

Abstract
Growth differentiation factor 8 (GDF8), a.k.a. myostatin, is a member of the larger TGFβ superfamily of signaling ligands. GDF8 has been well characterized as a negative regulator of muscle mass. After synthesis, GDF8 is held latent by a noncovalent complex between the N-terminal prodomain and the signaling ligand. Activation of latent GDF8 requires proteolytic cleavage of the prodomain at residue D99 by a member of the tolloid family of metalloproteases. While tolloid proteases cleave multiple substrates, they lack a conserved consensus sequence. Here, we investigate the tolloid cleavage site of the GDF8 prodomain to determine what residues contribute to tolloid recognition and subsequent proteolysis. Using sequential alanine mutations, we identified several residues adjacent to the scissile bond, including Y94, that when mutated, abolish tolloid-mediated activation of latent GDF8. Using the astacin domain of Tll1 (Tolloid Like 1) we determined that prodomain mutants were more resistant to proteolysis. Purified latent complexes harboring the prodomain mutations, D92A and Y94A, impeded activation by tolloid but could be fully activated under acidic conditions. Finally, we show that co-expression of GDF8 WT with prodomain mutants that were tolloid resistant, suppressed GDF8 activity. Taken together our data demonstrate that residues towards the N-terminus of the scissile bond are important for tolloid-mediated activation of GDF8 and that the tolloid-resistant version of the GDF8 prodomain can function dominant negative to WT GDF8.
Introduction
Growth differentiation factor 8 (GDF8), commonly known as myostatin, is a member of the larger transforming growth factor β (TGFβ) superfamily of signaling ligands and functions as a potent negative regulator of muscle mass [1–5]. Genetic deletion of GDF8 or use of GDF8 inhibitors results in a drastic increase in muscle mass [1,3,6–10]. In contrast, overexpression of GDF8 results in muscle atrophy [11–14]. Thus, over the last two decades significant effort has been put forth toward the development of therapies that can boost muscle mass by manipulating GDF8 signaling [8,15–21].
GDF8, like all TGFβ ligands, is synthesized as a precursor protein containing an N-terminal signal sequence and prodomain followed by the C-terminal component that dimerizes to form the signaling molecule or ligand (Figure 1A). The prodomain is essential for proper folding, localization, and plays an important role in the regulation of ligand activity [22–26]. During synthesis, the prodomain component is cleaved from the mature component by proprotein convertases (PCs) such as furin [22,23], during which, two mature domain chains are linked through a disulfide bond to form the final dimeric signaling molecule. These dimeric ligands assemble two type I and two type II serinethreonine kinase receptors that when brought into proximity will activate the type I receptor via phosphorylation by the type II receptor, in turn activating downstream SMAD transcription factors.
Figure 1. Latent GDF8 activation and Tll1 astacin domain structure.
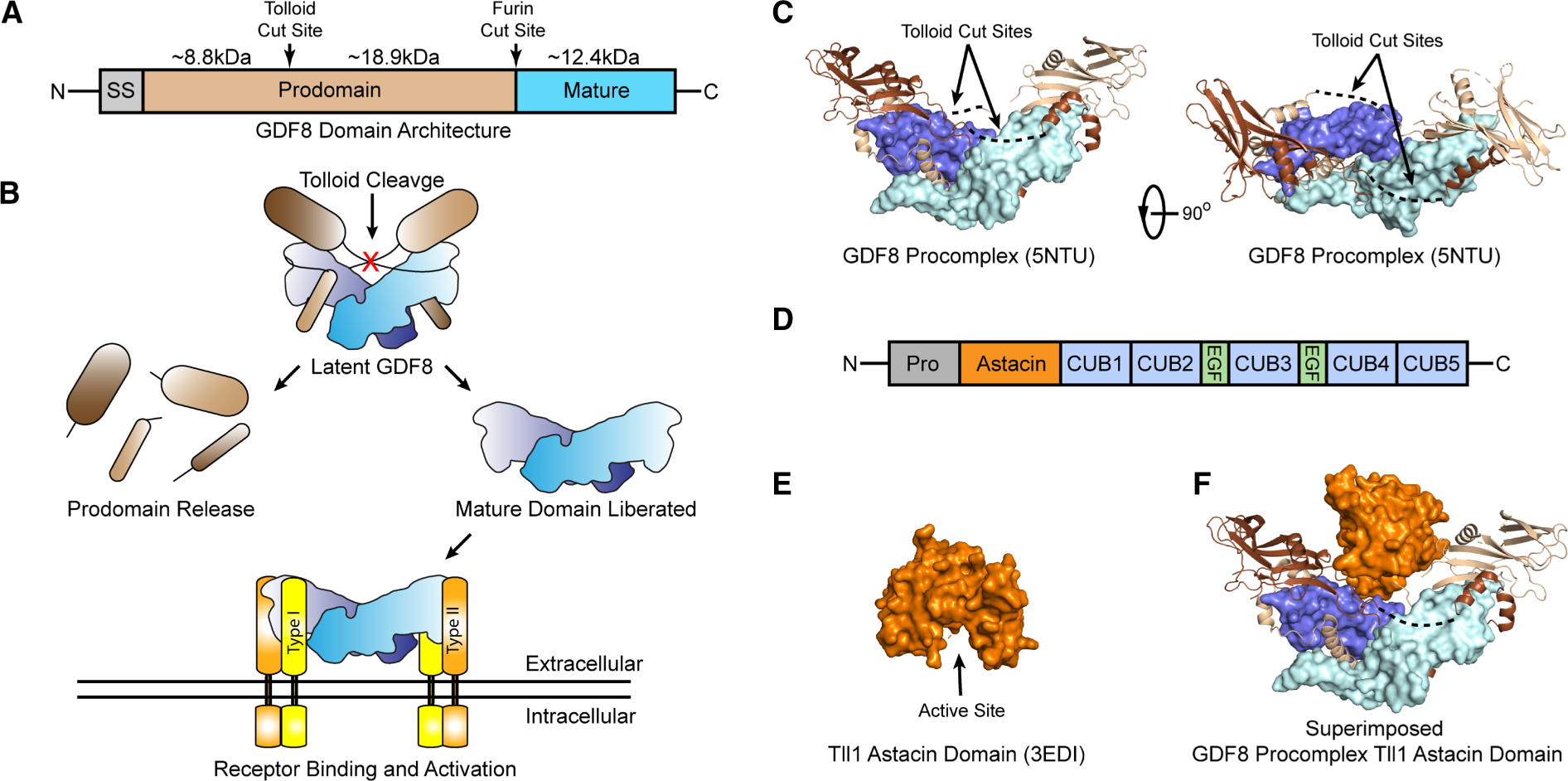
(A) Domain architecture of GDF8, signal sequence (SS), prodomain and mature domain. Tolloid and furin cut sites indicated with size in kDa of each fragment after furin and tolloid processing shown. (B) Schematic of latent GDF8 activation, including tolloid cleavage, mature domain release, and receptor binding. (C) Structure of the GDF8 procomplex [27]. Mature domain monomers in pale cyan and blue, prodomain monomers in light brown and brown. Dotted lines indicated resides not in density. Right panel is rotated 90° about the vertical axis. (D) Domain architecture of the tolloid family including the prodomain, astacin domain, CUB and EGF domains. (E) Structure of the Tll1 astacin domain, active site cleft shown [58]. (F) GDF8 prodomain structure shown in (c) superimposed with the astacin domain active site cleft positioned toward the scissile bond.
In most cases, after cleavage by furin, the prodomain remains non-covalently bound to the mature ligand dimer forming a procomplex. For the majority of the TGFβ superfamily, this procomplex is non-inhibitory and may even facilitate signaling. However, a handful of ligands including GDF8, GDF11, and the TGFβ subclass ligands form interactions with the prodomain that render the ligand inactive or latent [26–28]. These latent procomplexes require an additional activation event to liberate the ligand to allow signaling. For example, the procomplex of TGFβ1 is activated by mechanical force brought about by interactions with αVβ6 integrins and latent TGFβ binding proteins (LTBP1–3) or glycoprotein A repetitions predominant (GARP) [29–32]. For GDF8 and GDF11, procomplex activation requires an additional proteolytic cleavage event mediated by the tolloid family of metalloproteases [33–37]. Activation and signaling of latent GDF8 mediated by tolloid are shown schematically in Figure 1B.
Tolloids are metalloproteases characterized by having an astacin domain responsible for the proteolytic activity. The tolloid family shares a conserved domain architecture from Drosophila to humans, with an N-terminal prodomain, the proteolytic astacin domain, 2 tandem CUB (Compliment/Uegf/BMP1) domains (CUB1/2), an EGF (epidermal growth factor) domain (EGF1), a third CUB domain (CUB3), a second EGF domain (EGF2), and two more tandem CUB domains (CUB4/5) (Figure 1D). There are four members of the tolloid family: mammalian tolloid (mTLD), its splice form BMP1 (truncated at CUB3), tolloid like 1 (Tll1), and tolloid like 2 (Tll2). Tolloids are essential for the proper processing of extracellular matrix (ECM) components such as procollagen [34,38,39]. They also cleave Chordin, a BMP antagonist, to ensure proper axial patterning during embryogenesis [40–45]. Tll1/BMP1 homozygous null mice are embryonic lethal due to cardiac failure and exhibit abnormal collagen maturation [46–49].
Despite several known substrates of tolloid processing, very little is known about how tolloid family members selectively cleave different substrates. In part, this is because tolloid protease sites are highly variable with limited consensus [41,50]. When comparing sequences of known substrates, the most common residue is an aspartic acid in the P1′ site, directly C-terminal of the scissile bond [50]. For GDF8, tolloid cleavage occurs specifically at D99, despite multiple nearby aspartates [37]. Mutation of D99 to alanine abolishes tolloid activation of GDF8, resulting in increased muscle mass [35,37]. Beyond a preference for aspartate at position 99, little is known about the molecular mechanism that determines tolloid substrate recognition and processing. In terms of the tolloids, studies have shown that the non-catalytic domains can have a role in substrate recognition and/or activity [40,45,51,52]. Structural and functional studies showed that the C-terminal domains are involved in homodimer formation, restricting the activity of Tll1 and mTLD, but not in the mTLD splice form, BMP1, which lacks the domains involved in dimerization [40,45,52]. In contrast, removal of the CUB4 and CUB5 domains of Tll2 ablates chordin proteolysis suggesting, in this case, that the non-catalytic domains are important for substrate recognition [51].
