

Abstract
Objective
Paroxysmal epileptiform abnormalities on electroencephalography (EEG) are the hallmark of epilepsies, but it is uncertain to what extent epilepsy and background EEG oscillations share neurobiological underpinnings. Here, we aimed to assess the genetic correlation between epilepsy and background EEG oscillations.
Methods
Confounding factors, including the heterogeneous etiology of epilepsies and medication effects, hamper studies on background brain activity in people with epilepsy. To overcome this limitation, we compared genetic data from a genome‐wide association study (GWAS) on epilepsy (n = 12 803 people with epilepsy and 24 218 controls) with that from a GWAS on background EEG (n = 8425 subjects without epilepsy), in which background EEG oscillation power was quantified in four different frequency bands: alpha, beta, delta, and theta. We replicated our findings in an independent epilepsy replication dataset (n = 4851 people with epilepsy and 20 428 controls). To assess the genetic overlap between these phenotypes, we performed genetic correlation analyses using linkage disequilibrium score regression, polygenic risk scores, and Mendelian randomization analyses.
Results
Our analyses show strong genetic correlations of genetic generalized epilepsy (GGE) with background EEG oscillations, primarily in the beta frequency band. Furthermore, we show that subjects with higher beta and theta polygenic risk scores have a significantly higher risk of having generalized epilepsy. Mendelian randomization analyses suggest a causal effect of GGE genetic liability on beta oscillations.
Significance
Our results point to shared biological mechanisms underlying background EEG oscillations and the susceptibility for GGE, opening avenues to investigate the clinical utility of background EEG oscillations in the diagnostic workup of epilepsy.
Keywords: beta power, EEG, generalized epilepsy, GGE, oscillations, PRS
Key Points.
Genetic correlation studies show shared genetic underpinnings between GGE and power of background oscillations in the beta frequency band
Polygenic risk score analyses show that subjects with more beta power‐associated genetic variants have an increased risk of having GGE
Mendelian randomization analyses suggest a causal effect of GGE genetic liability on beta oscillations.
1. INTRODUCTION
The power of oscillations in background electroencephalogram (EEG) is a highly stable and heritable human trait. 1 It is easily acquired and can be automatically analyzed by software, rather than subjective interpretation. Epilepsy is highly heritable and is characterized by altered brain excitability. 2 , 3 Oscillatory activity is believed to serve an essential role in corticothalamic functioning, and can be measured as power of oscillations in background EEG at different broadband frequencies. 4 Neurophysiological relationships between background EEG and generalized epileptiform discharges have been well described. 5 , 6 , 7 , 8 However, it is currently unknown whether background oscillatory activity is itself associated with epilepsy, and whether background EEG and epilepsy have a shared neurobiological and genetic basis.
There have been some studies where background EEG oscillation measurements have been directly compared between people with epilepsy and healthy controls. However, such studies have yielded conflicting results, most likely because sample sizes were small and antiseizure drugs can strongly affect EEG measurements. 9 , 10 , 11 , 12 , 13 , 14 These limitations and bias can be overcome by large‐scale genetic studies, in which genetic determinants of background EEG measurements are assessed independently in healthy controls (presumably not taking antiseizure drugs). These genetic determinants can then be compared to genetic determinants of different epilepsy phenotypes, as assessed in a different study. Comparing these independent studies allows for a well‐powered and unbiased assessment of shared genetic determinants of epilepsy and EEG oscillations.
Here, we therefore assessed whether oscillatory background EEG is genetically correlated with focal and generalized epilepsy. The association between genetic variants and background brain activity was previously investigated in a genome‐wide association study (GWAS) on 8425 subjects without epilepsy. 15 We combined these data with our recently published large GWAS of epilepsy, 16 to examine genetic correlations between several types of epilepsy and oscillatory brain activity across frequency bands (delta, 1–3.75 Hz; theta, 4–7.75 Hz; alpha, 8‐12.75 Hz; and beta, 13–30 Hz). Next, we utilized polygenic risk scoring (PRS) to assess whether people with GGE have a genetic predisposition toward altered background brain activity. We then replicated genetic correlation and polygenic analyses using an independent cohort from the Epi25 Collaborative (n = 4851 people with epilepsy and 20 428 controls). Finally, we performed Mendelian randomization (MR) to gain insight into possible causal relationships between genetic variants associated with epilepsy and those associated with background EEG. We thus provide converging evidence for consistent cross‐trait genetic overlap between epilepsy and background EEG.
2. MATERIALS AND METHODS
2.1. Study population: Discovery dataset
The participants derived from the epilepsy GWAS 16 for the current analyses were Caucasian subjects. The epilepsy GWAS included 13 control cohorts. 16 Case/control ascertainment and diagnostic criteria were previously reported. 16 As described previously, 16 epilepsy specialists diagnosed people with epilepsy and ascertained phenotypic subtypes. Population‐based datasets, some of which had been screened to exclude neurological disorders, were used as controls. However, due to the relatively low prevalence of epilepsy in the general population (~0.5–1%), screening to exclude epilepsy in control cohorts will have only a minor effect on statistical power. Summary statistics from the recent epilepsy GWAS conducted by the International League Against Epilepsy (ILAE) Consortium on Complex Epilepsies GWAS were available for n = 12 803 cases (with either focal or generalized epilepsy) and 24 218 controls. 16 From those participants, the following subjects were excluded for those analyses requiring individual‐level genotype data: Finnish ancestry (none had genetic generalized epilepsy [GGE]) and the subset of the EPICURE‐SP1 cohort that lacked informed consent for the current analyses, resulting in subject‐level genotype data being available for 11 446 people with epilepsy and 22 078 controls. Subjects with epilepsy were stratified into GGE (n = 3122) and focal epilepsy (n = 8324); GGE was further subdivided into childhood absence epilepsy (CAE; n = 561), juvenile absence epilepsy (JAE; n = 311), juvenile myoclonic epilepsy (JME; n = 1000), and generalized tonic–clonic seizures only (GTCS only; n = 195). GGE subtype information was not available for 1055 people with epilepsy.
