

Abstract
Advanced single-molecule techniques have enabled tracking of nanometer-scale movements of DNA and RNA motor proteins in real time. Previously, we reported an ATP-independent diffusion of transactivation response RNA binding protein (TRBP) on dsRNA, yet the mechanistic details remain elusive. Using single-molecule fluorescence assays, we demonstrate that the diffusion activity of TRBP is coordinated by an independent movement of two subdomains, dsRBD1 and dsRBD2, in which the diffusion distance is determined by the length of a flexible linker domain that connects the two dsRBDs. When the linker is shortened, the diffusion distance is reduced proportionally, suggesting a ruler-like function of the linker domain. Diffusion stalls upon encountering a physical barrier in the form of an RNA:DNA hybrid segment or bulky secondary structures, indicating a dsRNA scanning mode of TRBP. The results display a plausible mechanism of TRBP in scanning for pre-miRNA or pre-siRNA as proper substrates for the RNAi pathway.
Graphical Abstract
INTRODUCTION
Double-stranded RNA (dsRNA) plays a critical role in cellular metabolism. RNAs that constitute ribosome, spliceosome, and a family of small regulatory RNAs including micro-RNA (miRNA), small interfering RNA (siRNA), and Piwi-interacting RNA (piRNA) all possess dsRNA as an essential structural and functional component.1 In addition, dsRNAs are replication intermediates for many RNA viruses such as hepatitis C, Ebola hemorrhagic fever, SARS, polio, and influenza.2,3 Accordingly, there are numerous cellular dsRNA binding proteins (dsRBPs) that are implicated in diverse cellular pathways including protein kinase R in antiviral immune response, ADAR1 in RNA editing, Dicer, transactivation response RNA binding protein (TRBP) and PACT in RNA interference (RNAi), and Staufen1 in mRNA transport and localization.4–10 Many dsRBPs harbor multiple dsRNA binding domains (dsRBDs) which enable highly specific binding to the A-form helix of dsRNA.11,12 It remains uncertain why some dsRBPs consist of multiple dsRBDs and how individual dsRBDs contribute to dsRNA binding and processing.
TRBP functions in HIV replication, PKR-mediated immune response, and RNAi. As an essential cofactor of Dicer, TRBP enhances dsRNA cleavage by Dicer13–15 and helps in recruiting Dicer complex to Argonaute for miRNA processing.16 TRBP consists of three dsRBDs in which the two N-terminal domains, dsRBD1 and dsRBD2, bind dsRNA with high affinity and dsRBD3 associates with Dicer.13,17 Earlier we reported that dsRBD2 of TRBP makes tight contact with three grooves and 2′-hydroxyl groups on the dsRNA backbone.11 Such contact was substantially lost on dsDNA or the DNA:RNA hybrid, underscoring the exclusive selectivity of TRBP toward dsRNA.11 We also demonstrated that TRBP exhibits repetitive diffusion on dsRNA without requiring ATP. Such motility of TRBP was sufficient to induce diffusion of the Dicer–TRBP complex on pre-miRNA or pre-siRNA substrates, leading to facilitated RNA dicing activity. Similar dynamics was observed in the orthologous proteins, PACT and R3D2.15 More recently, we reported that another member of the dsRBP family, Staufen, also diffuses on dsRNA, suggesting diffusion activity as a conserved mechanism in diverse dsRBPs.18 The diffusion of TRBP required a minimum of dsRBD1 and dsRBD2 as deletion of either domain abolished the activity. Nevertheless, it was not clear how the two dsRBDs coordinate the repetitive diffusion movement on dsRNA.
Here, using single-molecule fluorescence assays, we elucidate the underlying mechanism that orchestrates the repetitive diffusion of TRBP. We demonstrate that the two dsRBDs move separately, one domain at a time. Such movement involves stretching of a flexible linker that connects the two dsRBDs. Therefore, the linker sets a limit on the diffusion distance of the dsRBP, acting as a ruler that reaches up to a certain distance. We further confirmed the ruler-like function by demonstrating that reducing the linker length shortens the diffusion distance. In addition, when encountering a physical barrier in the form of a large bulge or long stretches of the DNA:RNA hybrid, the dsRBD displays substantial stalling before overcoming the barrier, suggesting a mechanism that allows for sensing and scanning of dsRNA. Together, we provide molecular insights into the mechanism that drives the repetitive diffusion of TRBP. This suggest a plausible role of TRBP in measuring the length of dsRNA while scanning for intact dsRNA. Such a mechanism likely enables discerning and selecting suitable RNA substrates for the RNAi pathway.
RESULTS
Difference in Diffusion Rate between dsRBD1 and dsRBD2 Reveals Independent Domain Movement.
TRBP consists of three dsRBDs, dsRBD1, 2, and 3. Previously, we established that TRBP diffuses on dsRNA independent of ATP, and the repetitive diffusion motion of TRBP on dsRNA is driven by the first two domains, dsRBD1 and dsRBD2.15 This is consistent with the role of dsRBD1 and 2 of TRBP in binding dsRNA and dsRBD3 in associating with Dicer.8,19,20 Here, we sought to examine how the two dsRBDs coordinate such repetitive diffusion along the axis of dsRNA. We generated a truncated TRBP protein, D12, in which dsRBD1 and 2 were intact, but dsRBD3 was deleted (Figure 1A). To probe the movement of individual dsRBDs, we labeled either dsRBD1 or dsRBD2 with Cy5 (red fluorescent dye) specifically. Unlike the nonspecific labeling of TRBP in our previous report,15 the site-specific labeling used here enables tracking the movement of individual dsRBD. Each labeled protein was applied to Cy3 (green)-labeled dsRNA (Figure 1B,C) in a single-molecule fluorescence resonance energy transfer (smFRET) assay. In both cases, smFRET traces exhibited repetitive FRET fluctuations, consistent with our previous report that verified dsRBD1 and 2 as necessary and sufficient for diffusion of TRBP (Figure 1D).15 Two plausible models of diffusion include two feet diffusing simultaneously as one body (Figure 1F) or independently (Figure 1G). To determine which mode D12 uses, we quantified the diffusion time of individual dsRBDs by performing an autocorrelation analysis on smFRET traces of over 100 diffusion events for each dsRBD. The rates calculated from an exponential fitting of each autocorrelation curve yielded 0.155 and 0.106 s of diffusion time for dsRBD1 and dsRBD2, respectively (Figure 1E). In this way, the diffusion constant represents the diffusion time for one round trip, that is, sliding back and forth. We note that the difference in diffusion constant is not due to Cy3 labeling on one stand of dsRNA because the protein binds to dsRNA in a sequence- and position-independent manner.15 The substantial difference (>30%) between the two diffusion times (Figure 1E) is likely caused by the mode where dsRBD1 and dsRBD2 move independently.
Figure 1.
