
Figure 2.
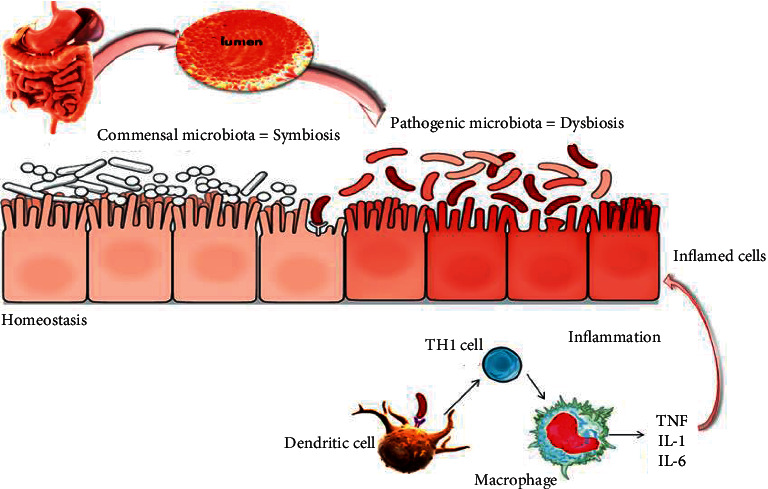
Mechanisms of inflammatory bowel disease. The intestine shelters a large diversity of microbiota that are in perfect balance (symbiosis); sometimes, this balance can be affected by many factors leading to the appearance of pathogenic bacteria that can alter the intestinal barrier and lead to the development of inflammatory bowel disease. The stimulation of the mucosal immune system may occur as a result of the penetration of bacterial products through the mucosal barrier, leading to their direct interaction with immune cells, especially dendritic cells and lymphocyte populations, to promote a classic adaptive immune response. Alternatively, bacterial products may stimulate the surface epithelium, possibly through receptors that are components of the innate immune-response system; the epithelium can, in turn, produce cytokines and chemokines that recruit and activate mucosal immune cells. Activation of classic antigen-presenting cells, such as dendritic cells, or direct stimulation through pattern-recognition receptors promotes the differentiation of type 1 helper T-cells (Th1) in patients with Crohn's disease (shown here) or, possibly, atypical type 2 helper T-cells in patients with ulcerative colitis. The stereotypical products of Th1 promote a self-sustaining cycle of activation with macrophages. In addition to producing the key cytokines that stimulate Th1 (interleukin-12, interleukin-18, and macrophage migration inhibitor factor), macrophages produce a mix of inflammatory cytokines, including interleukin-1, interleukin-6, and most notably tumor necrosis factor, which target a broad variety of other types of cells. The latter include endothelial cells, which then facilitate the recruitment of leukocytes to the mucosa from the vascular space. Most important, these functions may be altered either by genetically determined variants, as exemplified by germ-line mutations in the gene encoding NOD2, the product of the IBD1 locus, in some patients with Crohn's disease, or by environmental factors.