For GDF8, preliminary evidence suggests that all tolloids can remove latency through the processing of the prodomain [35,37]. However, it is not known what the role of the additional domains of the tolloid proteins play in the recognition of the GDF8 prodomain or if residues near scissile bond are important for tolloid-mediated GDF8 activation. To investigate this, we modified residues surrounding the scissile bond of GDF8 to determine their impact on tolloid-mediated activation. Our results indicate that residues toward the N-terminus of the scissile bond are important for tolloid-mediated activation. Furthermore, we demonstrate that the astacin domain alone is a potent activator of GDF8 latency, indicating that the additional domains of tolloids are not required for recognition of the GDF8 prodomain substrate.
Materials and methods
HEK293-(CAGA)12 luciferase-reporter assay
Luciferase assays were conducted, in large part, as previously described [35,53–57]. In short, HEK293-T cells stably transfected with a firefly luciferase-reporter gene under the control of the SMAD3-responsive (CAGA)12 promoter were seeded in growth media at 20 000 cells per well in a 96- well, poly-d lysine coated plate. Cells were then treated 24 h after seeding. Transfection-based luciferase assays in Figure 2 were conducted by transfecting 50 ng of full length, human Furin cloned into a pcDNA4 vector, 50 ng of the appropriate human tolloid, cloned into a pcDNA3 backbone and 100 ng of GDF8 DNA in pRK5. After 24 h growth media was swapped for serum-free media. Data were collected 24 h after the media swap and plotted using GraphPad Prism 5 software.
Figure 2. Transfection-based luciferase of GDF8 alanine screen.
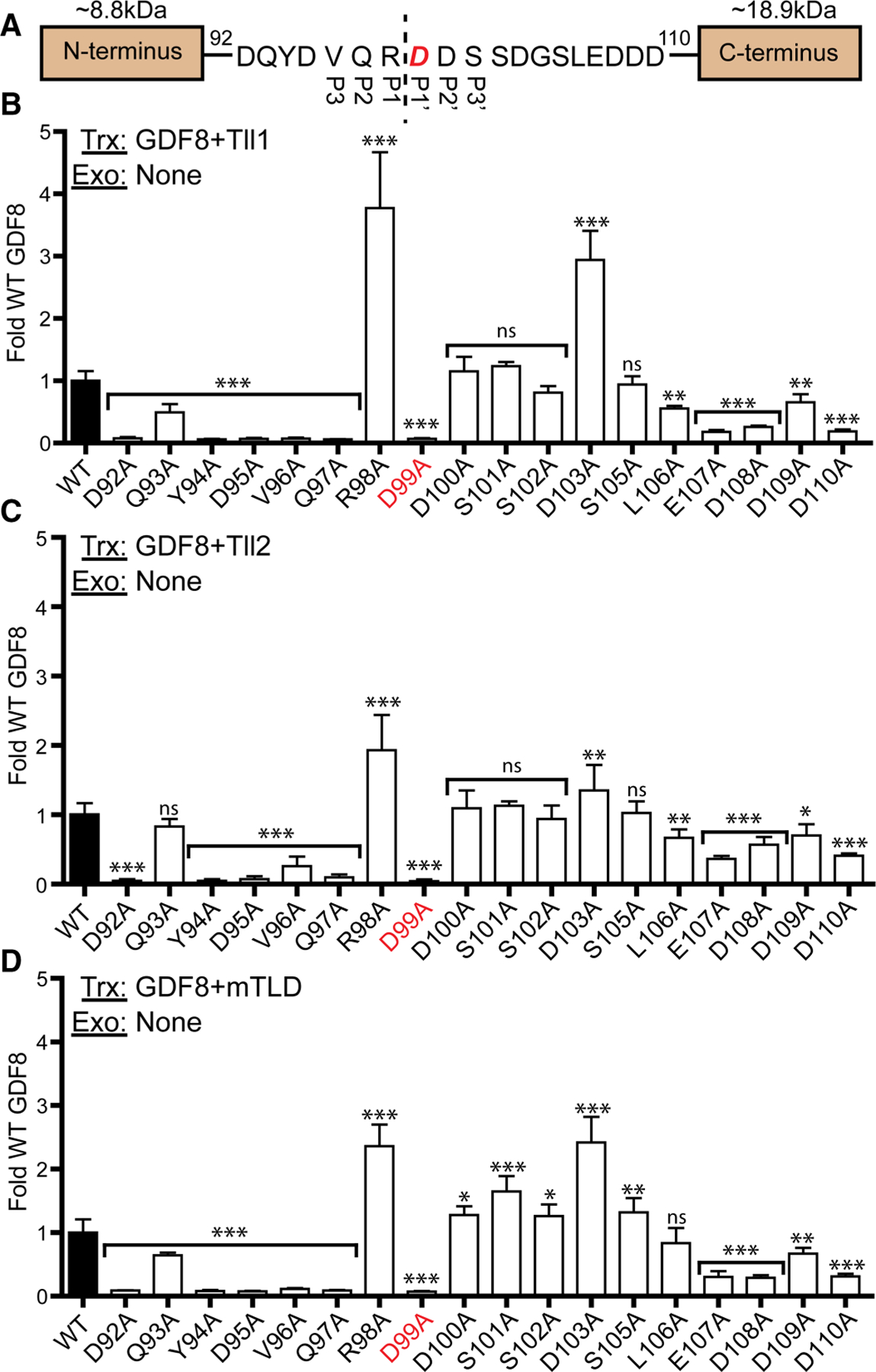
(A) Sequence of the GDF8 tolloid cut site with P3–P3′ residues denoted, and the essential D99 residue shown in red. (B) 100 ng of GDF8 WT or mutant DNA transfected with 50 ng of furin and Tll1 DNA. (C) Similar to (B) but with Tll2 DNA transfected. (D) Similar to (B,C) with mTLD DNA transfected. The DNA transfected (Trx) and exogenous (Exo) protein added (if applicable) is denoted in the top left each graph. Data plotted as mean ± SD and were conducted at least three times with experimental triplicate. Bar graphs were compared using one-way ANOVA with Bonferroni correction against WT GDF8 (*P < 0.05, **P < 0.01, ***P < 0.001).
EC50 curves in Figures 3B and 5B were generated using exogenous protein. Twenty-four hours after seeding, serum-free media containing a constant concentration of latent GDF8 (0.62 nM) was combined with the Tll1 astacin domain. Data were collected 24 h later and fit to a non-linear regression with variable slope via GraphPad Prism 5 software to generate the EC50 curves; 95% confidence intervals (C.I.) are reported in the text. Data in figure 2C were generated by treating cells with 0.62 nM of latent GDF8 (GDF8L), acid-activated GDF8, or mature GDF8 mixed with 1.86 nM of bacterially produced GDF8 prodomain with or without 175 nM of the Tll1 astacin domain. The EC50 curve in Figure 5A was generated by transfecting 100 ng of GDF8 DNA and 50 ng of furin. After 24 h growth media was swapped with serum-free media containing the Tll1 astacin domain. The resulting EC50 curve was fit using GraphPad Prism 5 software as before. The data shown in Figure 5C were generated by transfecting 50 ng of tolloid DNA and 50 ng of furin DNA. After 24 h media was replaced with serum-free containing 0.62 nM of latent recombinant GDF8 and plotted using GraphPad Prism 5 software. The data shown in Figure 6 were generated by transfecting in 50 ng furin and Tll2 DNA, with 50 ng of GDF8 DNA with either 50 ng of empty vector or 50 ng of the WT GDF8. Media was swapped 24 h later for serum-free and measured 24 h after media swap. In each applicable experiment, GDF8 concentrations refer to the dimeric protein.
Figure 3. Analysis of Tll1 astacin domain activity vs GDF8 prodomain states.
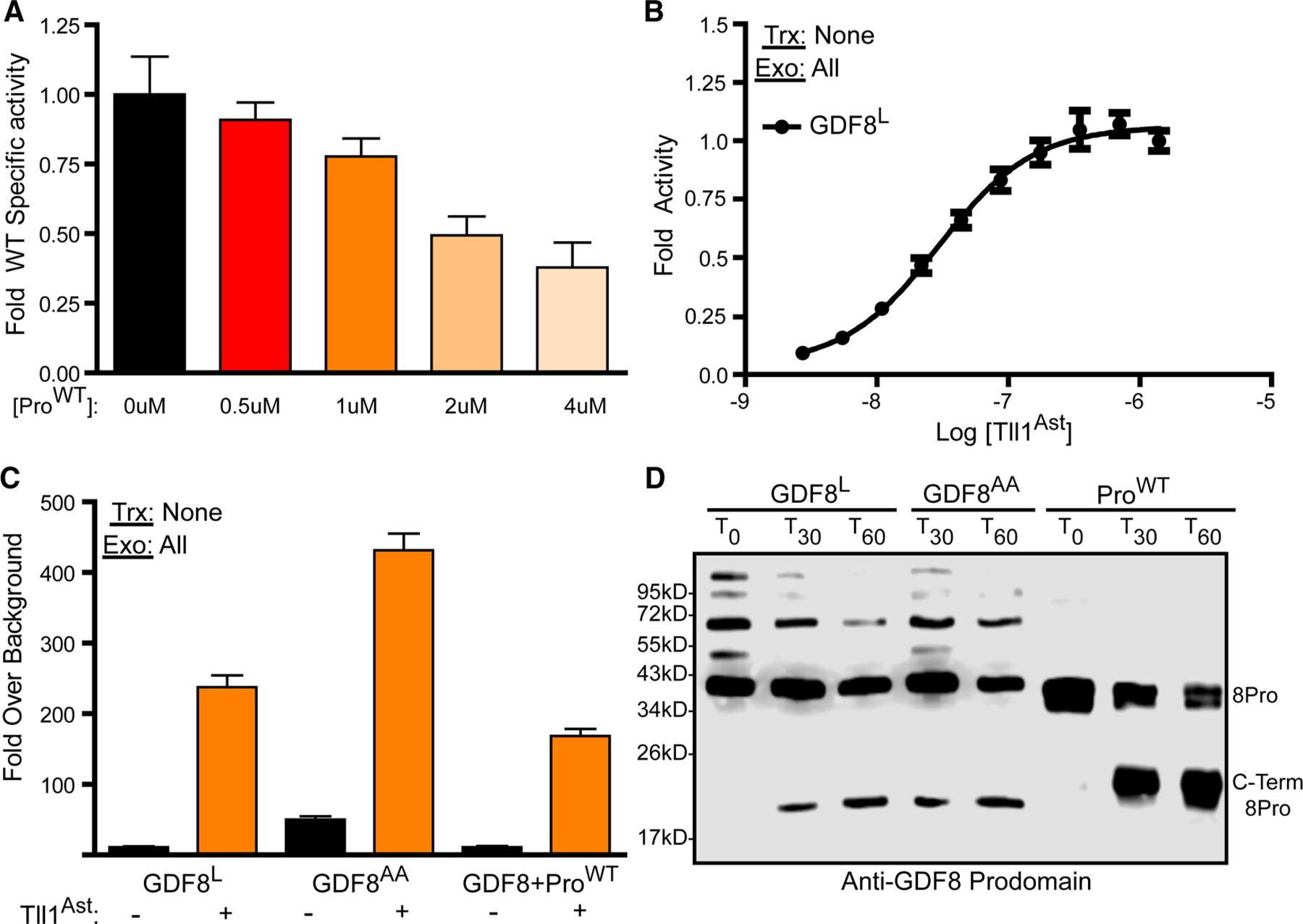
(A) Fluorogenic peptide incubated with the astacin domain and treated with a titration GDF8 prodomain, plotted as fold specific activity. (B) Latent GDF8 treated with a titration of the Tll1 astacin domain. Data were fit by non-linear regression with a variable slope to generate the EC50 curve. (C) Luciferase assay of latent GDF8 (GDF8L), acid-activated GDF8 procomplex (GDF8AA), and a mixture of GDF8 prodomain:mature domain in a 3 : 1 molar ratio, respectively, treated with or without the Tll1 astacin domain. The DNA transfected (Trx) and exogenous (Exo) protein added (if applicable) are denoted in the top left of each luciferase assay. (D) Anti-GDF8 prodomain western blot of GDF8L, GDF8AA, or GDF8 prodomain alone (ProWT) treated with the Tll1 astacin domain for 30 min or 1 h. Experiments a–c were conducted at least three times with experimental triplicate.