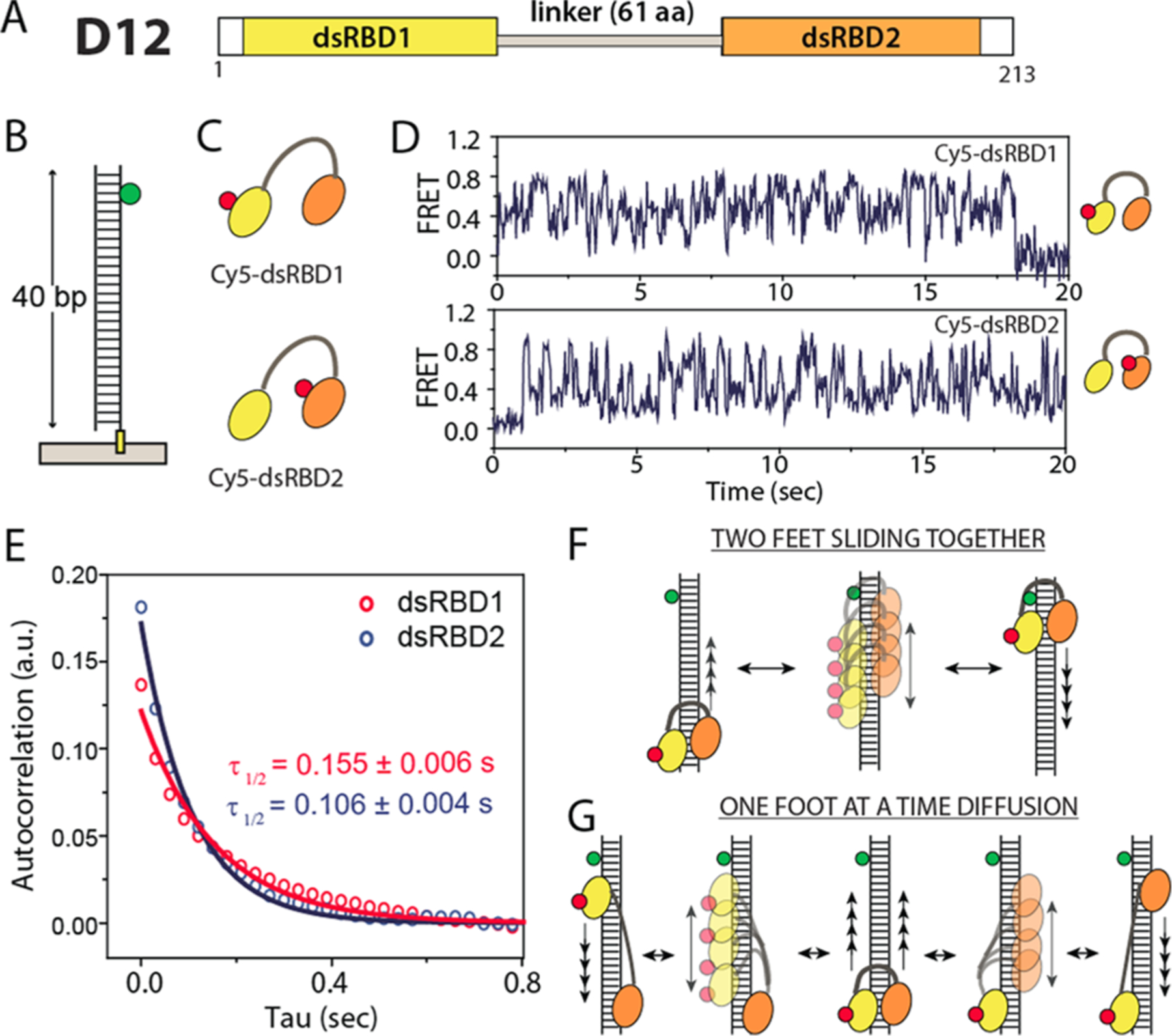
(A) TRBP truncation, dsRBD1 + dsRBD2 construct. (B) dsRNA (40 bp) labeled with Cy3 immobilized on a single-molecule surface. (C) Individually Cy5-labeled dsRBD1 and dsRBD2. (D) Representative smFRET traces from Cy5-dsRBD1 and Cy5-dsRBD2. (E) Autocorrelation fit of smFRET result. (F,G) Two plausible diffusion mechanisms.
Alternating FRET and PIFE Signals Provide Further Evidence for Independent Diffusion of Individual Domains.
To further test the independent movement mode, we probed the repetitive diffusion motion of dsRBP by the one color fluorescence assay termed “protein-induced fluorescence enhancement” (PIFE).15 Unlabeled protein induces signal enhancement of the fluorescent dye on dsRNA when in close proximity.21,22 In our current system, the repetitive diffusion of dsRBP produces intensity fluctuation of the Cy3 dye, reporting a periodic distance change between the protein and the dye at the end of dsRNA (Figure 2C). We performed the FRET experiment in which only one dsRBD was labeled with Cy5 nonspecifically. We observed two types of traces, both exhibiting an alternating PIFE and FRET signal (Figure 2A,B). One type of trace showed a long duration of PIFE fluctuation followed by a brief period of FRET and subsequent long duration of PIFE signal fluctuation (Figure 2A). The second pattern involved frequent exchange between PIFE and FRET signal fluctuation, in which the PIFE signals (green Cy3 spikes) are denoted by arrows (Figure 2B). The alternating signal change shown in Figure 2A,B is likely arising from one molecule of D12 rather than successive binding of different molecules for the following reasons. Based on our previous results, TRBP binds dsRNA as a monomer at the tested protein concentration (10 nM), and the off rate of TRBP (<0.008 s−1) is much lower than what is expected from the short spike of PIFE in Figure 2B.15 In this context, the pattern of alternating PIFE and FRET fluctuation cannot emerge if both dsRBD1 and dsRBD2 diffused together. This pattern can only be explained if one dsRBD diffuses at a time—along the Cy3-labeled RNA, diffusion of the unlabeled dsRBD of D12 TRBP produces the change in PIFE signal (Figure 2C), while diffusion of the Cy5-labeled dsRBD generates the FRET changes (Figure 2D). Although the position of the nondiffusing dsRBD cannot be pinpointed, our data indicate that it is located outside of the FRET-sensitive distance range (>8 nm), allowing the PIFE signal to be detected in the absence of FRET. The majority of the traces (>80%) display long-lived movement of one foot (dsRBD) followed by diffusion of the other foot (Figure 2A), while some exhibit rapidly exchanging two feet during diffusion (Figure 2B). We analyzed the frequency of the “changing foot” by counting the number of events where the PIFE signal converts to FRET and vice versa over many fluctuating events. While many successive steps occur without changing foot (tall bar at zero fraction, Figure 2E), there is approximately 25% (peak at 0.25 fraction) likelihood of changing foot during diffusion in the case of rapidly exchanging two feet and 12% in all cases (Figure 2E). This means that D12 changes feet approximately once every 10 diffusion events on average.
Figure 2.