We downloaded summary statistics of the ENIGMA‐EEG GWAS of resting state oscillation power in the delta (1–3.75 Hz), theta (4–7.75 Hz), alpha (8–12.75 Hz), and beta (13–30 Hz) bands at the vertex (Cz) electrode (n = 8425 participants). 15 This EEG GWAS was based on five cohorts from four cooperating centers. Although the selection criteria varied across cohorts, all adult cohorts included epilepsy and prolonged unconsciousness after head trauma as exclusion criteria, which were communicated at the time of recruitment or at the first laboratory visit; because neurological disorders were an exclusion criterium, we do not expect subjects to be taking antiseizure drugs (although this was no explicit exclusion criterion). All these were self‐ or parent‐reported retrospective questions. A full sample description and recording specifics are available in the supplement of the original study, 15 and the EEG analysis protocol is available online at http://enigma.ini.usc.edu/ongoing/enigma‐eeg‐working‐group/. In brief, eyes‐closed resting EEG was recorded or offline rereferenced to averaged earlobes, visually cleaned with standard criteria by local expert EEG analysts with rogue channels removed, and scanned for sleep transition (eye rolling, alpha dropout). Eye movement was removed using regression or independent component analysis. A minimum of 1 min of recording was required.
Approval for the source studies was obtained by all relevant institutional review boards, and all study participants provided written informed consent according to the Declaration of Helsinki.
2.2. Replication dataset
To replicate our findings, we used data from the Epi25 Collaborative (http://epi‐25.org/). This cohort currently comprises 4851 people with epilepsy, of whom 2612 have focal epilepsy and 2239 have GGE (no data on GGE subtypes were available). The cases were matched to a total of 20 428 controls from the Partners Healthcare Biobank (n = 14 857), the Epi25 Collaborative (n = 210), the Genetics and Personality consortium (n = 456), and an in‐house project on inflammatory bowel disease (n = 4905). The cohorts were genotyped on the Illumina Global Screening Array, with the exception of the Partners Healthcare Biobank participants, who were genotyped on the Illumina Multi‐Ethnic Screening Array. Approval was obtained by all relevant institutional review boards, and all study participants provided written informed consent according to the Declaration of Helsinki.
2.3. Genetic correlation analyses
Genetic correlations between epilepsy subtypes and oscillatory brain activity were computed using bivariate linkage disequilibrium score regression (LDSC). 17 For these analyses, as no individual‐level genotype data were available from the EEG dataset, we used published summary statistics of the EEG frequency bands (alpha, beta, delta, and theta; n = 8425 participants) and the epilepsy subtypes (focal, GGE, CAE, JAE, JME, and GTCS only; n = 12 803 cases suffering from either focal or generalized epilepsy and 24 218 controls) from the ILAE consortium as a discovery dataset. 16 For LDSC replication analyses, we used unpublished data from the Epi25 Collaborative (http://epi‐25.org/; n = 4851 people with epilepsy and 20 428 controls). For discovery and replication LDSC analyses, default settings of LDSC were used, with precomputed linkage disequilibrium (LD) score weights derived from the European subset of the 1000 Genomes project. 18 See Table S1 for the number of single nucleotide polymorphisms (SNPs) per LDSC analysis. The significance threshold was Bonferroni‐corrected for the two main epilepsy subtypes studied (GGE and focal) but not for the EEG power spectra, because these were all highly correlated at p < 10−17 (Table S2), resulting in a significance threshold of p = .05/2 = .025. Similarly, we did not correct for the individual GGE subtypes, which are phenotypically similar and genetically highly correlated. 16
2.4. PRS analyses
For PRS analyses, we used individual‐level genotype data derived from the epilepsy GWAS 16 and summary statistics from the EEG GWAS. 15 Quality control was performed as reported in the published epilepsy GWAS. 16 We then added a genotype filter for call rate greater than .99 and the exclusion of genetically related subjects to allow for highly conservative PRS estimates. Genetic interrelatedness was calculated with KING, 19 and one subject from each pair with third‐degree or higher relatedness (kinship coefficient > .0442) was excluded. PRSice 20 was used with default settings to assess whether subjects with epilepsy had different EEG frequency power PRSs compared to controls. In brief, to each SNP we assigned a weight proportional to its association in the four EEG GWASs (alpha, beta, delta, and theta). Next, individual PRSs were calculated as the sum of weighted effect alleles for every subject from the epilepsy cohort. These PRSs were standardized with a Z‐score transformation . SNPs were pruned to a subset of genetically uncorrelated SNPs (LD R 2 < .1), and PRS values were calculated using a number of different p‐value thresholds from .0001 to .5. Next, logistic regression analyses, corrected for sex and 10 genetic ancestry principal components (PCs), were performed to assess the association of these PRS scores with GGE. The PRS with the highest association with GGE was chosen as the "best fit," after which logistic regression analyses were repeated to assess the association of this PRS with the other epilepsy subtypes. We used a conservative p < .001 significance threshold to correct for multiple comparisons, as recommended for PRSice. 20 Explained variance represented by the Nagelkerke R 2 was computed using a logistic regression of the PRS, subtracted from the baseline model (covariates only: sex and four PCs). To quantify the association of beta power PRS with GGE, we used PRSice standard settings to divide subjects into 10 deciles based on their beta power PRS scores. We then performed logistic regression to compare the risk of having GGE between every decile, with the lowest (0%–10%) as a reference (corrected for sex and four PCs). We then repeated the analyses in the independent Epi25 cohort. This dataset contained approximately one third fewer GGE cases than the discovery cohort, providing insufficient power to exactly replicate our discovery PRS findings. We therefore performed quasireplication using a one‐sample test of the proportion to assess concordance effect directions between discovery and replication PRS analyses, computing Z‐scores that were converted into p‐values:
where the p = the sample proportion; H0 represents the null hypothesis: p = p 0; and the alternative hypothesis H1 is p ≠ p0.
2.5. Mendelian randomization
Two major limitations of observational studies and other types of studies are unmeasured confounding and uncertainties about cause and effect. MR has the potential to overcome these limitations, as MR leverages genetic instruments (most often SNPs) as exposures as well as outcomes. Because SNPs are not influenced by state‐dependent factors, MR has the potential to shed light on potential causal mechanisms between two traits; SNPs strongly associated with two or more traits index these traits without confounding. MR can be done in two directions for two given traits, with each MR analysis testing whether one trait has a potential effect on the other. However, here, we could only conduct one‐way MR due to lack of genome‐wide significant loci in the EEG GWAS. Several MR techniques are available, and the consensus is that results from different approaches show robustness and consistency of results across methods.