Figure 5. Analysis of fully latent GDF8 mutants.
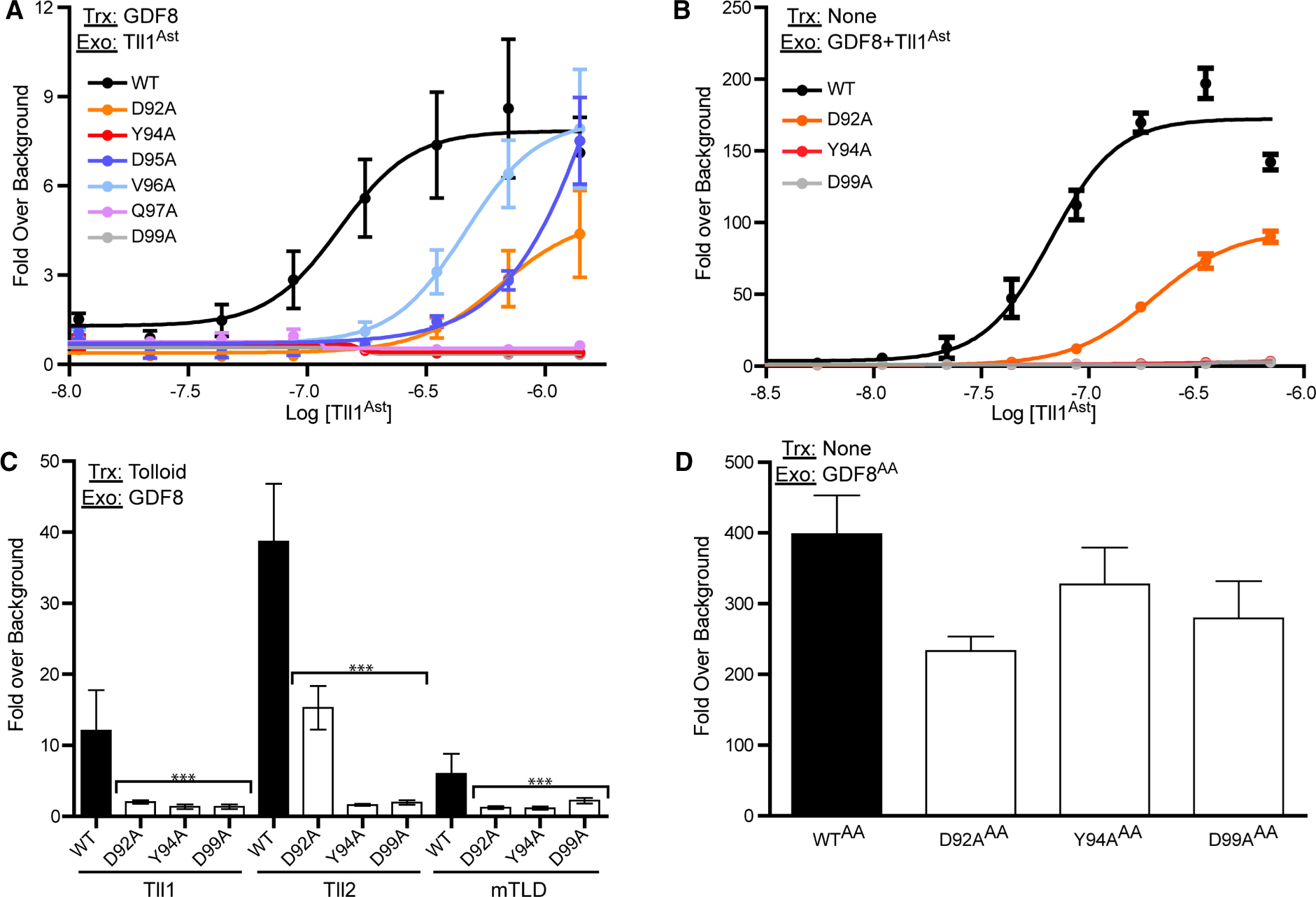
(A) 100 ng of GDF8 DNA and 50 ng Furin DNA were transfected into HEK293T (CAGA)12 cells and treated with a 1 : 2 serial dilution of the Tll1 astacin domain. (B) 0.62 nM of purified GDF8 procomplexes treated with a serial dilution of the Tll1 astacin domain, similar to (A) and added to HEK293T (CAGA)12 cells. Data from (A) and (B) were fit by non-linear regression with a variable slope to generate EC50 curves. (C) Transfection based luciferase assay were full-length Tll1, Tll2 or mTLD were transfected into (CAGA)12 cells. Cells were then treated with 0.62 nM of purified GDF8 procomplex. Bar graphs were compared using one-way ANOVA with Bonferroni correction against WT GDF8 (*P < 0.05, **P < 0.01, ***P < 0.001). (D) 20 nM GDF8 procomplex was acid activated by adding HCl and neutralized by NaOH addition. The acid-activated (AA) procomplex was then added to (CAGA)12 cells and plotted as fold-over serum-free background. The DNA transfected (Trx) and exogenous (Exo) protein added (if applicable) are denoted in the top left of each graph. Experiments were conducted at least three times with experimental triplicate and plotted as mean ± SD.
Figure 6. Transfection-based luciferase assay of dominant-negative GDF8.
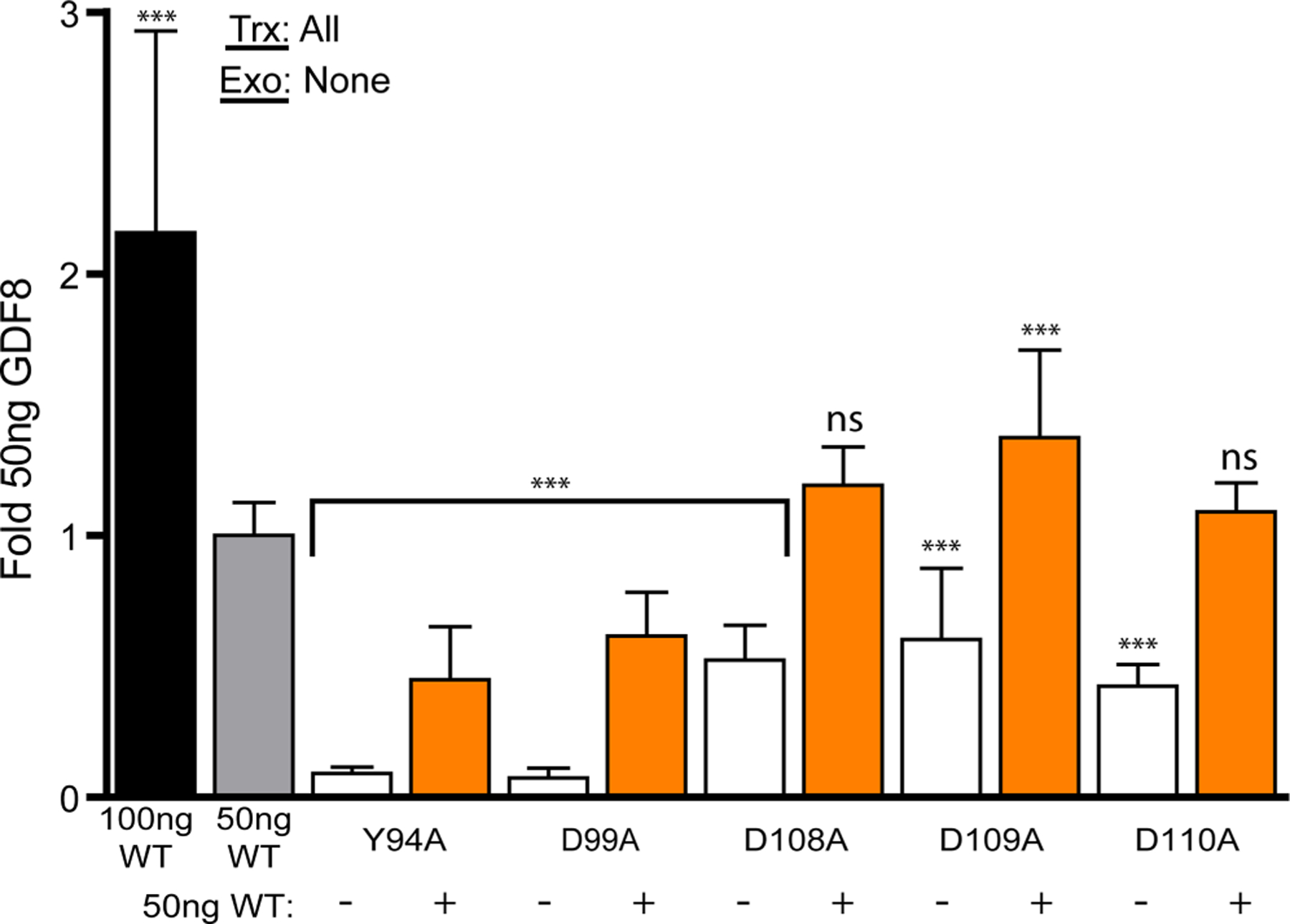
50 ng of furin and Tll2 DNA was transfected into HEK293 T (CAGA)12 cells with 50 ng of WT and/or 50 ng of mutant full-length GDF8 denoted under the X-axis. For wells not containing 100 ng of GDF8 DNA, an empty vector was used to normalize the total amount of DNA transfected into cells. Data plotted as mean ± SD and were conducted at least two times with experimental triplicate. Bar graphs were compared using one-way ANOVA with Bonferroni correction against WT GDF8 (*P < 0.05, **P < 0.01, ***P < 0.001). The DNA transfected (Trx) and exogenous (Exo) protein added (if applicable) are denoted in the top left of each graph.