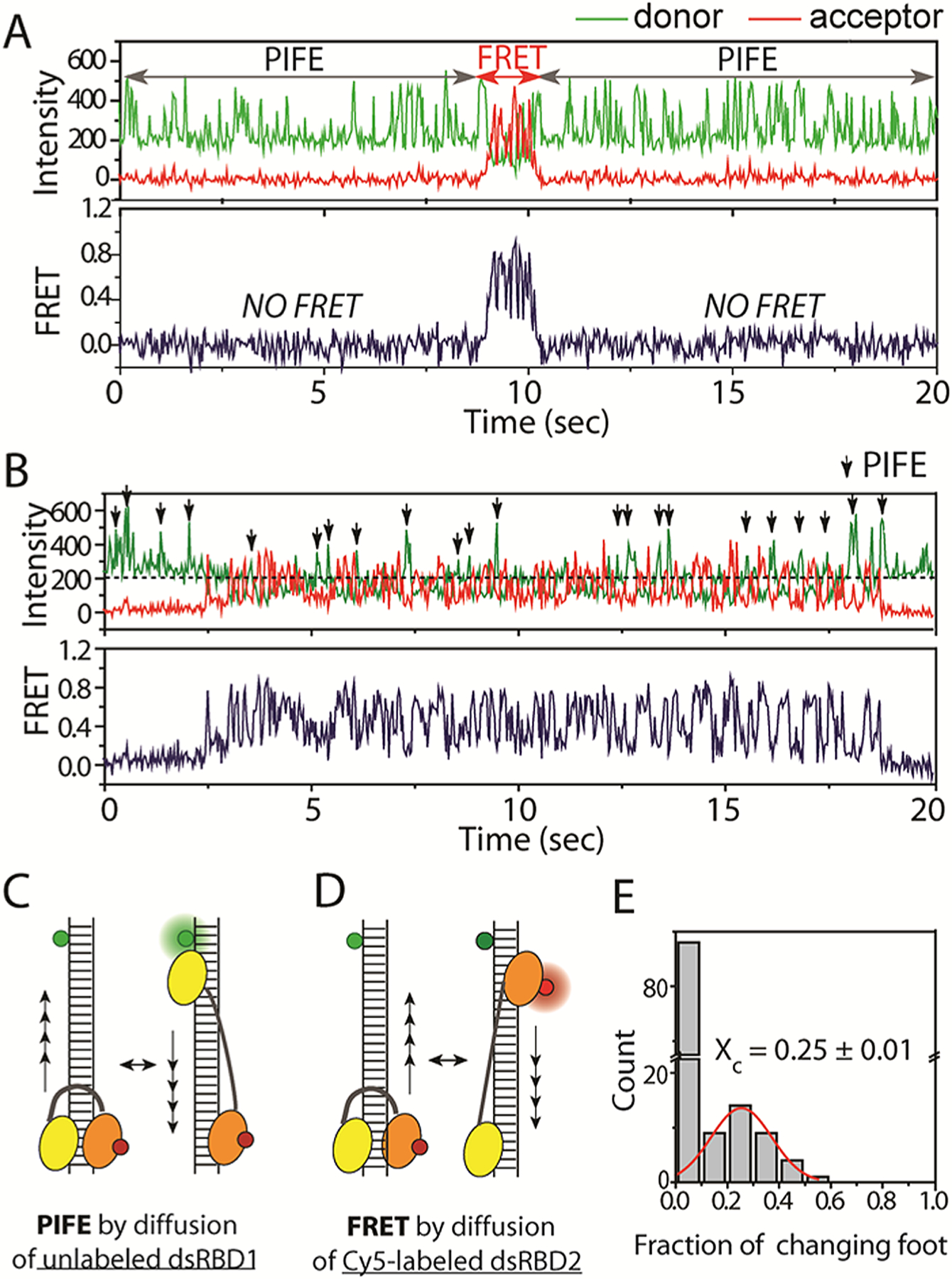
(A,B) smFRET traces which display mixture of PIFE and FRET fluctuations. (C,D) Detection scheme for PIFE and FRET. (E) Fraction of switching dsRBD1 and dsRBD2.
Double-Labeled Protein Displays Periodic Conformational Changes Concomitant with Repetitive Diffusion.
We reasoned that if TRBP diffusion was driven by one dsRBD at a time, the distance between the two domains would change periodically. On the other hand, if the two domains diffuse together, the distance may not undergo any change. To probe such conformational changes of TRBP during its diffusion along dsRNA, we prepared the double-labeled D12 protein by engineering two single cysteine mutations on individual dsRBDs (D12-DC in Table S1) and stochastically labeling with maleimide-conjugated Cy3 and Cy5 dye.23 Although we do not know which domain is labeled with Cy3 or Cy5, the dual-color labeling strategy is sufficient for monitoring a relative movement between the two dsRBDs. The protein with a single Cy3- or Cy5-labeled molecule can be easily distinguished from the double (Cy3 + Cy5)-labeled ones in the single-molecule analysis. The dsRBD labeled only with Cy3 (either one or two Cy3 dyes) would not yield any FRET, and proteins with Cy5 only (either one or two Cy5 dyes) would not produce any fluorescence signal with 532 nm laser excitation in our experimental setup. We have not observed any homoquenching in the case of two Cy3 labelings (Figure S1).
To first probe the conformation of D12 when it encounters dsRNA without diffusion, we prepared a 15 bp dsRNA substrate (Table S2), which is approximately the size of the footprint of one dsRBD (Figure 3A).15 We observed a short-lived high FRET signal, reflecting a transient interaction between 15 bp dsRNA and D12 in which two feet are close together (Figure 3B), reflecting a closed protein conformation. Consistent with the FRET time trace, the FRET histogram built from over 200 molecules shows a high FRET peak, indicating that most D12 molecules contact the short RNA in the closed conformation (Figure 3C). The FRET values collected at the initial binding moment (the first 100 ms) from the same experiment displays a sharp high FRET peak (Figure 3D), suggesting that the initial binding exhibits tightly closed conformation of D12. In contrast, when the double-labeled D12 was applied to a longer (38 bp) dsRNA substrate that may allow D12 diffusion (Figure 3E and Table S2), we observed repeated FRET fluctuations, which represents successive closing and opening of the two labeled dsRBDs (Figure 3F). This strongly suggests that the two domains do not diffuse together but rather independently, leading to a periodic distance change between them. The overall FRET histogram built from over 100 molecules displays two broad peaks, consistent with the fluctuating FRET between the closed (high FRET) and open (low FRET) states of D12 TRBP (Figure 3G). Unlike in the case of 15 bp dsRNA, the initial FRET values collected from D12 on 38 bp dsRNA exhibited multiple FRET peaks, reflecting nonuniform conformation of D12 when engaged with a longer length of dsRNA. To test if the conformation-driven FRET fluctuation correlates with the diffusion movement measured previously (Figure 1), we compared the time components of each FRET fluctuation calculated by the exponential fitting of autocorrelation analysis between the two measurements, conformational change versus diffusion. Two dsRNAs of different lengths, 19 and 38 bp, were tested, and the two time values from both sets of FRET data matched closely in both cases (Figure 3I), indicating that the conformational change between two dsRBDs correlates with the diffusion activity of D12 on dsRNA. Taken together, the diffusion of D12 arises from an independent movement of each dsRBD rather than a simultaneous movement of both dsRBDs (Figure 3J).