To explore possible causal effects of GGE genome‐wide loci (exposure) on EEG background oscillations (outcome), we thus conducted MR analyses using GGE and EEG summary statistics data. Two hundred twenty‐eight SNPs significantly associated with GGE (p < 5 × 10−8) were extracted from both the GGE and EEG GWASs. The summary statistics of 228 SNPs were harmonized to ensure the SNP effect direction corresponded with equal effect alleles across GGE and EEG. We used the “TwosampleMR” package 21 in R to perform fixed effects inverse variance‐weighted (IVW), weighted median, and MR Egger models. We then performed sensitivity analyses, including horizontal pleiotropic effects estimated by the intercept of MR Egger, residual heterogeneity due to pleiotropy estimated by Cochran Q test, 22 and leave‐one‐out analyses (for the fixed effects IVW model), to evaluate whether any single instrumental variable was driving the results. Generalized summary data‐based MR (GSMR) analyses were performed using the “GSMR” 23 package in R. To that end, first the LD matrix of the selected SNPs was calculated using PLINK 24 and GCTA 25 within 1000 Genomes Phase 3 data. 18 The minimum number of instrumental variables in the GSMR model was loosened from 10 to five as there were only eight independent (r 2 < .01, LD window = 10 Mb) significant loci identified in the GWAS of GGE (and none in the EEG GWAS). We used default options in GSMR with heterogeneity in dependent instruments (HEIDI) testing for instrumental outliers’ detection. At the end, we repeated GSMR with loosened LD prune thresholds (i.e., r 2 < .1, r 2 < .15, and r 2 < .2), because GSMR takes LD structure into account by adding the LD matrix. The significance threshold was Bonferroni corrected for all seven of these MR models (p = .05/7 = .007).
2.6. Data availability
GWAS summary statistics used for the current analyses are available online: http://enigma.ini.usc.edu/research/download‐enigma‐gwas‐results/; http://www.epigad.org/gwas_ilae2018_16loci.html.
3. RESULTS
3.1. Genetic correlations between epilepsy and oscillatory brain activity
In a total study population of 45 446 subjects (n = 8425 from the EEG and n = 37 021 from the epilepsy GWASs), we computed genetic correlations (Rg) of alpha, beta, delta, and theta oscillatory brain activity with focal epilepsy and GGE. We found significant correlations between GGE and beta power (Rg = 0.44 ± SE of .18, p = .01) and theta power (Rg = 0.25 ± 0.11, p = .02; Figure 1, upper panel, Table S1). This was further supported by the correlations between beta power and theta power with the GGE subtypes CAE, JAE, and JME; all had similarly high correlation coefficients. We found no genetic correlations between focal epilepsy and any of the EEG phenotypes. We then attempted to replicate the genetic correlations using the unpublished Epi25 dataset and found genetic correlations similar (in both sign and effect size) to the discovery analyses (Figure 1, lower panel); GGE correlated with beta power (Rg = 0.52 ± 0.21, p = .01), whereas the genetic correlation between theta power and GGE paralleled the discovery cohort (albeit not reaching significance: Rg = 0.16 ± 0.12, p = .18). All genetic correlation estimates with focal epilepsy were again nonsignificant. There were no data available for GGE subtypes.
FIGURE 1.
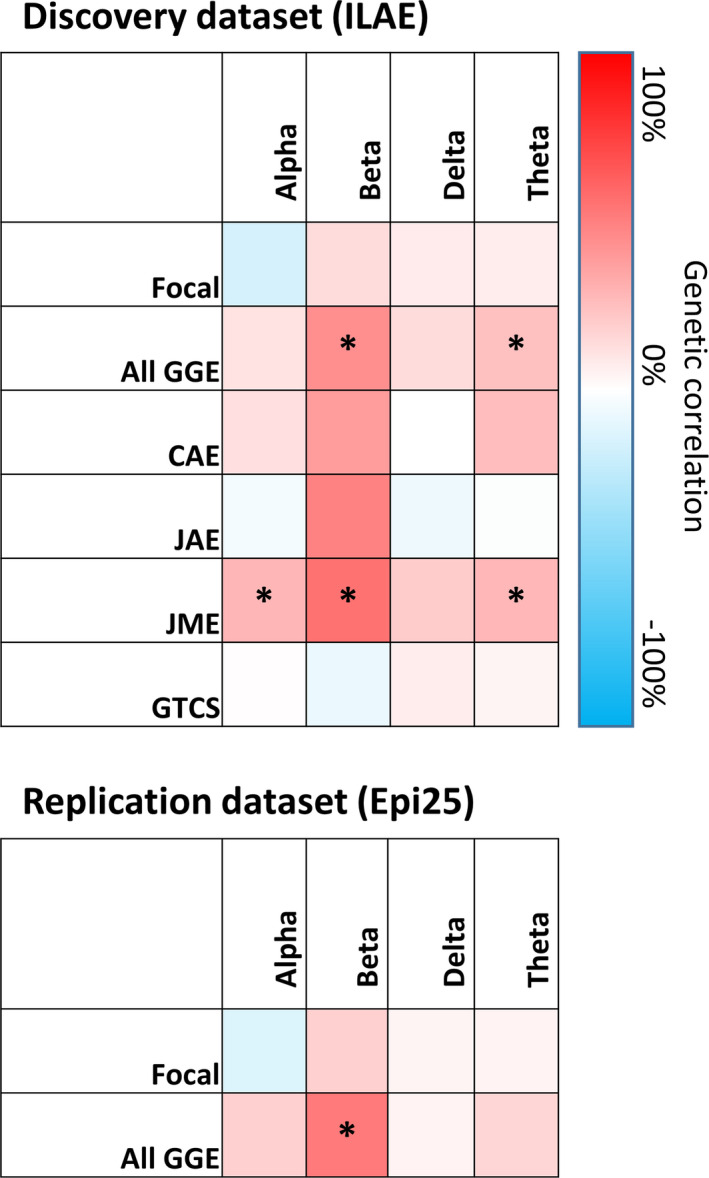
Genetic correlations between electroencephalographic (EEG) frequency bands and epilepsy subtypes. Genetic correlations were calculated by comparing the EEG frequency band genome‐wide association study (GWAS) with the International League Against Epilepsy (ILAE) GWAS (upper panel, discovery dataset) and the Epi25 GWAS (lower panel, replication dataset). * p < .05. CAE, childhood absence epilepsy; GGE, genetic generalized epilepsy; GTCS, generalized tonic–clonic seizures; JAE, juvenile absence epilepsy; JME, juvenile myoclonic epilepsy
3.2. Oscillatory brain activity polygenic scores are associated with generalized epilepsy
We used polygenic scoring to utilize the full distribution of background EEG‐associated SNPs to assess whether people with epilepsy have a different polygenic score for specific frequency bands compared to controls. We observed significant positive associations between beta and theta power PRSs with GGE, in line with the LDSC results (Figure 2). In particular, beta power PRSs were strongly associated with GGE (beta = .11, SE = .020, p = 5.3 × 10−8, explained variance = .21%; Figure 2), which was further supported by significant associations of beta power PRS with its subtypes CAE (beta = .15, SE = .044, p = 8.5 × 10−4) and JME (beta = .12, SE = .033, p = 3.6 × 10−4). Furthermore, of the participants in the GGE case–control cohort, those in the highest 10% decile of beta power PRS scores were 1.4‐fold more likely to have GGE compared to the people in the lowest 10% PRS decile (Figure 3; odds ratio [OR] = 1.40, 95% confidence interval [CI] = 1.18–1.67, p = 1.5 × 10−4). When using the independent Epi25 cohort as a replication dataset, we found that the directions of effect agreed with the discovery analyses for all associations between EEG PRSs and GGE (p one‐sided = .023, p two‐sided = .046; Figure S1). EEG PRSs were not significantly different between people with focal epilepsy and controls.
