

Abstract
Motivation
Understanding chemical–gene interactions (CGIs) is crucial for screening drugs. Wet experiments are usually costly and laborious, which limits relevant studies to a small scale. On the contrary, computational studies enable efficient in-silico exploration. For the CGI prediction problem, a common method is to perform systematic analyses on a heterogeneous network involving various biomedical entities. Recently, graph neural networks become popular in the field of relation prediction. However, the inherent heterogeneous complexity of biological interaction networks and the massive amount of data pose enormous challenges. This paper aims to develop a data-driven model that is capable of learning latent information from the interaction network and making correct predictions.
Results
We developed BioNet, a deep biological networkmodel with a graph encoder–decoder architecture. The graph encoder utilizes graph convolution to learn latent information embedded in complex interactions among chemicals, genes, diseases and biological pathways. The learning process is featured by two consecutive steps. Then, embedded information learnt by the encoder is then employed to make multi-type interaction predictions between chemicals and genes with a tensor decomposition decoder based on the RESCAL algorithm. BioNet includes 79 325 entities as nodes, and 34 005 501 relations as edges. To train such a massive deep graph model, BioNet introduces a parallel training algorithm utilizing multiple Graphics Processing Unit (GPUs). The evaluation experiments indicated that BioNet exhibits outstanding prediction performance with a best area under Receiver Operating Characteristic (ROC) curve of 0.952, which significantly surpasses state-of-theart methods. For further validation, top predicted CGIs of cancer and COVID-19 by BioNet were verified by external curated data and published literature.
Keywords: chemical–gene interaction, graph convolution network, heterogeneous biological network, parallel computing
Graphical abstract

Introduction
The traditional drug discovery process is time-consuming, and it could take years or even decades. However, in situations like the pandemic outbreak, we need to find drugs for urgent use within a short period of time. Therefore, the drug repurposing [1] approach becomes a feasible option, which attempts to screen the candidates from FDA-approved drugs and apply it to novel targets. It can significantly reduce the time to find therapeutics compared to a standard procedure from scratch. To be specific, most drugs are small-molecule chemicals that mainly act on single or multiple gene/protein targets to achieve satisfactory therapeutic effects. Therefore, it is worthwhile to investigate the interactions between chemicals and genes, also known as chemical–gene interactions (CGIs) [2].
Moreover, to better understand the complex biological mechanisms, it is important to disentangle the complicated relations among different types of biological entities besides chemicals and genes. Identifying the associations between diseases, chemicals, genes, and biological pathways have become a key step in understanding the cause of diseases and an indispensable step in finding effective therapeutics. Several studies have been proposed to represent interaction information by constructing a large-scale heterogeneous biological interaction network [3–5] from curated data or information extracted from literature. Methods like network pharmacology [6, 7] were utilized to analyze such a massive complex network. However, the scale of the current target network is overwhelming for any explicit analytical method. Therefore, data-driven methods become a better choice to mine valuable latent information from the interaction network.
Deep learning methods, especially graph-based models, have been widely used in link prediction between biological entities [8]. By utilizing the characteristics and the known interrelations, relation prediction model can extract latent relations between biological entities. Ran Wang et al. [9] constructed and decomposed a 3D tensor composed of the connection among drugs, targets and diseases to reuse drugs for cancer. NeurTN [10], Chemotext [11] and GNBR [8] each provides a powerful method to capture the non-linear relations among drugs, targets and diseases. PDGNet [12] used a deep neural network with multi-view features to excavate the potential interactions between diseases and genes. However, those methods cannot merge and embed the information of adjacent nodes, which limits the accuracy of prediction. Recently, the multilayer attention graph convolutional network (LAGCN) [13] was developed for drug–disease interaction prediction. I-RGCN [14] is a few-shot link prediction method for COVID-19 drug-repurposing. These two models use graph convolutional network (GCN) to get latent information, but the limited scale of utilized data prevents a comprehensive representation of available information, which restrict the performance of relation predictions. For instance, LAGCN only includes known drug–disease associations, drug–drug similarities and disease–disease similarities, while important information like pathogenic genes and pivotal biological pathways were neglected; I-RGCN only consider genes and drugs for COVID-19 and information for related diseases was not used.
Curated databases provide chances to include more entity data in graph-based models. However, training a large-scale data-driven model is computationally challenging. Therefore, it is also important to develop strategies for an efficient computing process.
To address the above problems, we proposed a scalable graph neural network model named BioNet to predict the relations between chemicals and genes. Our major contributions can be summarized as follows:
We constructed a comprehensive and large-scale heterogeneous biological interaction network by integrating curated datasets related to chemicals, genes, pathways and diseases.
We proposed a deep graph neural network model named BioNet based on an encoder-decoder architecture, which utilizes a graph convolution encoder to learn entity embeddings from subgraphs and employs a tensor decomposition decoder to predict CGIs.
We developed a parallel strategy to boost the learning process and improved the model’s ability to handle large-scale data.
We exemplified the value of BioNet by evaluating the CGIs of cancer [15] and COVID-19 [16], which prioritizes chemicals with higher potential for effective therapeutics.
Materials and methods
Model architecture of BioNet
Given a set of nodes 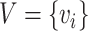 , a set of edges
, a set of edges 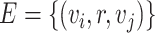 , where r is the type of the edge, the graph
, where r is the type of the edge, the graph  can be denoted as
can be denoted as 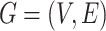 . The goal of our model is to compute the probability of the interesting edge
. The goal of our model is to compute the probability of the interesting edge 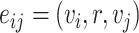 . To achieve this goal, BioNet adopts an encoder–decoder architecture (Figure 1). The encoder is equipped with the graph convolutional networks, whereas the decoder adopts a tensor factorization model. The following sections give the details of BioNet.
. To achieve this goal, BioNet adopts an encoder–decoder architecture (Figure 1). The encoder is equipped with the graph convolutional networks, whereas the decoder adopts a tensor factorization model. The following sections give the details of BioNet.
Figure 1.