HEK293t expression test of GDF8 prodomain mutants
A 1.5 μg of GDF8 DNA, and 0.75 μg furin DNA was transfected into HEK293T cells in a 6-well format with 0.75 μg of empty vector or Tll2 DNA to replicate 96-well conditions. Media was swapped 24 h later and conditioned media (CM) was harvested 24 h after the media swap. CM was then concentrated and analyzed by western blots under reducing conditions using antibodies for GDF8 prodomain antibody (AF1539 RnD, Lot UTG0119091) and Sheep IgG (HAF016 RnD, Lot XDP1319051).
Tll1 astacin domain production and refolding from Escherichia coli
The astacin domain of Tll1 was produced and refolded as previously described [58]. In short, the astacin domain of Tll1, residues 148–347, was cloned into the pET28a(+) expression vector without N- or C-terminal tags and used to transform Rosetta™ (DE3) competent cells. Cells were cultured until an OD of 0.8 at 600 nm then subjected to induction where cells are placed in an ice bath and EtOH is added to a final concentration of 2% (v/v). After approximately10 min cells are induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for culture at 20°C overnight. Cells were harvested by centrifugation and suspended in 5 mM EDTA, 5 mM benzamidine, and 50 mM Tris–HCl pH 8.0 (lysis buffer) before sonication for cell lysis. The inclusion body containing the astacin domain was then isolated by centrifugation and washed three time with lysis buffer followed by a final wash with 4 M urea, 1% (v/v) Triton-X100, 5 mM EDTA, 50 mM Tris–HCl pH 8.0. Inclusion bodies were solubilized to a final concentration of ~5 mg/ml in 8 M guanidinium chloride, 100 mM DTT, and 50 mM Tris–HCL pH 8.0 as previously described [58]. The solubilized astacin domain was then refolded by rapid dilution in 50 mM Tris–HCl pH 8.5, 0.8 M l-arginine, 125 mM NaCl, 1 mM CaCl2, 10 mM ZnCl2, 1 mM reduced glutathione, 0.25 mM oxidized glutathione to a final concentration of 50 μg/ml and stirred at 4°C for 14 days. The refolded astacin domain was then extensively dialyzed into 20 mM Hepes pH 8.0, 125 mM NaCl, 1 mM CaCl2, 10 mM ZnCl2 at 4°C. Dialyzed material was then concentrated, filtered to remove misfolded material and tested for activity using the RnD systems fluorogenic peptide (Cat#: ES007).
Astacin domain processing of fluorogenic peptide (MCA-YVADAPK(DNP)-OH)
A commercially available fluorogenic peptide from RnD systems (Cat#:ES007) was used to analyze astacin domain activity and conducted via manufacturer protocol. The fluorogenic peptide consists of an Mca fluorophore linked by a short peptide (YVADAPK) to a DNP (OH) quencher. The peptide can be cleaved by tolloid, releasing the Mca from the quencher allowing fluorescence with an excitation/emission of 320/405. In short, 250 nM of Tll1 astacin domain was mixed with 2.5 μM of the fluorogenic peptide with or without increasing amounts of bacterially produced GDF8 prodomain using the following buffer conditions: 25 mM Hepes, 0.1% Brij-35 (w/v), pH 7.5. Fluorescence was then measured continuously for 5 min following the mixing of each component. Specific activity was then calculated using the below equation and plotted as a bar graph using graph pad Prism software:
*Calibration standard MCA-P-L-OH (Bachem Catalog#: M-1975)
Production and purification of GDF8 prodomain from Escherichia coli
The production and purification of the GDF8 prodomain from E. coli was conducted as previously published [35]. In short, GDF8 prodomain mutants were cloned into a modified pET28a expression vector containing an N-terminal 6x-His-tag, a maltose-binding protein (MBP), followed by an HRV-3C protease cleavage site. The four cysteine residues within the GDF8 prodomain (C39/C41/C137/C138) were mutated to serine residues to improve solubility. E. coli Rosetta™ (DE3) cells transformed with the corresponding WT or mutant GDF8 prodomain construct were grown to an OD of 0.8 at 600 nm and induced with 0.5 mM IPTG at 20°C overnight. Cells were lysed via sonication and soluble 6xHis-MBP-GDF8 prodomain was applied to a nickel affinity column (GE Lifesciences) followed by elution with 20 mM Tris pH 7.5, 500 mM NaCl, 500 mM imidazole. The eluates were dialyzed into 20 mM Tris pH 7.5, 500 mM NaCl, 1 mM DTT, 4% ethylene glycol (v/v) and subsequently treated with HRV-3C protease. Following cleavage, the protein was dialyzed into 10 mM HCl and applied to a C4 reverse-phase column (Sepax) and eluted with a linear acetonitrile gradient (0–95% in 0.1% TFA) over 30 column volumes. Prodomain containing fractions were identified through SDS–PAGE and pooled.
Production and purification of recombinant latent WT GDF8 and mutants
Latent GDF8 featured in Figure 2 was purified from a stably transfected CHO cell line. Latent GDF8 and GDF8 mutants were transiently co-transfected with furin into expi293 cells (Life Technologies) and purified from CM. Purification from CHO CM is as follows: CM was concentrated 10-fold and dialyzed into 50 mM Tris, pH 7.4, and 500 mM NaCl and applied to Lentil Lectin Sepharose 4B (Amersham Biosciences). Elution was conducted using the same buffer with 500 mM Methyl Mannose. The procomplex was then separated using size-exclusion chromatography with a running buffer consisting of 20 mM Hepes, pH 7.4, 500 mM NaCl. Purification of GDF8 from expi293 cells was conducted using the same method as CHO CM without the initial concentration. Quantification was conducted as previously described using western blot analysis and quantified using a GDF8 mature standard under non-reduced conditions [35,53,56,59,60].
Western blot processing of the GDF8 prodomains
Western blot analysis of astacin domain processing of the GDF8 prodomain was conducted by mixing 50 ng of GDF8 prodomain, alone or in complex, with 50 ng of the astacin domain for prodomain alone experiments or 100 ng for prodomain complexes, 1.2 : 1 or 2.4 : 1 molar ratio (protease : prodomain), respectively, in 25 mM HEPES pH 7.5, 0.1% Brij-35. For the experiments in Figure 1D, 100 ng of prodomain was used with a 2.4 : 1 molar ratio of astacin : prodomain. Processing via BMP1 was conducted in the same manner as the Tll1 astacin domain. The mixture was incubated at 37°C for 1 h. For fully latent complexes overnight incubation with BMP1 and the astacin domain was needed to observe processing. SDS–PAGE gels were analyzed under non-reducing conditions and detected by western blot using the anti-GDF8 prodomain antibody, AF1539 (RnD, Lot UTG0119091). Western blot signal was measured using the C-DiGit blot scanner (Li-COR).
Acid activation
Acid activation of latent complexes was conducted as previously described [35]. In short, latent GDF8 was rapidly acidified through the addition of 100% HCl to a concentration of 1 M and incubated for 10 min; 10 M NaOH was then added to a final concentration of 1 M to neutralize the solution. The acid-activated material was then used for western blot analysis of astacin processing and luciferase assays.
Results
Recent structural studies have revealed the GDF8 procomplex adopts a different conformation relative to that of TGFβ1, giving insight into tolloid-mediated processing. Unlike latent TGFβ1, GDF8 adopts a more elongated ‘open’ conformation resembling a ‘V’ (Figure 1B) [27,28]. The tolloid cut for the GDF8 prodomain is positioned at the center of the procomplex, in a crevice adjacent to the ligand (Figure 1B,C). Using the crystal structure of the astacin domain alone, which is unbound by substrate protein, it is clear that this open conformation would be necessary for tolloid to gain access to the scissile bond (Figure 1E,F). However, the structure of the GDF8 procomplex lacks electron density for the scissile bond, including the adjacent residues spanning from V96-L106 indicating that this region is solvent exposed and likely flexible. Sequence alignments show that the residues adjacent to the D99 (P1′) cut site are highly conserved across different species (Supplementary Table S1). Thus, we wanted to investigate whether these residues were important for tolloid-mediated activation of GDF8.