FIGURE 2.
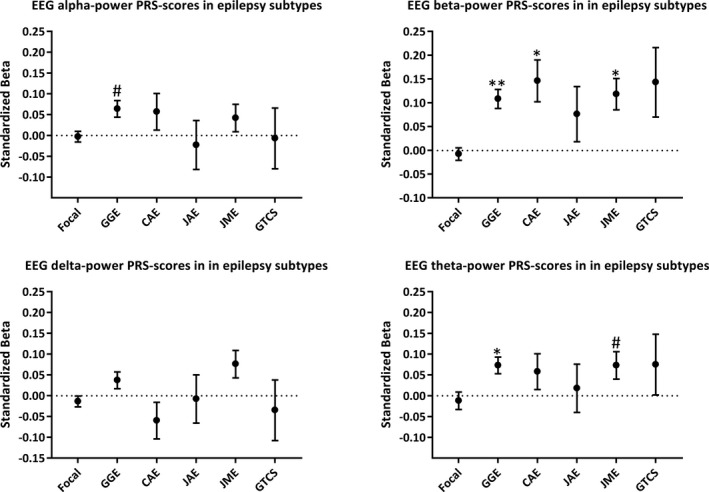
(A) Beta and theta power electroencephalographic (EEG) oscillation polygenic risk scores (PRSs) are associated with generalized epilepsy but not with focal epilepsy. The "best‐fit" p‐value threshold (p t) was chosen based on the most significant association with genetic generalized epilepsy (GGE), which was then applied to all other epilepsy subtypes. The numbers of single nucleotide polymorphisms included in each model were 2670 for alpha power (p t = .0105), 10 861 for beta power (p t = .06245), 8182 for delta power (p t = .0446), and 3833 for theta power (p t = .01665). Logistic regression analyses were performed to assess the association between the PRSs and the different epilepsy subtypes, corrected for sex and 10 principal components. #p < .05, *p < .001, **p < 10−7. Childhood absence epilepsy (CAE), generalized tonic–clonic seizures only (GTCS), juvenile absence epilepsy (JAE), and juvenile myoclonic epilepsy (JME) are GGE subtypes. Focal, focal epilepsy
FIGURE 3.
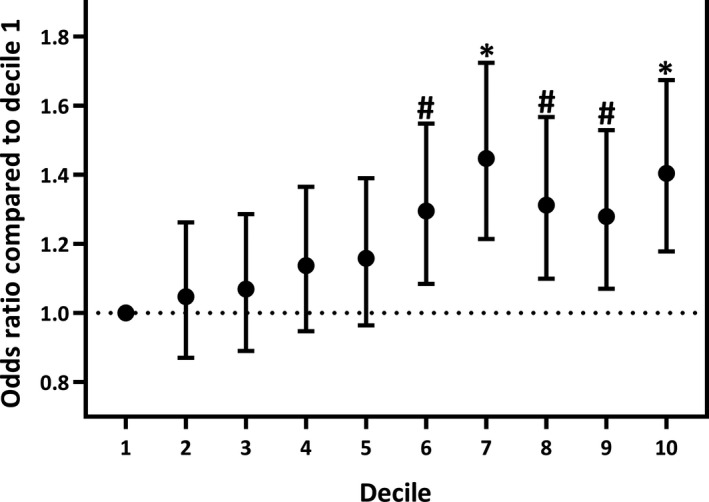
Polygenic risk score (PRS) analyses show that higher beta‐power PRS is associated with an increased likelihood of having GGE. All subjects were divided into 10 deciles based on their beta‐power PRS scores. Logistic regression analyses were performed to quantify the increased risk of having GGE between every decile compared to the lowest decile (0–10%) as a reference. The odds ratios of these analyses are displayed on the Y‐axis. #p<.05; *p<.001
3.3. MR analyses
MR analyses were performed to assess potential causative relationships between background EEG and GGE. Eight GGE‐associated SNPs were selected as instrumental variables at a strict LD prune threshold (r 2 < .01, LD window = 10 Mb). These were used in fixed effects IVW, weighted median, MR Egger, and GSMR (r 2 < .01) models. After loosening the LD threshold, 11 (r 2 < .1), 12 (r 2 < .15), and 14 (r 2 < .2) SNPs were selected as instrumental variables for GSMR models. Causal effects of GGE loci on beta oscillations were found at the LD r 2 < .15 and r 2 < .2 thresholds (OR = 1.79, 95% CI = 1.189–2.707, p = 5.2 × 10−3 and OR = 1.723, 95% CI = 1.180–2.516, p = 4.8 × 10−3, respectively; Table S4, Figure S2). Significant heterogeneity was detected in the fixed effects IVW model (Q‐statistic = 18.188, df = 7, p = .01) and MR Egger model (Q‐statistic = 14.594, df = 6, p = .02). No SNPs altered the pooled β coefficient in the leave‐one‐out sensitivity analysis (β = .374, p = .314) in the fixed effects IVW model. We found no evidence of horizontal pleiotropic effects. Similarly, the HEIDI test detected no SNPs as pleiotropic outliers.