Figure 3.
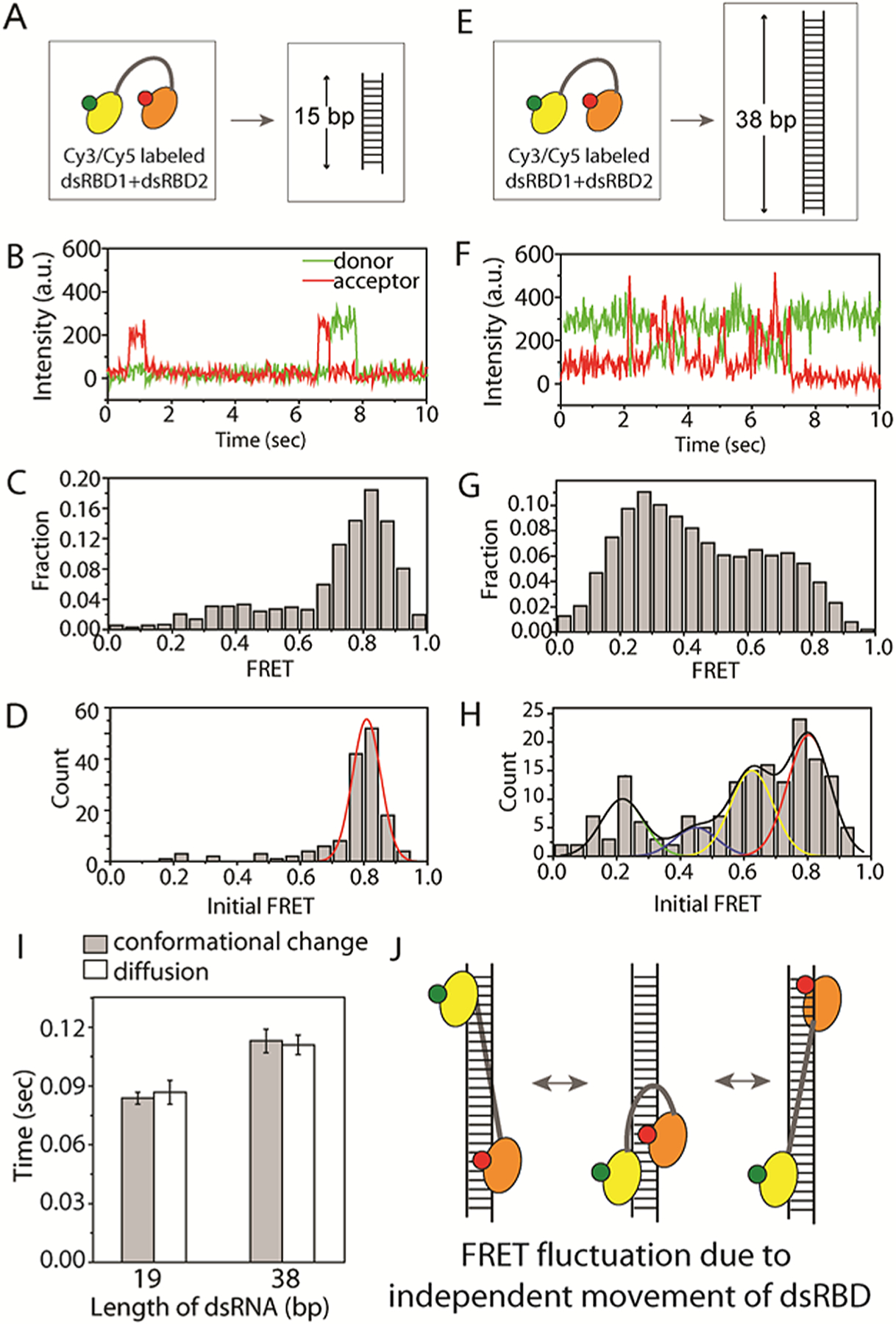
(A,E) Doubly labeled D12 protein subjected to (A) 15 bp and (E) 38 bp RNA. (B,F) Representative smFRET traces obtained from experiment (A,E). (C,G) Overall FRET histogram from two experiments. (D,H) FRET histogram from initial binding moments. (I) Time collected from exponential fitting of autocorrelation curve of FRET fluctuation probing conformation and diffusion for two different RNA substrates, 19 and 38 bp. (J) Schematic depicting the independent movement of dsRBD1 and dsRBD2.
Linker Length Controls Diffusion Distance.
The dsRBD1 and dsRBD2 in D12 are interconnected by a long and flexible linker of 61 amino acids.24 Based on the independent domain movement we observed above, we hypothesized that the linker between the two domains may control how far the two domains can move apart from each other. To test this hypothesis, we created two truncation mutants, D12-L37 and D12-L17, in which the linker length was reduced from 61 to 37 and 17 amino acids, respectively (Figure 4A and Table S1). The Cy5-labeled proteins of L37 and L17 were each applied to 55 bp dsRNA labeled with Cy3 (Figure 4B). We analyzed all the FRET traces regardless of two FRET patterns in Figure 2. The FRET histograms obtained for the D12-L61 (full length) displayed two distinct peaks centered at 0.25 and 0.8. In contrast, the L37 and L17 mutants yielded FRET peaks ranging 0.4–0.8 and 0.55–0.8, respectively (Figure 4C, left column). The reduced FRET peak separation in the two mutants suggests shortened diffusion distance corresponding to the shortened linker length between the two dsRBDs. The two distinct peaks are likely due to slightly longer time the protein spends at both ends of the diffusion, indicated by greater dwell times at high and low FRET state in smFRET traces (Figure 4C). In agreement, the smFRET traces showed FRET fluctuation exhibiting the greatest amplitude change for L61 but substantially reduced changes for L37 and L17 mutants (Figure 4C, right column). The same experiment performed by PIFE shows a similar pattern for L17 and L61, likely due to the short distance sensitivity of PIFE (Figure S2). Taken together, our results suggest that the linker length sets the diffusion distance between the two dsRBDs; that is, when the linker length shortens, the diffusion distance diminishes proportionally (Figure 4E,F). When we plotted the linker length versus the diffusion distance calculated by FRET change, we obtained a linear fit with an R2 value of 0.998, reflecting a positive correlation between the linker length and the diffusion distance (Figure 4D). Our data suggest that the linker between dsRBD1 and dsRBD2 functions like a ruler that measures and sets a limit on the diffusion distance. Consistently, PACT containing a shorter linker length between two dsRBDs (29 aa vs 61 aa of TRBP) exhibited a shorter diffusion range compared to TRBP (Figure S3).
Figure 4.