Luciferase assays screening GDF8 alanine mutations
In previous studies, we established a cell-based assay where plasmids containing GDF8, furin and tolloid could be transfected into HEK293 cells and shown to produce a GDF8-mediated luciferase signal via the SMAD2/3 responsive (CAGA)12 promoter [35]. Using a similar assay format, we tested whether residues adjacent to the D99 (P1′) are important to GDF8 activity. For this, we made individual alanine mutations from D92 and D110 and measured their activity in the HEK293T (CAGA)12 GDF8-responsive transfection luciferase assay using different Tolloid family members, Tll1, Tll2, or mTLD (Figure 2A). Results of the point mutations are plotted as fold activation compared with wild type (WT) GDF8 in Figure 2B–D. For clarity, all luciferase assays within this study include a legend in the top left denoting what genes were transfected (Trx) or what proteins were added exogenously (Exo). As expected, mutating the P1′ site, D99, to an alanine completely abolishes GDF8 activity [35,37]. Interestingly, residues N-terminal to D99, including D92A, Y94A, D95A, V96A, and Q97A, significantly reduced GDF8 activity when co-transfected with Tll1, Tll2, or mTLD (Figure 2B–D). On the other hand, residues C-terminal to the cut-site had less of an impact. Four C-terminal mutants E107A, D108A, D109A, and D110A had reduced GDF8 activity, while D100A, S101A, S102A, and S105A had little effect with a modest increase when co-transfected with mTLD. Curiously, two mutants R98A and D103A had significantly increased GDF8 activity across all three tolloids. To ensure the impact of signaling was not a result of differences in expression, WT, D92A, Y94A, D95A, V96A, Q97A, and D99A DNA were transfected into HEK293T cells with furin and analyzed by an anti-GDF8 prodomain western blot (Supplementary Figure S1). These results show that loss of GDF8 signaling is not from loss of protein expression. Since Tll2 was previously shown to yield the highest activation of GDF8 compared with other tolloids [35] we also monitored the processing of each GDF8 prodomain mutation (Supplementary Figure S1). Cleavage of the prodomain by tolloid at D99 results in an 8.8 kDa N-terminal fragment and 18.9 kDa C-terminal fragment (Figure 1A). The latter fragments are detectable by Western blot analysis and can be used to monitor the cleavage of the GDF8 prodomain by tolloid. These results show that a reduction in Tll2 mediated prodomain processing occurred as noted by a reduction in the C-terminal fragment occurred for several of the point mutations, including D92A, Y94A, Q97A, and D99A when compared with WT GDF8 (Supplementary Figure S1). On the other hand, the R98A and D103A mutation generated prodomains more susceptible to tolloid processing (Supplementary Figure S1), consistent with the increase in activity observed in the transfection assay (Figure 2).
The proteolytic astacin domain is sufficient for latent GDF8 activation
The consistent results across the different tolloids for both WT and the GDF8 mutations tested suggested that the astacin domain alone might be sufficient for processing the prodomain and activating GDF8. Using previously established refolding protocols, we generated an active Tll1 astacin domain and showed it was capable of processing the commercially available Mca-YVADAPK(Dnp)-OH fluorogenic substrate with a specific activity of 35.3 ± 6.96 pmol/min/μg [58]. Using a similar assay format, we first wanted to determine if the prodomain alone could serve as a competitive substrate against the Mca-YVADAPK(Dnp)-OH fluorogenic substrate. Prodomain (ProWT) was produced in bacteria as previously published [35] and titrated against a constant concentration of the fluorogenic peptide (2.5 μM) and the astacin domain (250 nM) (Figure 3A). The addition of ProWT shows a clear reduction in fluorescence indicating that the astacin domain activity for the fluorogenic peptide is being inhibited.
Next, we wanted to determine if the astacin domain could activate latent GDF8. Previously, we were able to produce and purify latent GDF8, which has minimal activity when administered to (CAGA)12-luc cells [35,53,55,56,59]. To test this, we titrated latent GDF8 (GDF8L) with increasing amounts of the astacin domain and measured GDF8 signaling (Figure 3B). GDF8L was activated in a concentration-dependent manner by the astacin domain yielding an EC50 of 29.6 nM (C.I. 23.6–33.2 nM). This result indicates that the astacin domain is sufficient for latent GDF8 activation.
Previous studies by our laboratory and others have shown that the procomplex of GDF8, in addition to the fully latent state, can also exist in a partially active state [35]. This was shown by incubating the latent procomplex under acidic conditions to activate signaling. This is referred to as an acid-activated (GDF8AA) procomplex where the prodomain remains associated but the ligand can still signal, albeit significantly reduced to that of fully liberated GDF8. Figure 3C shows a comparison of the activity from treating both GDF8L and GDF8AA with the astacin domain. As expected, without the astacin domain GDF8AA shows an increase in activity relative to GDF8L (Figure 3C). Treatment of either GDF8 L or GDF8AA with the astacin enhances activation with the GDF8AA achieving a higher level of activity (Figure 3C). Similar results to GDF8L were observed when reconstituting the procomplex by combining excess ProWT with mature GDF8 at a 3 : 1 molar ratio (Figure 3C). Cleavage analysis by Western Blot showed that both GDF8L and GDF8AA were processed by the astacin domain. Interestingly, the processing of the procomplex was much less efficient than the prodomain alone, indicating that processing is hindered when the prodomain is bound by the GDF8 ligand (Figure 3D). See Supplementary Figure S2 for a schematic representation of different GDF8 states.
Direct comparison of astacin domain mediated processing of GDF8 prodomain mutants
We next wanted to determine if residues adjacent to the scissile bond, when mutated, had a direct impact on astacin-mediated proteolysis. GDF8 prodomain and select mutants, namely D92A, Y94A, D95A, V96A, and D99A were expressed and purified from E. coli. The extent of prodomain processing of just the prodomain alone was monitored by Western blot analysis (Figure 4A). As expected, the uncut GDF8 prodomain (ProWT) migrates at 34 kD, while incubation with the astacin domain results in fragmentation as noted by the appearance of a band ~18.9 kD. The data in Figure 4A show that all mutants showed slight to complete reduction in processing when compared with ProWT. D92A, Y94A, D95A, and V96A were processed to a much lesser extent than WT prodomain, while D99A showed no processing. We next analyzed the processing of the prodomain after reconstituting with a ligand. Due to recombinant protein availability, these experiments were conducted with GDF11 instead of GDF8. GDF11 is 90% similar to GDF8 and binds both bind to the prodomain of GDF8 with high affinity. After mixing, the prodomain ligands complex(es) were purified through size-exclusion chromatography. The reconstituted procomplexes showed reduced processing by the astacin domain, indicating that the reconstituted procomplex is more difficult to proteolyze than the prodomain alone (Figure 4B). Consistent with the prodomain alone results, mutants in the reconstituted procomplex were processed less efficiently than WT using either the astacin domain alone or (Figure 4B) full-length tolloid (BMP1) (Figure 4C,D). While similar trends were observed for both the astacin domain and BMP1, the reconstituted procomplex was less efficiently processed by BMP1. Collectively, these results demonstrate that residues N-terminal to the scissile bond directly impact astacin domain mediated processing of the prodomain, both alone and in the presence of the ligand, and that processing is dramatically impacted by the presence of the ligand.
Figure 4. Western blot analysis of GDF8 prodomain mutant processing by astacin domain and BMP1.
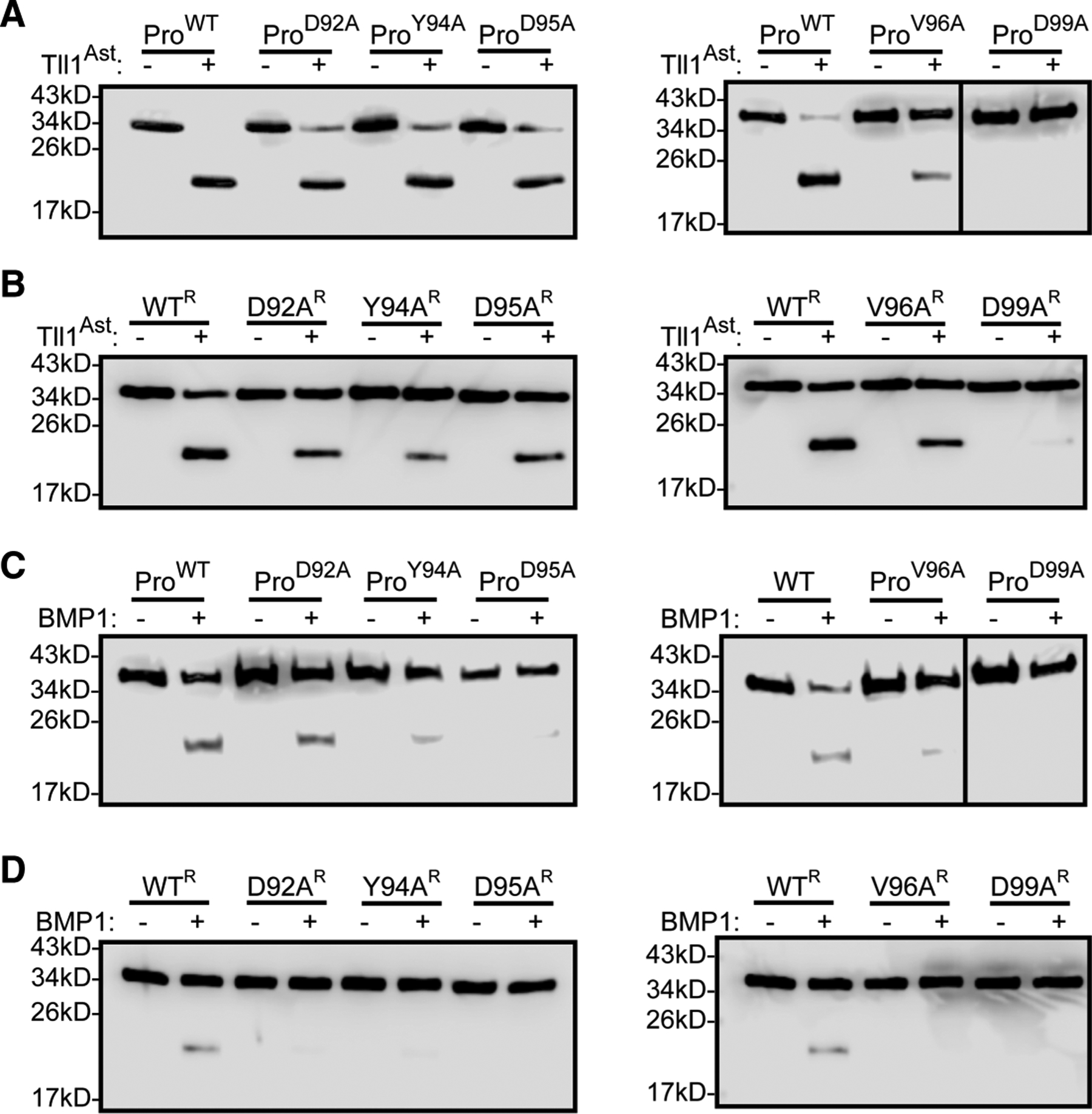
All westerns are anti-GDF8 prodomain blots (A) Bacterially produced GDF8 prodomain treated with astacin domain in a 1.2 : 1 molar ratio (astacin : prodomain) for 1 h. (B) Reconstituted complexes, of bacterially produced GDF8 prodomain and GDF11 treated with astacin domain in a 2.4 : 1 molar ratio (astacin:prodomain) for 1 h. (C) Bacterially produced GDF8 prodomain treated with BMP1 in a 1.2 : 1 molar ratio (BMP1:prodomain) for 1 h. (D) Reformed complexes of bacterially produced GDF8 prodomain and GDF11 treated with BMP1 in a 2.4 : 1 molar ratio (BMP1:prodomain) for 1 h.