4. DISCUSSION
Here, we leveraged the largest currently available GWASs to assess shared genetic underpinnings of epilepsy and of background EEG oscillations. In particular, we found strong genetic relationships between GGE and beta power oscillations, which were replicated in an independent sample.
Previous studies comparing EEG background oscillations between people with epilepsy and controls are inconsistent; some show increased power in all frequency bands (alpha, beta, delta, theta), whereas others show only increases in specific frequency bands or even decreases in power. 9 , 10 , 11 , 12 , 13 , 14 This heterogeneity likely reflects multiple variables that are difficult to control for in clinical studies, such as antiepileptic drug (AED) usage, sleep deprivation, influence of (inter‐)ictal epileptic brain activity, EEG processing, and electrode placement. We overcame such limitations by determining the genetic underpinnings of EEG frequency bands in people without epilepsy who are AED‐naive, and with consistent electrode placement and signal processing. We applied several statistical models to assess this overlap and found that people with generalized, but not focal, epilepsy carry a relative abundance of genetic variation associated with higher beta oscillations. MR analyses pointed to causal effects of genetic liability to GGE on beta power.
We did not find genetic correlations between background EEG and focal epilepsy. Although power was limited for this analysis, this finding is consistent with the low contribution of common genetic variants in focal epilepsy and the lack of genetic overlap between focal and generalized epilepsy. 16 Focal epilepsy is likely to represent a more heterogenous group of different causes of epilepsy, many of which do not have a primary genetic cause (e.g., symptomatic epilepsy after traumatic brain injury). Moreover, focal epilepsy by definition only affects one part of the brain and is therefore less likely to be associated with germline genetic variation and background EEG oscillations, which most likely affect the whole brain. Although we found associations of common variants with focal epilepsy in our latest GWAS, the overall polygenic burden and SNP‐based heritability was modest compared to GGE. 16 This suggests that further studies assessing common genetic variants in focal epilepsy are less likely to yield major advances. Perhaps further studies on smaller, more homogenous focal epilepsy cohorts or studies assessing rare genetic variants could yield more insights into its pathophysiology. In contrast to focal epilepsy, the EEG discharges that characterize generalized epilepsy are dependent on the thalomocortical system. 5 , 26 Similarly, background oscillations have been functionally attributed to the thalamocortical system, 27 , 28 suggesting that thalamocortical functioning could represent a common neurobiological mechanism reflecting overall brain excitability, which influences both GGE risk and (beta power) background oscillations.
Our results should be interpreted in the light of several limitations. First, we are aware of the possible advantages of using genome complex trait analysis (GCTA) relative to LDSC, but because no subject‐level genotype data are available for the EEG GWAS, we restricted our genetic correlation estimates to LDSC, which is based on summary statistics. LDSC has proven to be a reliable method for genetic correlation estimates, and results between LDSC and GCTA have proven consistent. Second, we found that the same genetic variants underlie both GGE and beta power oscillations, but our study does not prove that people with GGE have altered background oscillations, because we did not have EEG measurements of people with epilepsy in this study. Third, only one‐way MR analyses were performed due to lack of genome‐wide significant loci in the EEG GWAS. Our results suggest that GGE causally influences beta power oscillations. However, we cannot exclude the possibility of bidirectional causality between EEG and GGE, and thus it could also be possible that beta power has a causal effect on GGE risk. Fourth, we had insufficient data available to carry out subgroup analyses on subjects with nonlesional focal epilepsy.
Altogether, our results point to shared biological mechanisms underlying background EEG oscillations and the susceptibility for generalized seizures. Our findings thus open avenues to investigate the clinical utility of background oscillations in genetic generalized epilepsy. Potentially, prospective studies could confirm whether altered beta oscillatons could be a prodromal state of GGE or whether aberrant beta oscillations constitute a feature of epilepsy. Future studies may also integrate transcranial magnetic stimulation–EEG and/or event‐related potentials to examine whether beta and theta powers correlate with altered brain excitability in subjects with high epilepsy liability. We hypothesize that the genetic correlation between GGE and background oscillations will be reflected by measurable differences in background EEG measures between people with and without GGE, which could be used in the diagnostic workup after a first suspected seizure. This information can be used in machine‐learning studies by integrating background EEG with other sources of clinical and demographic data, which may one day increase the accuracy of epilepsy diagnosis.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