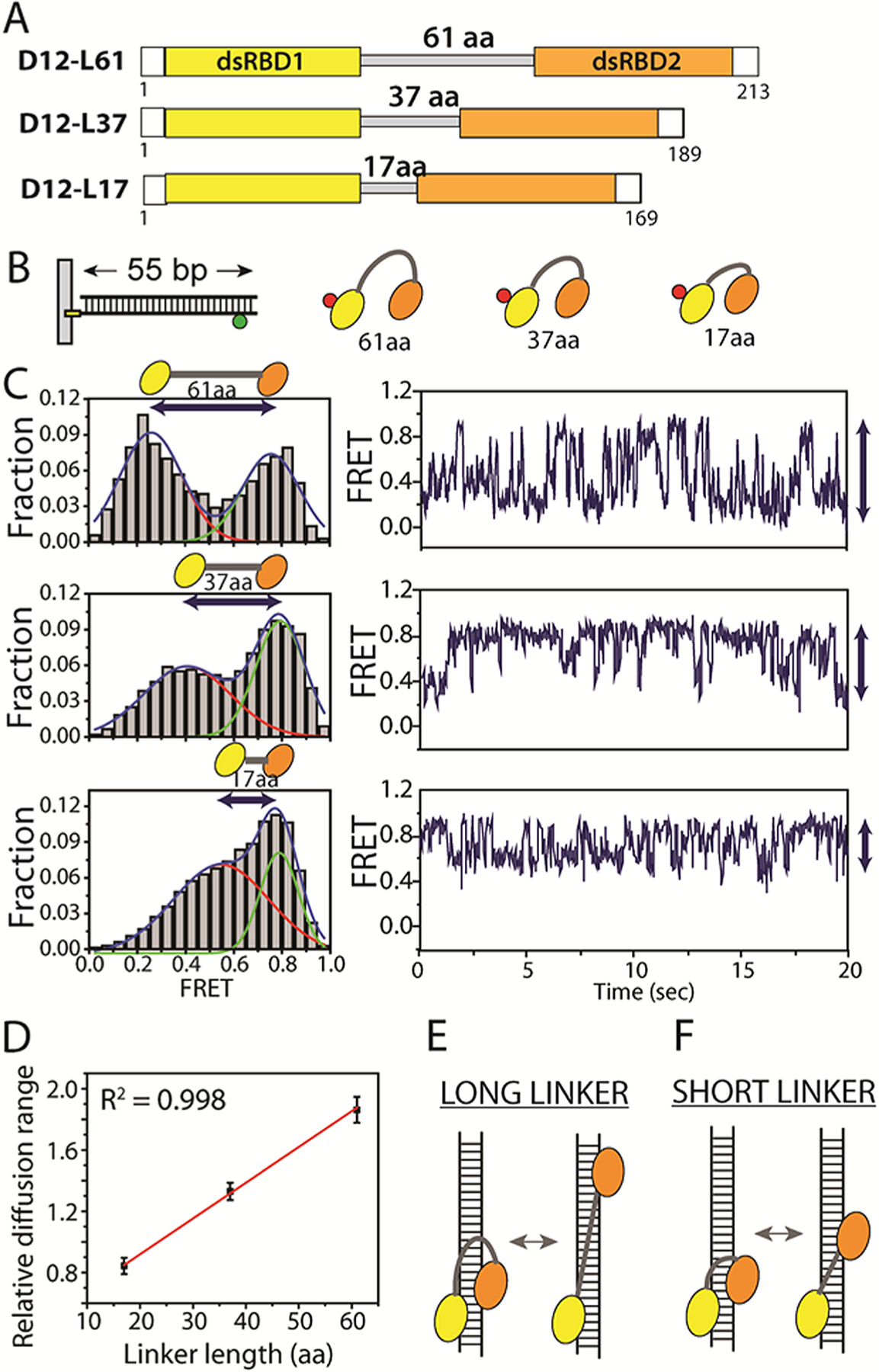
(A) D12 (D12-L61) and two linker length variants, D12-L37 and D12-L17. (B) Experimental scheme. (C) FRET histogram obtained from three measurements with varying linker lengths. (D) Linker length plotted against diffusion distance deduced from FRET values. (E,F) Schematic representation of D12 diffusion for long vs short linker length.
dsRNA Scanning by Diffusion.
Next, we asked if this diffusion mechanism of TRBP enables dsRNA scanning by sensing a non-dsRNA segment as a physical barrier. Previously, we demonstrated that TRBP diffusion tolerates small-sized mismatches on the dsRNA stem but not excessive secondary structures with substantial bulges and mismatches.15 To test the barrier effect more systematically, we inserted varying lengths (3–9 bp) of the DNA:RNA hybrid in the middle of dsRNA substrates (Figure 5A) based on our previous result that TRBP does not bind to the DNA:RNA hybrid structure.15 We termed the substrates DI3, DI6, and DI9 for DNA insertion of 3, 6, and 9 nt (Figure 5A and Table S2). This allows us to test the barrier effect in a length-dependent manner. We analyzed all of the FRET traces regardless of two FRET patterns in Figure 2. The overall FRET histogram for DI3 displays two broad peaks approximately centered at 0.3 and 0.8, similar to that of dsRNA with no interruptions (Figure 5B). In DI6 and DI9, the increase in the middle FRET peak (peak 2) was evident, whereas the other two peaks, 1 and 3, decreased (Figure 5C,D). To quantify, we fitted three FRET peaks with Gaussian distribution to distinguish peak 1 (green), peak 2 (red), and peak 3 (blue), representing the low, middle, and high FRET peaks, respectively. Peaks 1 and 3 represent the positions where Cy5-labeled dsRBD is farthest and closest to Cy3 on dsRNA, respectively. Peak 2 at mid-FRET represents the “stall” position of the protein on the substrate, corresponding to the location of the DI3–DI9 barrier in the substrate (Figure 5E). Thus, the size of peak 2 relative to 1 or 3 reflects how long and often molecules stall at the barrier position compared to the other two end positions on RNA. Therefore, the relative distribution of peak 2 over the other two peaks can serve as a proxy for the extent of the barrier effect. For this comparison, we plotted the area of peak 1 (green bars) and peak 2 (red bars) for each RNA. While peak 1 is slightly higher than peak 2 for DI3, the trend is reversed in DI6 and such an effect is more pronounced in DI9, suggesting that the barrier effect is the greatest for the longest DNA insertion, DI9 (Figure 5F). This effect is evident in smFRET trace for DI9, which exhibits frequent stalling at the mid-FRET level (displayed as a pink bar in Figure 5G) and returning to high FRET without getting through the barrier to low FRET (Figure 5G). In contrast, the DI3 traces display the full range of FRET fluctuation without much interruption. With a further extended barrier of DI12, we observed that TRBP could rarely bypass the barrier, severely limiting its diffusion range to a half of the full length of dsRNA. Infrequent visit to the other half of dsRNA gave rise to a small population of the low FRET range represented as peak 1 (Figure S4). We also show that other types of barriers, such as mismatches and bulges, produce a similar barrier effect as seen in DI6 and 9 (Figure S5).
Figure 5.