Fully latent GDF8 mutant processing by tolloid
In a previous study, we showed that purified procomplex from mammalian cell culture is fully latent whereas, reconstitution of the procomplex from purified components retains significant activity [35]. Therefore, we wanted to investigate if the prodomain mutations had an impact on the fully latent GDF8 procomplex purified from CHO cells. We first tested whether exogenous delivery of the astacin domain could activate GDF8 or the corresponding mutants when transfected in HEK293 cells. Using the (CAGA)12 reporter cell line we generated EC50 curves for the titration of the astacin domain for each of the following mutants: WT, D92A, Y94A, D95A, V96A, Q97A, and D99A (Figure 5A). While WT GDF8 was effectively activated by the addition of the astacin domain (EC50 137 nM (C.I. 115–164 nM)), a significant reduction in luciferase activity was observed for the prodomain mutations, consistent with the data in Figure 2 where the tolloids were transfected. In fact, exogenous addition of increasing amounts of the astacin domain up to 1.4 μM failed to activate Y94A, Q97A, and D99A. Furthermore, significant reductions were observed for D92A (EC50 648 nM; C.I. 496–846 nM), D95A (EC50 1.97 μM; C.I. 420–530 nM) and V96A (EC50 462 nM; C.I. 164–23.7 μM), respectively. Analysis showed the differences are not a result of different levels of procomplex formed during transfection (Supplementary Figure S1).
These results promoted us to express and purify latent complexes from Expi293F and to measure their cleavage and activation by the astacin domain. As expected, the purified latent complexes had little to no activity in the (CAGA)12 reporter cell line. Titration of the astacin domain was used to activate either purified latent WT GDF8 or the mutants D92A, Y94A, and D99A (Figure 5B). The WT procomplex was readily activated by the astacin domain with an EC50 of 66.5 nM (C.I. 59–75 nM). Both Y94A and D99A were not activated by the astacin domain (up to 700 nM), whereas D92A had an EC50 of 201 nM (C.I. 191–212 nM). Furthermore, the maximum signal for D92A was approximately half that of WT. Western blot analysis also showed that each mutation was not processed by the astacin domain under conditions where the WT was readily processed (24 h at 37C in an excess of astacin) (Supplementary Figure S3).
We next tested if the exogenous latent complexes could be activated by full-length tolloids. Here, we transfected full-length Tll1, Tll2, and mTLD and added purified latent GDF8 procomplexes to the HEK293 (CAGA)12 reporter cell line (Figure 5C). WT latent complex was activated by all three tolloids, with Tll2 resulting in the strongest activation. Y94A and D99A remained latent and were not activated by the different tolloids whereas D92A was only activated by Tll2 (Figure 5C). Again, of the fully latent complexes only WT showed significant processing via western blot when using recombinant BMP1 (Tll1, Tll2, or mTLD are not yet available commercially) (Supplementary Figure S3). To rule out the possibility that lack of signaling was a result of improper folding, we acid-activated the purified latent complexes and tested their activity in (CAGA)12 cells (Figure 5D). As expected, acid-activation of WT latent procomplex resulted in robust signals. Furthermore, acid-activation of D92A, Y94A, and D99A similarly resulted in robust activation, demonstrating that the GDF8 procomplex are indeed latent and can be activated, but are resistant to tolloid-mediated activation
GDF8 mutants defective in tolloid activation can suppress WT GDF8
Since GDF8 is dimeric, we next wanted to investigate what would happen if the WT construct was co-expressed with a mutant version of the prodomain that lacked the ability to be activated by tolloid. We hypothesized that the mutant prodomain GDF8 would function in a dominant-negative manner and suppress WT signaling. This hypothesis was partly based on previous results which showed that human mutations in the furin cleavage site of GDF11 could suppress WT GDF11, giving rise to orofacial clefting [63]. For this experiment, we selected D99A and Y94A, both of which exhibit significant disruption in tolloid cleavage. In addition, we tested mutants D108A, D109A, and D110A which had a modest decrease in activity in Figure 1B,D but not to the extent of Y94A or D99A. For the analysis, we transfected WT GDF8 and mutant GDF8 DNA into HEK293T cells, individually or in a 1 : 1 ratio and measured activation of the (CAGA)12 promoter. As expected, WT GDF8 without mutant DNA was able to signal while both Y94A and D99A did not produce a signal. D108A, D109A, and D110A had approximately half the signaling of WT (Figure 6). WT GDF8 was transfected with D108A and D110A there was no significant change in signaling while transfection with D109A resulted in a significant increase. In contrast, when WT GDF8 was transfected with Y94A or D99A a ~50% reduction in signaling was observed (Figure 6). Titration of transfected Y94A demonstrated that this observed dominant-negative effect was dose dependent, where wt GDF8 signaling was abolished with 4-fold excess Y94A plasmid DNA (Supplementary Figure S4). Thus, co-expression of WT with mutations in the prodomain that are resistant to tolloid activation can suppress WT GDF8 signaling.
Discussion
Tolloids are essential for the proper maintenance of ECM components and regulate the activity of different TGFβ superfamily ligands. Despite the prevalence of tolloid activity in vivo, little is known about the molecular mechanisms that dictate substrate selection. Previous studies have shown that the non-catalytic domains of Tll1 and mTLD serve a negative regulatory role while in contrast, Tll2 non-catalytic domains were shown to enhance binding and cleavage of the BMP antagonist, chordin [45,52,61]. Examining different substrate cut sites reveals no concrete consensus sequence, outside of the regular occurrence of an aspartic acid in the P1′ site, however, tolloid cleavage is highly specific [41,50]. Thus, individual substrates require a more focused analysis to ascertain the molecular requirements for specific tolloid-mediated cleavage. In this study, we investigated the molecular requirements of tolloid-mediated cleavage of the GDF8 prodomain, and the subsequent activation from latency.
Unlike other substrates, latent GDF8 can be activated by all four tolloid family members, Tll1, Tll2, mTLD, and BMP1. Sequence alignment of 21 different species reveals that residues near the tolloid cut site (aa89–aa112) are 87% identical (Supplementary Table S1). Recent structural analysis of the GDF8 procomplex shows that this region is disordered. Given the high sequence conservation of the residues near the tolloid cut site implies a functional role, especially given that they are solvent exposed and more likely to be prone to substitution. Thus, we hypothesized that residues in the vicinity of the scissile bond are important for tolloid recognition and activation of latent GDF8. High-throughput transfection-based luciferase assays revealed that mutants made N-terminal of the tolloid cut site abolished activation by all three family members tested, Tll1, Tll2, and mTLD (Figure 2B,D). Due to the proximity of the cut site and that single point mutations were able to disrupt activation by all family members suggests that mutants likely impaired processing by the astacin domain. In support, mutations in the prodomain were less efficiently processed by the astacin domain alone. It is possible that these mutations could disrupt direct interactions with the astacin domain or possibly alter the conformation of the prodomain loop making it less prone to proteolysis. Interestingly, mutation of R98A, and to a lesser extent D103A, increased tolloid processing, resulting in enhanced activation of GDF8 signaling. This suggests these residues are not optimal for tolloid recognition. For example, comparison across tolloid substrates indicates that at the P1 position (R98), alanine and glycine have a higher occurrence frequency [50]. Interestingly, the closely related ligand, GDF11, which also undergoes a similar tolloid dependent activation, has a glycine in the P1 position. This implies that GDF8 and GDF11 might exhibit significant differences in susceptibility to tolloid-mediated activation. This indicates that residues around the tolloid consensus sequence can impact the sensitivity to tolloid processing, consistent with the high variation observed across tolloid substrates.
Given that all tolloids had similar responses to the panel of mutations, suggests that the astacin domain, which is 88% identical across the different tolloids, is the primary factor driving proteolysis. This is supported by the recent structure of the GDF8 procomplex, which shows a conformation that could accommodate astacin domain binding [27]. Thus, we asked whether the astacin domain alone was sufficient for processing the prodomain and activating latent GDF8. Using purified astacin domain from Tll1 we demonstrated that indeed the astacin domain is sufficient for not only GDF8 prodomain cleavage but latent GDF8 activation (Figure 3B,D). While the non-catalytic domains might fine-tune cleavage, they are not required for latent GDF8 recognition and activation. Interestingly, the prodomain alone was cleaved more efficiently than either the latent procomplex or the acid-activated form implying that the prodomain in complex with the ligand dampens tolloid proteolysis. Since the astacin domain can process the prodomain alone suggests that the procomplex is not required for tolloid activation, and the recognition might be more dependent on the primary sequence. However, it is also possible that the prodomain alone retains necessary structural elements for astacin recognition and cleavage even in the absence of ligand.
Using recombinant prodomains expressed and purified from E. coli. we showed that the mutants with reduced GDF8 activity also displayed a decrease in sensitivity to astacin domain proteolysis. As expected, complete loss of processing occurred when D99 (P1′ site) was altered to an alanine. Other mutations, D92A, Y94A, D95A, and V96A all showed a significant decrease in astacin domain processing when not in complex with the GDF8 ligand (Figure 4). Again, indicating that these residues directly impact the ability of the astacin domain to cleave the prodomain even in the absence of ligand.
Comparison of the astacin domain to the full-length BMP1 showed even a reduction in processing, indicating that the non-catalytic domains attenuate processing. This is similar to other studies that show BMP1 is less efficient at processing the collagen prodomain when compared with the astacin domain alone [61]. Nevertheless, prodomain mutants D92A, Y94A, D95A, and V96A still resulted in less proteolysis by BMP1 than the WT prodomain either alone or in complex with the ligand. In addition, these results consistently show that the WT or prodomain mutants alone are more readily processed than the corresponding procomplexes, and that the astacin domain is more potent than BMP1.