R.S., J.J.L., D.Sm., and B.P.C.K. contributed to the conception and design of the study. R.S., J.J.L., B.D.L., D.Sm., and B.P.C.K. contributed to the acquisition and analysis of data. R.S., J.J.L., B.D.L., C.L., D.L., A.S., D.Sc., J.A.C., K.R., R.A.R.B., O.D., K.P.J.B., F.E.J., D.Sm., and B.P.C..K. contributed to the drafting of the manuscript and preparing the figures. Members of the ILAE Consortium on Complex Epilepsies and EPI25 Collaborative contributed clinical and genetic data.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the people with epilepsy and volunteers who participated in this research. We thank the following clinicians and research scientists for their contribution through sample collection (cases and controls), data analysis, and project support: Geka Ackerhans, Muna Alwaidh, R. E. Appleton, Willem Frans Arts, Guiliano Avanzini, Paul Boon, Sarah Borror, Kees Braun, Oebele Brouwer, Hans Carpay, Karen Carter, Peter Cleland, Oliver C. Cockerell, Paul Cooper, Celia Cramp, Emily de los Reyes, Chris French, Catharine Freyer, William Gallentine, Michel Georges, Peter Goulding, Micheline Gravel, Rhian Gwilliam, Lori Hamiwka, Steven J. Howell, Adrian Hughes, Aatif Husain, Monica Islam, Floor Jansen, Mary Karn, Mark Kellett, Ditte B. Kjelgaard, Karl Martin Klein, Donna Kring, Annie W. C. Kung, Mark Lawden, Jo Ellen Lee, Benjamin Legros, Leanne Lehwald, Edouard Louis, Colin H. T. Lui, Zelko Matkovic, Jennifer McKinney, Brendan McLean, Mohamad Mikati, Bethanie Morgan‐Followell, Wim Van Paesschen, Anup Patel, Manuela Pendziwiat, Marcus Reuber, Richard Roberts, Guy Rouleau, Cathy Schumer, B. Sharack, Kevin Shianna, N. C. Sin, Saurabh Sinha, Laurel Slaughter, Sally Steward, Deborah Terry, Chang‐Yong Tsao, T. H. Tsoi, Patrick Tugendhaft, Jaime‐Dawn Twanow, Jorge Vidaurre, Sarah Weckhuysen, Pedro Weisleder, Kathleen White, Virginia Wong, Raju Yerra, Jacqueline Yinger, and all contributing clinicians from the Department of Clinical and Experimental Epilepsy at the National Hospital for Neurology and Neurosurgery and University College London Institute of Neurology. We would like to thank the Ming Fund for providing funding for R.S. This work was in part supported by a Translational Research Scholars award from the Health Research Board of Ireland (Christopher D. Whelan) and by research grants from Science Foundation Ireland (16/RC/3948 and 13/CDA/2223), and cofunded under the European Regional Development Fund and by FutureNeuro industry partners. Further funding sources include Wellcome Trust (grant 084730); Epilepsy Society, UK, National Institute for Health Research (NIHR; 08‐08‐SCC); GIHE, National Institutes of Health (NIH) R01‐NS‐49306‐01 (Russell J. Buono); NIH R01‐NS‐053998 (Daniel H. Lowenstein); GSCFE, NIH R01‐NS‐064154‐01 (Russell J. Buono, Hakon Hakonarson); NIH UL1TR001070, Development Fund from the Children’s Hospital of Philadelphia (Hakon Hakonarson); National Health and Medical Research Council program grant 1091593 (Samuel F. Berkovic, Ingrid E. Scheffer, Karen L. Oliver, Katja E. Boysen); Royal Melbourne Hospital Foundation Lottery Grant (Slavé Petrovski); Royal Melbourne Hospital Neuroscience Foundation (Terence J. O'Brien); European Union’s Seventh Framework Programme (FP7/2007‐2013) under grant agreements 279062 (EpiPGX) and 602102, Department of Health NIHR Biomedical Research Centres funding scheme, European Community (EC; FP6 project EPICURE: LSHM‐CT2006‐037315); German Research Foundation (DFG; SA434/4‐1/4‐26‐1 (Thomas Sander), WE4896/3‐1); EuroEPINOMICS Consortium (European Science Foundation/DFG: SA434/5‐1, NU50/8‐1, LE1030/11‐1, HE5415/3‐1 [Thomas Sander, Peter Nürnberg, Holger Lerche, Ingo Helbig], RO 3396/2‐ 1); German Federal Ministry of Education and Research, National Genome Research Network (NGFNplus/EMINet: 01GS08120, and 01GS08123 [Thomas Sander, Holger Lerche]; IntenC, TUR 09/I10 [Thomas Sander]); Netherlands National Epilepsy Fund (grant 04‐08); EC (FP7 project EpiPGX 279062); and Research Grants Council of the Hong Kong Special Administrative Region, China project numbers HKU7623/08 M (Stacey S. Cherny, Patrick Kwan, Larry Baum, Pak C. Sham), HKU7747/ 07 M (Stacey S. Cherny., Pak C. Sham), and CUHK4466/06 M (Patrick Kwan, Larry Baum). Collection of Belgian cases was supported by the Fonds National de la Recherche Scientifique, Fondation Erasme, Université Libre de Bruxelles. GlaxoSmithKline funded the recruitment and data collection for the GenEpA Consortium samples. We acknowledge the support of Nationwide Children’s Hospital in Columbus, Ohio, USA. The Wellcome Trust (WT066056) and the NIHR Biomedical Research Centres Scheme (P31753) supported UK contributions. Further support was received through the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (contract N01HD33348). The project was also supported by the popgen 2.0 network through a grant from the German Ministry for Education and Research (01EY1103). Parts of the analysis of this work were performed on resources of the High Performance Center of the University of Luxembourg and Elixir‐Luxembourg. The KORA study was initiated and financed by the Helmholtz Zentrum München–German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences, Ludwig Maximilian University, as part of LMUinnovativ. The ILAE facilitated the Consortium on Complex Epilepsies through the Commission on Genetics and by financial support; however, the opinions expressed in the article do not necessarily represent the policy or position of the ILAE.
Remi Stevelink and Jurjen J. Luykx contributed equally to this work.
Dirk J. A. Smit and Bobby P. C. Koeleman jointly directed this work.
Contributor Information
Jurjen J. Luykx, Email: J.Luykx@umcutrecht.nl.