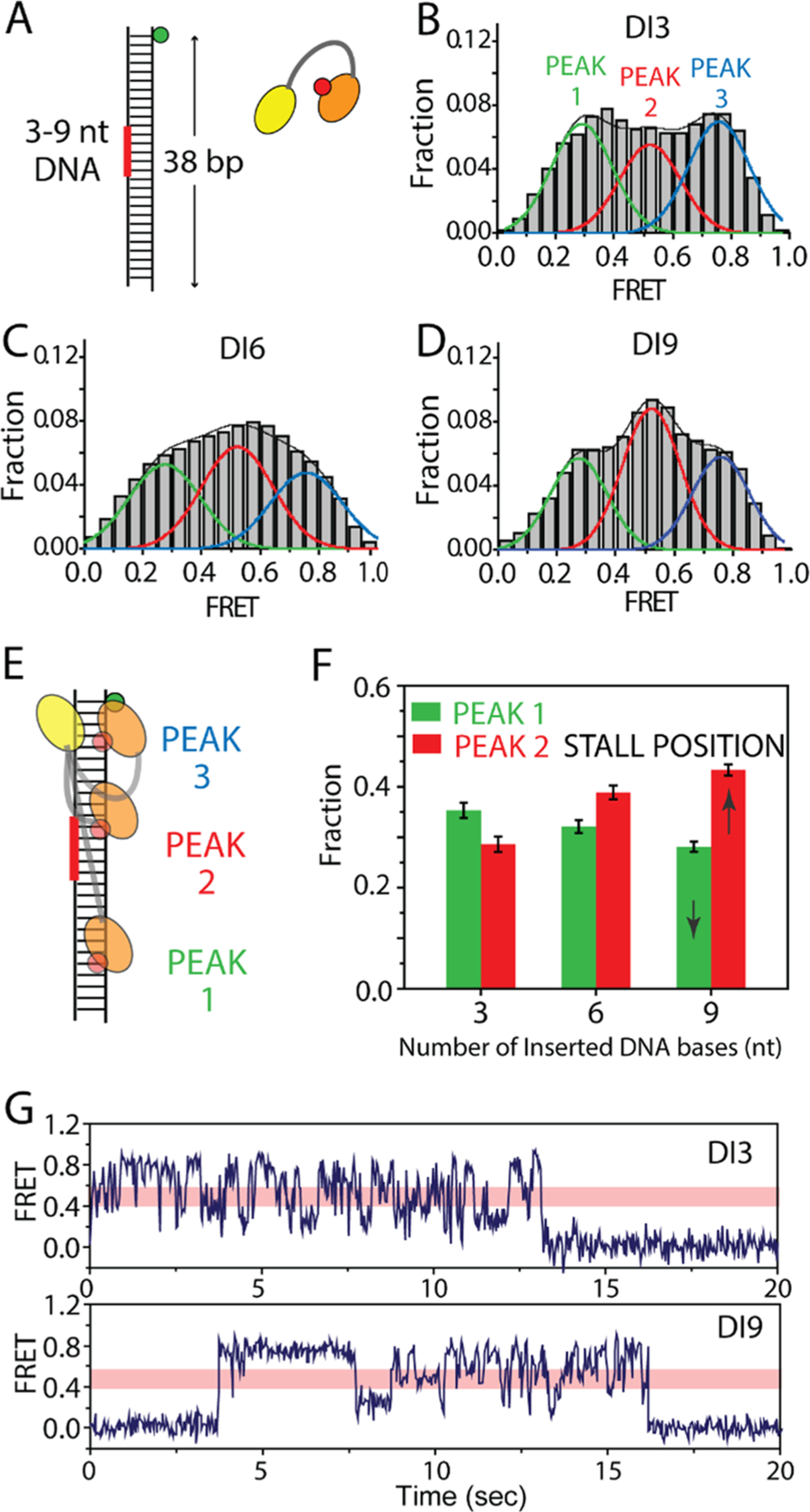
(A) dsRNA substrate engineered with varying length of DNA:RNA hybrid inserted in the middle as a physical blockade. (B–D) FRET histograms taken for DI3, DI6, and DI9. (E) FRET peak positions in terms of location on dsRNA. (F) Molecule fractions of peak 1 and peak 2 for DI3, DI6, and DI9. (G) Representative smFRET traces of DI3 and DI9.
DISCUSSION
dsRBPs are responsible for recognizing and processing dsRNA in cells. They function in translational control, mRNA storage, small RNA-induced silencing, antiviral immune response, and viral RNA replication. Based on biochemical and structural analysis, the protein–dsRNA interactions have been considered static, for the most part. Our previous report challenged this view by providing a clear evidence that the TRBP–dsRNA interaction is highly dynamic, and the mobility likely plays a functional role in enhancing Dicer’s RNA cleavage activity.15 In the context of pre-miRNA processing, such mobility of Dicer–TRBP can facilitate the search for a correct cleavage site along dsRNA, thereby accelerating the rate of RNA cleavage. Similar mobility detected in other dsRBPs including PACT, R3D2, and Staufen15,18 suggests that the diffusion activity of dsRBP on dsRNA may be a conserved interaction mode that serves a functional role in cellular metabolism. Here, we sought to dissect the molecular mechanism responsible for the diffusion motility of TRBP by examining the motions of the two domains, dsRBD1 and dsRBD2.
The first question we posed was whether the two domains diffuse simultaneously or independently. In order to distinguish the two scenarios, we performed several complementary assays. First, we labeled dsRBD1 and dsRBD2 independently and analyzed dwell times of individually labeled proteins. In our previous report, we mostly used a full-length TRBP, which was nonspecifically, yet singly labeled. Therefore, the result of both the dsRBD1 and dsRBD2 was collected together in our single-molecule data. They were not distinguishable because the FRET fluctuation range is similar regardless of the labeling position. In contrast, in this study, when the dsRBD3 was removed and dsRBD1 and dsRBD2 were individually labeled and tested for diffusion, we observed a discrete difference in the diffusion kinetics, providing evidence against a simultaneous movement of the two domains.
Second, we dissected the single-molecule data to decouple FRET and PIFE events. The distinct exchange between the FRET and PIFE signal exhibited in single-molecule traces further supported an independent motion by the two domains. So far, we have employed the PIFE method mostly when applying unlabeled protein to Cy3-labeled substrates.15,18,21,22,25,26 Based on the time-separated PIFE and FRET signals in our current scheme, we were able to interpret the PIFE signal within the FRET experiment. The dsRBD-induced PIFE signal separated distinctly from the FRET signal change, likely because the movement of the unlabeled domain and labeled domain occur independently. It is important to understand that the continuous signal fluctuation from both PIFE and FRET come from one binding event, not from successive binding of multiple protein units as TRBP binds dsRNA as a monomer at the tested protein concentration, and the off rate of TRBP is much slower than the time scale of PIFE or FRET fluctuation.15 Based on the signals obtained in single-molecule traces, PIFE signal fluctuation occurs in the absence of FRET fluctuation; that is, the Cy3 (green) signal fluctuates without any anticorrelated change in Cy5 signal. It is not due to photobleaching of the Cy5 dye because the Cy5 signal comes back when there is FRET fluctuation (Figure 2A,B). As a result, the emerging model is that the two domain/feet diffuse on the dsRNA axis one foot at a time.