To further investigate the activation of the GDF8 procomplex we determined that exogenous delivery of the astacin domain could readily activate HEK293T (CAGA)12 cells transfected with WT GDF8 (Figure 5A). Consistent with previous results, transfection of Y94A, Q97A, and D99A GDF8 could not be activated by the exogenous astacin domain. Other mutants, D95A and V96A could be activated but with a higher EC50 than WT. To further characterize select prodomain mutants, D92A, Y94A, and D99A we purified recombinant versions of the fully latent complex and assessed their ability to be activated by the astacin domain. Titration of recombinant procomplexes with astacin domain resulted in GDF8 signaling activity with an EC50 similar to transfected WT GDF8. However, the astacin domain could not activate Y94A or D99A, and was significantly less effective in activating D92A. In addition, all mutant versions displayed a reduced sensitivity to proteolysis by the astacin domain. These results further support that mutations in the prodomain hindered activation by suppressing tolloid proteolysis. Most importantly, we observed a significant reduction in signaling when treating HEK293T (CAGA)12 cells with recombinant versions of the procomplexes transfected with different tolloid family members. Critically, we show that the purified mutant procomplexes can be acid-activated, indicating that a lack of signaling is not a result of improperly folded GDF8. While previous work has described the importance of D99, this is the first time that additional residues in the prodomain have been identified to render the procomplex tolloid resistant.
A current strategy for a therapeutic strategy for boosting muscle mass has been through the inhibition of tolloid-mediate activation of GDF8 [21,62]. Studies show that administering D99A alone, without the mature domain, results in muscle growth [63,64]. Recently, an antibody has been developed that binds the GDF8 procomplex and prevents prodomain cleavage by tolloid [62]. Interestingly, the antibody does not bind directly to the tolloid cut site, but on the top of the prodomain proximal to the ligand. Antibody binding appears to either inhibit tolloid-mediated processing through the exclusion of the protease or allosterically by restricting the conformation of the prodomain. Thus, it appears that the conformation of the loop that harbors the scissile bond might be sensitive to conformational disruption and is consistent with our findings that the conservation of these residues is important.
While most strategies rely on inhibiting GDF8 post-translationally, recent studies have implied that non-functional variants of GDF11 can have a dominant-negative effect on WT GDF11 [65]. Because TGFβ ligands are covalent dimers, this is likely due to the non-functional copy combining with the WT version, forming a less functional procomplex. In this study, we wanted to test if WT GDF8 could be inhibited when co-expressed with a tolloid-resistant GDF8. Indeed, our results show that co-transfection of the GDF8 prodomain mutations that are tolloid resistant with WT GDF8 suppressed GDF8 signaling activity. This might provide another option for suppressing GDF8 in vivo which could target endogenously produced GDF8 before secretion.
Prior to this study only D99 of the GDF8 prodomain was known to be required for tolloid-mediated activation. Since a tolloid consensus sequence is not known we investigated the conserved residues near the scissile bond and identified additional residues important for proteolysis and activation. We show that the astacin domain alone is sufficient for processing the prodomain and activating GDF8 procomplex. Further evidence suggests that the prodomain processing is attenuated when bound to the ligand and, further, that the astacin domain is more effective in activating GDF8 procomplex than the full-length tolloids. Due to the widespread roles of tolloids in ECM maintenance, wound healing, and controlling the activity of other TGFβ ligands, it is important to understand how individual substrates are recognized and processed by the tolloid family [34,42,66]. Collectively, this study provides insight into the specific mechanism of GDF8 activation by the tolloid family of proteases.
Supplementary Material
Acknowledgements
We would like to thank Kierra Ware for help with the production and purification of the prodomain mutant V96A from E. coli cells.
Funding
Funding was provided by the National Institute of General Medical Sciences (NIGMS) R01 GM114640 and R35 GM134923 to T.B.T. and AHA 19PRE33990312 to J.C.M.
Abbreviations
- AA
acid activated
- Ast
astacin
- ECM
extracellular matrix
- GDF
growth differentiation factor
- mTLD
mammalian tolloid
- TGF
transforming growth factor
- Tll1
Tolloid like 1
- Tll2
Tolloid like 2
Footnotes
Conflicts of Interest
Thomas B. Thompson is a consultant for Acceleron Pharma.
CRediT Author Contribution
Thomas B. Thompson: Conceptualization, resources, data curation, supervision, funding acquisition, project administration, writing — review and editing. Jason C. McCoy: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing — original draft, writing — review and editing. Erich J. Goebel: Data curation, investigation, writing — review and editing.
Data Availability
All data generated during this study are included in this article and the corresponding supplementary file.
References
- 1.McPherron AC and Lee S-J (1997) Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl Acad. Sci. U.S.A 94, 12457–12461 10.1073/pnas.94.23.12457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee SJ and McPherron AC (2001) Regulation of myostatin activity and muscle growth. Proc. Natl Acad. Sci. U.S.A 98, 9306–9311 10.1073/pnas.151270098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McPherron AC, Lawler AM and Lee S-J (1997) Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387, 83–90 10.1038/387083a0 [DOI] [PubMed] [Google Scholar]
- 4.Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J et al. (2000) Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem 275, 40235–40243 10.1074/jbc.M004356200 [DOI] [PubMed] [Google Scholar]
- 5.Lee S-J (2010) Extracellular regulation of myostatin: a molecular rheostat for muscle mass. Immunol. Endocr. Metab Agents Med. Chem 10, 183–194 10.2174/187152210793663748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y et al. (2008) Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 22, 477–487 10.1096/fj.07-8673com [DOI] [PubMed] [Google Scholar]
- 7.Parenté A, Boukredine A, Baraige F, Duprat N, Gondran-Tellier V, Magnol L et al. (2020) GASP-2 overexpressing mice exhibit a hypermuscular phenotype with contrasting molecular effects compared to GASP-1 transgenics. FASEB J. 34, 4026–4040 10.1096/fj.201901220R [DOI] [PubMed] [Google Scholar]
- 8.Puolakkainen T, Ma H, Kainulainen H, Pasternack A, Rantalainen T, Ritvos O et al. (2017) Treatment with soluble activin type IIB-receptor improves bone mass and strength in a mouse model of Duchenne muscular dystrophy. BMC Musculoskelet. Disord 18, 20 10.1186/s12891-016-1366-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee S-J, Lee Y-S, Zimmers TA, Soleimani A, Matzuk MM, Tsuchida K et al. (2010) Regulation of muscle mass by follistatin and activins. Mol. Endocrinol 24, 1998–2008 10.1210/me.2010-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monestier O, Brun C, Heu K, Passet B, Malhouroux M, Magnol L et al. (2012) Ubiquitous Gasp1 overexpression in mice leads mainly to a hypermuscular phenotype. BMC Genomics 13, 541 10.1186/1471-2164-13-541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funkenstein B and Rebhan Y (2007) Expression, purification, renaturation and activation of fish myostatin expressed in Escherichia coli: facilitation of refolding and activity inhibition by myostatin prodomain. Protein Expr. Purif 54, 54–65 10.1016/j.pep.2007.02.002 [DOI] [PubMed] [Google Scholar]
- 12.Sakuma K, Aoi W and Yamaguchi A (2017) Molecular mechanism of sarcopenia and cachexia: recent research advances. Pflug. Arch. Eur. J. Physiol 469, 573–591 10.1007/s00424-016-1933-3 [DOI] [PubMed] [Google Scholar]
- 13.Costelli P, Muscaritoli M, Bonetto A, Penna F, Reffo P, Bossola M et al. (2008) Muscle myostatin signalling is enhanced in experimental cancer cachexia. Eur. J. Clin. Invest 38, 531–538 10.1111/j.1365-2362.2008.01970.x [DOI] [PubMed] [Google Scholar]
- 14.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN et al. (2002) Induction of cachexia in mice by systemically administered myostatin. Science 296, 1486–1488 10.1126/science.1069525 [DOI] [PubMed] [Google Scholar]
- 15.Singh P, Rong H, Gordi T, Bosley J and Bhattacharya I (2016) Translational pharmacokinetic/pharmacodynamic analysis of MYO-029 antibody for muscular dystrophy. Clin. Transl. Sci 9, 302–310 10.1111/cts.12420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC and Lach-Trifilieff E (2016) ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet Muscle 6, 26 10.1186/s13395-016-0098-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendell JR, Sahenk Z, Malik V, Gomez AM, Flanigan KM, Lowes LP et al. (2015) A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther 23, 192–201 10.1038/mt.2014.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuchida K (2008) Targeting myostatin for therapies against muscle-wasting disorders. Curr. Opin. Drug. Discov. Devel 11, 487–494 [PubMed] [Google Scholar]
- 19.Smith RC and Lin BK (2013) Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr. Opin. Support. Palliat. Care 7, 352–360 10.1097/SPC.0000000000000013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JL, Walton KL, Al-Musawi SL, Kelly EK, Qian H, La M et al. (2015) Development of novel activin-targeted therapeutics. Mol. Ther 23, 434–444 10.1038/mt.2014.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pirruccello-Straub M, Jackson J, Wawersik S, Webster MT, Salta L, Long K et al. (2018) Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Sci. Rep 8, 2292 10.1038/s41598-018-20524-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrison CA, Al-Musawi SL and Walton KL (2011) Prodomains regulate the synthesis, extracellular localisation and activity of TGF-β superfamily ligands. Growth Factors 29, 174–186 10.3109/08977194.2011.608666 [DOI] [PubMed] [Google Scholar]
- 23.Constam DB (2014) Regulation of TGFβ and related signals by precursor processing. Semin. Cell Dev. Biol 32, 85–97 10.1016/j.semcdb.2014.01.008 [DOI] [PubMed] [Google Scholar]