International League Against Epilepsy Consortium on Complex Epilepsies:
Bassel Abou‐Khalil, Pauls Auce, Andreja Avbersek, Melanie Bahlo, David J. Balding, Thomas Bast, Larry Baum, Albert J. Becker, Felicitas Becker, Bianca Berghuis, Samuel F. Berkovic, Katja E. Boysen, Jonathan P. Bradfield, Lawrence C. Brody, Russell J. Buono, Ellen Campbell, Gregory D. Cascino, Claudia B. Catarino, Gianpiero L. Cavalleri, Stacey S. Cherny, Krishna Chinthapalli, Alison J. Coffey, Alastair Compston, Antonietta Coppola, Patrick Cossette, John J. Craig, Gerrit‐Jan de Haan, Peter De Jonghe, Carolien G. F. de Kovel, Norman Delanty, Chantal Depondt, Dennis J. Dlugos, Colin P. Doherty, Christian E. Elger, Johan G. Eriksson, Thomas N. Ferraro, Martha Feucht, Ben Francis, Andre Franke, Jacqueline A. French, Saskia Freytag, Verena Gaus, Eric B. Geller, Christian Gieger, Tracy Glauser, Simon Glynn, David B. Goldstein, Hongsheng Gui, Youling Guo, Kevin F. Haas, Hakon Hakonarson, Kerstin Hallmann, Sheryl Haut, Erin L. Heinzen, Ingo Helbig, Christian Hengsbach, Helle Hjalgrim, Michele Iacomino, Andrés Ingason, Jennifer Jamnadas‐Khoda, Michael R. Johnson, Reetta Kälviäinen, Anne‐Mari Kantanen, Dalia Kasperavičiūte, Dorothee Kasteleijn‐Nolst Trenite, Heidi E. Kirsch, Robert C. Knowlton, Roland Krause, Martin Krenn, Wolfram S. Kunz, Ruben Kuzniecky, Patrick Kwan, Yu‐Lung Lau, Anna‐Elina Lehesjoki, Holger Lerche, Wolfgang Lieb, Dick Lindhout, Warren D. Lo, Iscia Lopes‐Cendes, Daniel H. Lowenstein, Alberto Malovini, Anthony G. Marson, Thomas Mayer, Mark McCormack, James L. Mills, Nasir Mirza, Martina Moerzinger, Rikke S. Møller, Anne M. Molloy, Hiltrud Muhle, Mark Newton, Ping‐Wing Ng, Markus M. Nöthen, Peter Nürnberg, Terence J. O’Brien, Karen L. Oliver, Aarno Palotie, Faith Pangilinan, Sarah Peter, Slavé Petrovski, Annapurna Poduri, Michael Privitera, Rodney Radtke, Sarah Rau, Philipp S. Reif, Eva M. Reinthaler, Felix Rosenow, Josemir W. Sander, Thomas Sander, Theresa Scattergood, Steven C. Schachter, Christoph J. Schankin, Ingrid E. Scheffer, Bettina Schmitz, Susanne Schoch, Pak C. Sham, Jerry J. Shih, Graeme J. Sills, Sanjay M. Sisodiya, Lisa Slattery, David F. Smith, Michael C. Smith, Philip E. Smith, Anja C. M. Sonsma, Doug Speed, Michael R. Sperling, Bernhard J. Steinhoff, Ulrich Stephani, Konstantin Strauch, Pasquale Striano, Hans Stroink, Rainer Surges, K. Meng Tan, Liu Lin Thio, G. Neil Thomas, Marian Todaro, Rossana Tozzi, Maria S. Vari, Eileen P. G. Vining, Frank Visscher, Sarah von Spiczak, Nicole M. Walley, Yvonne G. Weber, Zhi Wei, Ruta Mameniskiene, Judith Weisenberg, Christopher D. Whelan, Peter Widdess‐Walsh, Markus Wolff, Stefan Wolking, Wanling Yang, Federico Zara, and Fritz Zimprich
Epi25 Collaborative:
Yen‐Chen Anne Feng, Daniel P. Howrigan, Liam E. Abbott, Katherine Tashman, Felecia Cerrato, Dennis Lal, Claire Churchhouse, Namrata Gupta, Stacey B. Gabriel, Mark J. Daly, Eric S. Lander, Benjamin M. Neale, Samuel F. Berkovic, Holger Lerche, David B. Goldstein, Daniel H. Lowenstein, Gianpiero L. Cavalleri, Patrick Cossette, Chris Cotsapas, Peter De Jonghe, Tracy Dixon‐Salazar, Renzo Guerrini, Hakon Hakonarson, Erin L. Heinzen, Ingo Helbig, Patrick Kwan, Slavé Petrovski, Sitharthan Kamalakaran, Sanjay M. Sisodiya, Randy Stewart, Sarah Weckhuysen, Dennis J. Dlugos, Ingrid E. Scheffer, Pasquale Striano, Catharine Freyer, Roland Krause, Patrick May, Kevin McKenna, Brigid M. Regan, Susannah T. Bellows, Caitlin A. Bennett, Esther M.C. Johns, Alexandra Macdonald, Hannah Shilling, Rosemary Burgess, Dorien Weckhuysen, Melanie Bahlo, Terence J. O'Brien, Marian Todaro, Hannah Stamberger, Chantal Depondt, Danielle M. Andrade, Tara R. Sadoway, Kelly Mo, Heinz Krestel, Sabina Gallati, Savvas S. Papacostas, Ioanna Kousiappa, George A. Tanteles, Katalin Štěrbová, Markéta Vlčková, Lucie Sedláčková, Petra Laššuthová, Karl Martin Klein, Felix Rosenow, Philipp S. Reif, Susanne Knake, Wolfram S. Kunz, Gábor Zsurka, Christian E. Elger, Jürgen Bauer, Michael Rademacher, Manuela Pendziwiat, Hiltrud Muhle, Annika Rademacher, Andreas van Baalen, Sarah von Spiczak, Ulrich Stephani, Zaid Afawi, Amos D. Korczyn, Moien Kanaan, Christina Canavati, Gerhard Kurlemann, Karen Müller‐Schlüter, Gerhard Kluger, Martin Häusler, Ilan Blatt, Johannes R. Lemke, Ilona Krey, Yvonne G. Weber, Stefan Wolking, Felicitas Becker, Christian Hengsbach, Sarah Rau, Ana F. Maisch, Bernhard J. Steinhoff, Andreas Schulze‐Bonhage, Susanne Schubert‐Bast, Herbert Schreiber, Ingo Borggräfe, Christoph J. Schankin, Thomas Mayer, Rudolf Korinthenberg, Knut Brockmann, Dieter Dennig, Rene Madeleyn, Reetta Kälviäinen, Pia Auvinen, Anni Saarela, Tarja Linnankivi, Anna‐Elina Lehesjoki, Mark I. Rees, Seo‐Kyung Chung, William O. Pickrell, Robert Powell, Natascha Schneider, Simona Balestrini, Sara Zagaglia, Vera Braatz, Anthony G. Marson, Michael R. Johnson, Pauls Auce, Graeme J. Sills, Larry W. Baum, Pak C. Sham, Stacey S. Cherny, Colin H.T. Lui, Nina Barišić, Norman Delanty, Colin P. Doherty, Arif Shukralla, Mark McCormack, Hany El‐Naggar, Laura Canafoglia, Silvana Franceschetti, Barbara Castellotti, Tiziana Granata, Federico Zara, Michele Iacomino, Francesca Madia, Maria Stella Vari, Maria Margherita Mancardi, Vincenzo Salpietro, Francesca Bisulli, Paolo Tinuper, Laura Licchetta, Tommaso Pippucci, Carlotta Stipa, Raffaella Minardi, Antonio Gambardella, Angelo Labate, Grazia Annesi, Lorella Manna, Monica Gagliardi, Elena Parrini, Davide Mei, Annalisa Vetro, Claudia Bianchini, Martino Montomoli, Viola Doccini, Carla Marini, Toshimitsu Suzuki, Yushi Inoue, Kazuhiro Yamakawa, Birute Tumiene, Lynette G. Sadleir, Chontelle King, Emily Mountier, S. Hande Caglayan, Mutluay Arslan, Zuhal Yapıcı, Uluc Yis, Pınar Topaloglu, Bulent Kara, Dilsad Turkdogan, Aslı Gundogdu‐Eken, Nerses Bebek, Sibel Uğur‐İşeri, Betül Baykan, Barış Salman, Garen Haryanyan, Emrah Yücesan, Yeşim Kesim, Çiğdem Özkara, Annapurna Poduri, Russell J. Buono, Thomas N. Ferraro, Michael R. Sperling, Warren Lo, Michael Privitera, Jacqueline A. French, Steven Schachter, Ruben I. Kuzniecky, Manu Hegde, Pouya Khankhanian, Katherine L. Helbig, and Colin A. Ellis