Third, to gain further insight into our proposed model, we examined the interdomain movement by the double-labeled D12 protein. When the double-labeled D12 bound to a short dsRNA (15 bp), we obtained static high FRET, indicating that both domains engage with the RNA substrates in a closed conformation. When applied to a longer dsRNA (19 or 38 bp), we obtained FRET fluctuation, indicating a repetitive opening and closing of the two dsRBDs. Taken together, the results strongly suggest that dsRBD1 and dsRBD2 diffuse not together but as independent units.
The independent movement of each dsRBD for the diffusion activity suggests that the linker between two domains may regulate the diffusion. The linker truncation mutants revealed the function of this flexible linker as a ruler that measures and limits the distance separation between the two domains, thereby setting a restriction in the diffusion distance (Figure 6A, B). In light of TRBP-induced mobility of the Dicer–TRBP complex on pre-miRNA, the limited diffusion distance is useful for measuring the finite length of 25–30 bp of the relevant substrates.
Figure 6.
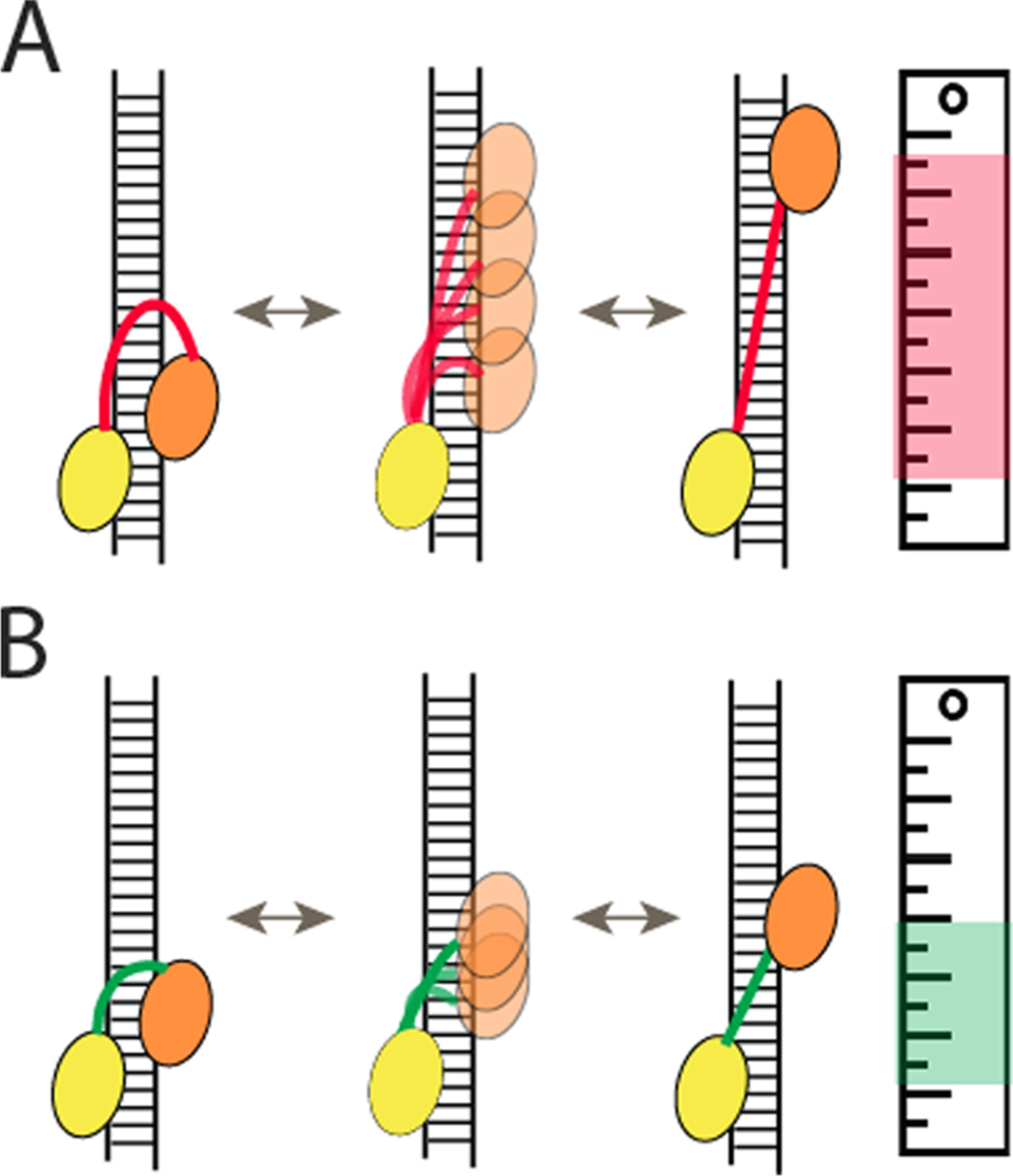
(A,B) TRBP scans dsRNA by diffusing on RNA, one foot at a time. The diffusion distance scales with linker length.
The test on the DNA:RNA hybrid barriers suggests that while a small disruption on dsRNA can be tolerated, long stretches of non-dsRNA impediment causes frequent and extended stalling at the barrier. Based on the diffusion mechanism elucidated here, we propose a model in which TRBP serves a scanning function to identify suitable pre-miRNA substrates. The scanning may be achieved by (i) checking for the A-form dsRNA, (ii) measuring the length of dsRNA, and (iii) reading out the topology/disruption along dsRNA. Based on the similar composition of many dsRBPs, where multiple dsRBDs are interconnected by variable length of linker domain, it is plausible that a similar mechanism is at work in other dsRBPs. Depending on the type of dsRBDs and length of linker, they may exhibit different substrate selectivity suited for particular pathways.
SUMMARY
We elucidated a molecular mechanism underlying a diffusion movement of dsRBP on dsRNA. By taking advantage of site-specific fluorescent labeling and high temporal and spatial resolution of single-molecule fluorescence assays, we resolved the independent movement of two subdomains that drives the diffusive motion. These results provide new insights into the role of dsRBPs as a molecular machine that scans RNA with a built-in ruler to search for the optimal substrates, thereby providing functional significance of having multiple dsRBDs in these proteins.
EXPERIMENTAL SECTION
Protein Mutation and Labeling.
We prepared TRBP mutants (i) lacking dsRBD3 (D12) that possess three different linker lengths (17, 37, and 61 aa) between dsRBD1 and dsRBD2, (ii) single cysteine mutation at either dsRBD1 or dsRBD2, and (iii) double cysteine mutation at both dsRBD1 and dsRBD2. The TRBP mutants were cloned as cleavable N-terminal His6–maltose binding protein fusions. Each was purified separately from bacterial overexpression using methods previously described.16 The amino acid sequence of TRBP mutants is in Table S1.
