeTime: Maximum‐likelihood phylodynamic analysis. Virus Evol. 2018;4(1):042. 10.1093/ve/vex042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aksamentov I, Roemer C, Hodcroft EB & Neher RA Nextclade: clade assignment, mutation calling and quality control for viral genomes. 2021. doi:10.5281/zenodo.5607694
- 15. Arora P, Kempf A, Nehlmeier I, et al. Delta variant (B.1.617.2) sublineages do not show increased neutralization resistance. Cell Mol Immunol. 2021;18(11):2557‐2559. 10.1038/s41423-021-00772-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
Newly generated SARS‐CoV‐2 sequences have been deposited in GISAID under accession numbers EPI_ISL_4498838, EPI_ISL_4498842, EPI_ISL_4571292, EPI_ISL_4884907, EPI_ISL_4884919, EPI_ISL_4884920, EPI_ISL_5163108, EPI_ISL_5163112, EPI_ISL_5163673, and EPI_ISL_5421318.