REFERENCES
- 1. Smit DJA, Posthuma D, Boomsma DI, De Geus EJC. Heritability of background EEG across the power spectrum. Psychophysiology. 2005;42:691–7. [DOI] [PubMed] [Google Scholar]
- 2. Devinsky O, Vezzani A, O'Brien TJ, Jette N, Scheffer IE, de Curtis M, et al. Epilepsy. Nat Rev Dis Primers. 2018;4:18024. [DOI] [PubMed] [Google Scholar]
- 3. Thomas RH, Berkovic SF. The hidden genetics of epilepsy—a clinically important new paradigm. Nat Rev Neurol. 2014;10:283–92. [DOI] [PubMed] [Google Scholar]
- 4. Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101:243–76. [DOI] [PubMed] [Google Scholar]
- 5. Gloor P. Generalized epilepsy with bilateral synchronous spike and wave discharge. New findings concerning its physiological mechanisms. Electroencephalogr Clin Neurophysiol Suppl. 1978;34:245–9. [PubMed] [Google Scholar]
- 6. Gloor P. Generalized epilepsy with spike‐and‐wave discharge: a reinterpretation of its electrographic and clinical manifestations. The 1977 William G. Lennox Lecture, American Epilepsy Society. Epilepsia. 1979;20:571– 88. [DOI] [PubMed] [Google Scholar]
- 7. Gloor P, Fariello RG. Generalized epilepsy: some of its cellular mechanisms differ from those of focal epilepsy. Trends Neurosci. 1988;11:63–8. [DOI] [PubMed] [Google Scholar]
- 8. Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28:317–24. [DOI] [PubMed] [Google Scholar]
- 9. Clemens B, Szigeti G, Barta Z. EEG frequency profiles of idiopathic generalised epilepsy syndromes. Epilepsy Res. 2000;42:105–15. [DOI] [PubMed] [Google Scholar]
- 10. Clemens B. Pathological theta oscillations in idiopathic generalised epilepsy. Clin Neurophysiol. 2004;115:1436–41. [DOI] [PubMed] [Google Scholar]
- 11. Clemens B, Bessenyei M, Piros P, Tóth M, Seress L, Kondákor I. Characteristic distribution of interictal brain electrical activity in idiopathic generalized epilepsy. Epilepsia. 2007;48:941–9. [DOI] [PubMed] [Google Scholar]
- 12. Clemens B. Valproate decreases EEG synchronization in a use‐dependent manner in idiopathic generalized epilepsy. Seizure. 2008;17:224–33. [DOI] [PubMed] [Google Scholar]
- 13. Clemens B, Puskás S, Besenyei M, Kovács NZS, Spisák T, Kis SA, et al. Valproate treatment normalizes EEG functional connectivity in successfully treated idiopathic generalized epilepsy patients. Epilepsy Res. 2014;108:1896–903. [DOI] [PubMed] [Google Scholar]
- 14. Tikka SK, Goyal N, Umesh S, Nizamie SH. Juvenile myoclonic epilepsy: clinical characteristics, standard and quantitative electroencephalography analyses. J Pediatr Neurosci. 2013;8:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smit DJA, Wright MJ, Meyers JL, Martin NG, Ho YYW, Malone SM, et al. Genome‐wide association analysis links multiple psychiatric liability genes to oscillatory brain activity. Hum Brain Mapp. 2018;39:4183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. International League Against Epilepsy Consortium on Complex Epilepsies . Genome‐wide mega‐analysis identifies 16 loci and s diverse biological mechanisms in the common epilepsies. Nat Commun. 2018;9(1):5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bulik‐Sullivan BK, Loh P‐R, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD score regression distinguishes confounding from polygenicity in genome‐wide association studies. Nat Genet. 2015;47:291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. 1000 Genomes Project Consortium , Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526 : 68– 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome‐wide association studies. Bioinformatics. 2010;26:2867–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Euesden J, Lewis CM, O'Reilly PF. PRSice: polygenic risk score software. Bioinformatics. 2015;31:1466–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR‐Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30:543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu Z, Zheng Z, Zhang F, Wu Y, Trzaskowski M, Maier R, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun. 2018;9:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome‐wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steriade M, Contreras D. Spike‐wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol. 1998;80:1439–55. [DOI] [PubMed] [Google Scholar]
- 27. Steriade M, Amzica F, Contreras D. Synchronization of fast (30–40 Hz) spontaneous cortical rhythms during brain activation. J Neurosci. 1996;16:392–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steriade M, Contreras D, Amzica F, Timofeev I. Synchronization of fast (30–40 Hz) spontaneous oscillations in intrathalamic and thalamocortical networks. J Neurosci. 1996;16:2788–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
GWAS summary statistics used for the current analyses are available online: http://enigma.ini.usc.edu/research/download‐enigma‐gwas‐results/; http://www.epigad.org/gwas_ilae2018_16loci.html.