For labeling single cysteine and double cysteine TRBP mutants, we mixed fluorophores (Cy5 for single cysteine mutant and a mixture of Cy3 and Cy5 for double cysteine mutant) with the mutants, incubated them for 30 min at room temperature, and removed the excess dye using a Biorad P-6 column. The labeling efficiency was calculated by measuring absorbance of protein and fluorophores.
RNA Substrate Preparation.
The sequences of all RNA substrates are listed in Table S2, which were synthesized and HPLC-purified by Integrated DNA Technology Inc. with proper chemical modification such as biotin and amino modification for immobilization and dye labeling, respectively. For labeling of RNA, we mixed 10–20 μL of 100 μM of ssRNA containing amino modification with 0.1 mg of NHS ester-conjugated Cy3 dye in 100 mM NaHCO3 buffer, pH 8.5, incubated the mixture overnight, and removed the excess dye using ethanol precipitation. If the labeling efficiency was lower than 90%, we reran the labeling reaction using the poorly labeled RNA to improve the labeling efficiency. The complementary ssRNAs were annealed together by mixing each strand in an annealing buffer (10 mM Tris, pH 8, and 100 mM NaCl), incubating at 90 °C for 2 min, and cooling slowly to room temperature.
Single-Molecule Fluorescence Assays.
We used our home-built total internal reflection fluorescence microscope for single-molecule fluorescence assays. To measure the FRET signal between Cy3 or DY547 and Cy5, we excited our samples with a solid-state 532 nm laser (Spectra Physics), separated the emission by two, Cy3 or DY547 and Cy5, using a dichroic mirror (cutoff = 630 nm), and detected it using an EMCCD camera (Andor). To study the diffusion of dsRBPs on dsRNA, we applied Cy5-labeled dsRBPs to Cy3- or DY547-labeled dsRNAs, which were immobilized on a polyethyleneglycol-coated quartz surface via biotin–neutravidin linkage. To measure the conformational change of two dsRBDs, we applied doubly labeled dsRBPs to nonlabeled dsRNAs. All of the single-molecule measurements were performed in 20 mM Tris, pH 7.5, 25 mM NaCl, 1 mM DTT, and 0.1 mg/mL BSA with an oxygen scavenging system (0.5 w/v % glucose, 1 mg/mL glucose oxidase, and 88 U/mL catalase in 5–10 mM trolox) to stabilize fluorophores.
Diffusion Range Calculation Using FRET.
When Cy5-labeled D12 was applied to Cy3-labeled dsRNA, we obtained a FRET histogram showing two peaks (low and high), reflecting the closest and farthest position of D12 relative to the end of dsRNA with Cy3. The diffusion range can be calculated using the two distances at the closest and farthest positions that were deducted by FRET efficiency of two peaks through the equation shown below:
where R0 represents Forster radius and E is the FRET efficiency.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the American Cancer Society RSG-12-066-01-DMC, NIH 1DP2GM105453, Human Frontier Science Program RGP0007/2012, and the National Science Foundation Physics Frontiers Center Program (0822613) through the Center for the Physics of Living Cells to H.K. and S.M.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b10387.
Amino acid sequences of all protein variants and RNA sequences used in the study; data for PACT and RNA barrier effects (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Kim VN; Han J; Siomi MC Nat. Rev. Mol. Cell Biol 2009, 10, 126. [DOI] [PubMed] [Google Scholar]
- (2).Meister G; Tuschl T Nature 2004, 431, 343. [DOI] [PubMed] [Google Scholar]
- (3).Heyam A; Lagos D; Plevin M Wiley Interdiscip Rev. RNA 2015, 6, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Stark GR; Kerr IM; Williams BR; Silverman RH; Schreiber RD Annu. Rev. Biochem 1998, 67, 227. [DOI] [PubMed] [Google Scholar]
- (5).Nishikura K Annu. Rev. Biochem 2010, 79, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hutvagner G; McLachlan J; Pasquinelli AE; Balint E; Tuschl T; Zamore PD Science 2001, 293, 834. [DOI] [PubMed] [Google Scholar]
- (7).Chendrimada TP; Gregory RI; Kumaraswamy E; Norman J; Cooch N; Nishikura K; Shiekhattar R Nature 2005, 436, 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lee Y; Hur I; Park SY; Kim YK; Suh MR; Kim VN EMBO J. 2006, 25, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tang SJ; Meulemans D; Vazquez L; Colaco N; Schuman E Neuron 2001, 32, 463. [DOI] [PubMed] [Google Scholar]
- (10).Krichevsky AM; Kosik KS Neuron 2001, 32, 683. [DOI] [PubMed] [Google Scholar]
- (11).Vukovic L; Koh HR; Myong S; Schulten K Biochemistry 2014, 53, 3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Benoit MP; Imbert L; Palencia A; Perard J; Ebel C; Boisbouvier J; Plevin MJ Nucleic Acids Res. 2013, 41, 4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Haase AD; Jaskiewicz L; Zhang H; Laine S; Sack R; Gatignol A; Filipowicz W EMBO Rep. 2005, 6, 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chakravarthy S; Sternberg SH; Kellenberger CA; Doudna JA J. Mol. Biol 2010, 404, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Koh HR; Kidwell MA; Ragunathan K; Doudna JA; Myong S Proc. Natl. Acad. Sci. U. S. A 2013, 110, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).MacRae IJ; Ma E; Zhou M; Robinson CV; Doudna JA Proc. Natl. Acad. Sci. U. S. A 2008, 105, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Parker GS; Maity TS; Bass BL J. Mol. Biol 2008, 384, 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang X; Vukovic L; Koh HR; Schulten K; Myong S Nucleic Acids Res. 2015, 43, 7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lee JY; Kim H; Ryu CH; Kim JY; Choi BH; Lim Y; Huh PW; Kim YH; Lee KH; Jun TY; Rha HK; Kang JK; Choi CR J. Biol. Chem 2004, 279, 30265. [DOI] [PubMed] [Google Scholar]
- (20).Yamashita S; Nagata T; Kawazoe M; Takemoto C; Kigawa T; Guntert P; Kobayashi N; Terada T; Shirouzu M; Wakiyama M; Muto Y; Yokoyama S Protein Sci. 2011, 20, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hwang H; Kim H; Myong S Proc. Natl. Acad. Sci. U. S. A 2011, 108, 7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hwang H; Myong S Chem. Soc. Rev 2014, 43, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Myong S; Rasnik I; Joo C; Lohman TM; Ha T Nature 2005, 437, 1321. [DOI] [PubMed] [Google Scholar]
- (24).Benoit MP; Plevin MJ Biomol. NMR Assignments 2013, 7, 229. [DOI] [PubMed] [Google Scholar]
- (25).Myong S; Cui S; Cornish PV; Kirchhofer A; Gack MU; Jung JU; Hopfner KP; Ha T Science 2009, 323, 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Qiu Y; Antony E; Doganay S; Koh HR; Lohman TM; Myong S Nat. Commun 2013, 4, 2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.