- 24.Sengle G, Ono RN, Sasaki T and Sakai LY (2011) Prodomains of transforming growth factor β (TGFβ) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem 286, 5087–5099 10.1074/jbc.M110.188615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goebel EJ, Hart KN, McCoy JC and Thompson TB (2019) Structural biology of the TGFβ family. Exp. Biol. Med 244, 1530–1546 10.1177/1535370219880894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinck AP, Mueller TD and Springer TA (2016) Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol 8, 1–52 10.1101/cshperspect.a022103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cotton TR, Fischer G, Wang X, McCoy JC, Czepnik M, Thompson TB et al. (2018) Structure of the human myostatin precursor and determinants of growth factor latency. EMBO J. 37, 367–383 10.15252/embj.201797883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T et al. (2011) Latent TGF-β structure and activation. Nature 474, 343–349 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stockis J, Dedobbeleer O and Lucas S (2017) Role of GARP in the activation of latent TGF-β1. Mol. BioSyst 13, 1925–1935 10.1039/C7MB00251C [DOI] [PubMed] [Google Scholar]
- 30.Wang R, Zhu J, Dong X, Shi M, Lu C and Springer TA (2012) GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell 23, 1129–1139 10.1091/mbc.e11-12-1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C et al. (2017) Force interacts with macromolecular structure in activation of TGF-β. Nature 542, 55–59 10.1038/nature21035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarrazy V, Koehler A, Chow ML, Zimina E, Li CX, Kato H et al. (2014) Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res 102, 407–417 10.1093/cvr/cvu053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge G, Hopkins DR, Ho W-B and Greenspan DS (2005) GDF11 forms a bone morphogenetic protein 1-activated latent complex that can modulate nerve growth factor-induced differentiation of PC12 cells. Mol. Cell. Biol 25, 5846–5858 10.1128/MCB.25.14.5846-5858.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ge G and Greenspan DS (2006) Developmental roles of the BMP1/TLD metalloproteinases. Birth Defects Res. C - Embryo Today 78, 47–68 10.1002/bdrc.20060 [DOI] [PubMed] [Google Scholar]
- 35.Walker RG, McCoy JC, Czepnik M, Mills MJ, Hagg A, Walton KL et al. (2018) Molecular characterization of latent GDF8 reveals mechanisms of activation. Proc. Natl Acad. Sci. U.S.A 115, E866–E875 10.1073/pnas.1714622115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le VQ, Iacob RE, Tian Y, McConaughy W, Jackson J, Su Y et al. (2018) Tolloid cleavage activates latent GDF8 by priming the pro-complex for dissociation. EMBO J. 37, 384–397 10.15252/embj.201797931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolfman NM, McPherron AC, Pappano WN, Davies MV, Song K, Tomkinson KN et al. (2003) Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc. Natl Acad. Sci. U.S.A 100, 15842–15846 10.1073/pnas.2534946100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott IC, Blitz IL, Pappano WN, Imamura Y, Clark TG, Steiglitz BM et al. (1999) Mammalian BMP-1/Tolloid-related metalloproteinases, including novel family member mammalian Tolloid-like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev. Biol 213, 283–300 10.1006/dbio.1999.9383 [DOI] [PubMed] [Google Scholar]
- 39.Goff SV-L, Hulmes DJS and Moali C (2015) BMP-1/tolloid-like proteinases synchronize matrix assembly with growth factor activation to promote morphogenesis and tissue remodeling. Matrix Biol. 44–46, 14–23 10.1016/j.matbio.2015.02.006 [DOI] [PubMed] [Google Scholar]
- 40.Petropoulou V, Garrigue-Antar L and Kadleri KE (2005) Identification of the minimal domain structure of bone morphogenetic protein-1 (BMP-1) for chordinase activity: chordinase activity is not enhanced by procollagen C-proteinase enhancer-1 (PCPE-1). J. Biol. Chem 280, 22616–22623 10.1074/jbc.M413468200 [DOI] [PubMed] [Google Scholar]
- 41.Yang RT, Lim GL, Yee CT, Fuller RS and Ritchie HH (2014) Site specificity of DSP-PP cleavage by BMP1. Connective Tissue Res. 55, 142–145 10.3109/03008207.2014.923863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HX, Mendes FA, Plouhinec JL and De Robertis EM (2009) Enzymatic regulation of pattern: BMP4 binds CUB domains of tolloids and inhibits proteinase activity. Genes Dev. 23, 2551–2562 10.1101/gad.1839309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ploper D, Lee HX and De robertis EM (2011) Dorsal-ventral patterning: crescent is a dorsally secreted Frizzled-related protein that competitively inhibits tolloid proteases. Dev. Biol 352, 317–328 10.1016/j.ydbio.2011.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Canty EG Garrigue-Antar L and Kadler KE (2006) A complete domain structure of Drosophila tolloid is required for cleavage of short gastrulation. J. Biol. Chem 281, 13258–13267 10.1074/jbc.M510483200 [DOI] [PubMed] [Google Scholar]
- 45.Berry R, Jowitt TA, Garrigue-Antar L, Kadler KE and Baldock C (2010) Structural and functional evidence for a substrate exclusion mechanism in mammalian tolloid like-1 (TLL-1) proteinase. FEBS Lett. 584, 657–661 10.1016/j.febslet.2009.12.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muir AM, Ren Y, Butz DH, Davis NA, Blank RD, Birk DE et al. (2014) Induced ablation of Bmp1 and Tll1 produces osteogenesis imperfecta in mice. Hum. Mol. Genet 23, 3085–3101 10.1093/hmg/ddu013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pappano WN, Steiglitz BM, Scott IC, Keene DR and Greenspan DS (2003) Use of Bmp1/Tll1 doubly homozygous null mice and proteomics to identify and validate in vivo substrates of bone morphogenetic protein 1/tolloid-like metalloproteinases. Mol. Cell. Biol 23, 4428–4438 10.1128/MCB.23.13.4428-4438.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark TG, Conway SJ, Scott IC, Labosky PA, Winnier G, Bundy J et al. (1999) The mammalian Tolloid-like 1 gene, Tll1, is necessary for normal septation and positioning of the heart. Development 126, 2631–2642 [DOI] [PubMed] [Google Scholar]
- 49.Suzuki N, Labosky PA, Furuta Y, Hargett L, Dunn R, Fogo AB et al. (1996) Failure of ventral body wall closure in mouse embryos lacking a procollagen C-proteinase encoded by Bmp1, a mammalian gene related to Drosophila tolloid. Development 122, 3587–3595 [DOI] [PubMed] [Google Scholar]
- 50.Hopkins DR, Keles S and Greenspan DS (2007) The bone morphogenetic protein 1/Tolloid-like metalloproteinases. Matrix Biol. 26, 508–523 10.1016/j.matbio.2007.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bayley CP, Ruiz Nivia HD, Dajani R, Jowitt TA, Collins RF, Rada H et al. (2016) Diversity between mammalian tolloid proteinases: oligomerisation and non-catalytic domains influence activity and specificity. Sci. Rep 6, 21456 10.1038/srep21456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berry R, Jowitt TA, Ferrand J, Roessle M, Grossmann JG, Canty-Laird EG et al. (2009) Role of dimerization and substrate exclusion in the regulation of bone morphogenetic protein-1 and mammalian tolloid. Proc. Natl Acad. Sci. U.S.A 106, 8561–8566 10.1073/pnas.0812178106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cash JN, Angerman EB, Kattamuri C, Nolan K, Zhao H, Sidis Y et al. (2012) Structure of myostatin·follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J. Biol. Chem 287, 1043–1053 10.1074/jbc.M111.270801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cash JN, Angerman EB, Kirby RJ, Merck L, Seibel WL, Wortman MD et al. (2013) Development of a small-molecule screening method for inhibitors of cellular response to myostatin and activin A. J. Biomol. Screen 18, 837–844 10.1177/1087057113482585 [DOI] [PubMed] [Google Scholar]
- 55.Walker RG, Angerman EB, Kattamuri C, Lee YS, Lee SJ and Thompson TB (2015) Alternative binding modes identified for growth and differentiation factor-associated serum protein (GASP) family antagonism of myostatin. J. Biol. Chem 290, 7506–7516 10.1074/jbc.M114.624130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walker RG, Czepnik M, Goebel EJ, McCoy JC, Vujic A, Cho M et al. (2017) Structural basis for potency differences between GDF8 and GDF11. BMC Biol. 15, 19 10.1186/s12915-017-0350-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCoy JC, Walker RG, Murray NH and Thompson TB (2019) Crystal structure of the WFIKKN2 follistatin domain reveals insight into how it inhibits growth differentiation factor 8 (GDF8) and GDF11. J. Biol. Chem 294, 6333–6343 10.1074/jbc.RA118.005831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sweeney AM, Gil-Parrado S, Vinzenz D, Bernardi A, Hein A, Bodendorf U et al. (2008) Structural basis for the substrate specificity of bone morphogenetic protein 1/Tolloid-like metalloproteases. J. Mol. Biol 384, 228–239 10.1016/j.jmb.2008.09.029 [DOI] [PubMed] [Google Scholar]
- 59.Cash JN, Rejon CA, McPherron AC, Bernard DJ and Thompson TB (2009) The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J. 28, 2662–2676 10.1038/emboj.2009.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee Y-S and Lee S-J (2013) Regulation of GDF-11 and myostatin activity by GASP-1 and GASP-2. Proc. Natl Acad. Sci. U.S.A 110, E3713–E3722 10.1073/pnas.1309907110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wermter C, Höwel M, Hintze V, Bombosch B, Aufenvenne K, Yiallouros I et al. (2007) The protease domain of procollagen C-proteinase (BMP1) lacks substrate selectivity, which is conferred by non-proteolytic domains. Biol. Chem 388, 513–521 10.1515/BC.2007.054 [DOI] [PubMed] [Google Scholar]
- 62.Dagbay KB, Treece E, Streich FC, Jackson JW, Faucette RR, Nikiforov A et al. (2020) Structural basis of specific inhibition of extracellular activation of pro- or latent myostatin by the monoclonal antibody SRK-015. J. Biol. Chem 295, 5404–5418 10.1074/jbc.RA119.012293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morine KJ, Bish LT, Pendrak K, Sleeper MM, Barton ER and Sweeney HL (2010) Systemic myostatin inhibition via liver-targeted gene transfer in normal and dystrophic mice. PLoS One 5, e9176 10.1371/journal.pone.0009176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen C, Hu S, Sheng J, Sun Y, Cao X and Qiao J (2010) Enhanced muscle growth by plasmid-mediated delivery of myostatin propeptide. J. Biomed. Biotechnol 2010, 862591 10.1155/2010/862591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cox TC, Lidral AC, McCoy JC, Liu H, Cox LL, Zhu Y et al. (2019) Mutations in GDF11 and the extracellular antagonist, Follistatin, as a likely cause of Mendelian forms of orofacial clefting in humans. Hum. Mutat 40, 1813–1825 10.1002/humu.23793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muir AM, Massoudi D, Nguyen N, Keene DR, Lee SJ, Birk DE et al. (2016) BMP1-like proteinases are essential to the structure and wound healing of skin. Matrix Biol. 56, 114–131 10.1016/j.matbio.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated during this study are included in this article and the corresponding supplementary file.