The panorama of BioNet. Network construction → graph representation → graph convolutional encoder → target node embedding → tensor factorization decoder → the probabilities of interactions.
Network construction
In this paper, we constructed a graph containing seven subgraphs: chemical–chemical subgraph (CC-graph), gene–gene subgraph (GG-graph), chemical–path subgraph (CP-graph), gene–pathway subgraph (GP-graph), chemical–gene subgraph (CG-graph), chemical–disease subgraph (CD-graph) and gene–disease subgraph (GD-graph). There are 720 155 chemical–chemical interactions, 713 471 gene–gene interactions, 1 285 158 chemical–pathway interactions, 135 809 gene–pathway interactions and 1 798 796 CGIs from the STITCH database [17], the SNAP Graph Library [18] and the Comparative Toxicogenomics Database (CTD) [19].
Especially, we introduced diseases as interaction entities. The basic idea is that curated databases have collected a lot of data on interactions of gene–disease and chemical–disease, which can provide an extra and valuable context for predicting CGIs. On the one hand, many diseases can be closely attributed to abnormal changes in genes. On the other hand, a specific chemical may be used to treat many diseases while a given disease might be cured by different chemicals. Therefore, we built the CD-graph and GD-graph with 2 686 187 chemical–disease interactions and 26 663 499 gene–disease interactions from the CTD database. Table 1 shows the data source and data type of the final integrated multi-relational graph.
Table 1.
Statistics and data source of the final integrated multi-type interaction graph
| Subgraph | # of Entity1 | # of Entity2 | Interaction | # of Edges | Data source |
|---|---|---|---|---|---|
| CC-graph | Chemical: 389 393 | Chemical: 389 393 | Chemicals associate with target chemicals | 720 155 | The STITCH database [17] |
| GG-graph | Gene: 19 081 | Gene: 19 081 | Genes associate with target genes | 713 471 | SNAP Graph Library [18] |
| CP-graph | Chemical: 10 034 | pathways: 2185 | Chemicals associate with target pathways | 1 285 158 | CTD [19] |
| GP-graph | Gene: 11 588 | Pathways: 2363 | Genes associate with target pathways | 135 809 | CTD [19] |
| CG-graph | Chemical: 13 488 | Gene: 50 876 | Chemicals interact with target genes | 1 801 222 | CTD [19] |
| CD-graph | Chemical: 16 146 | Disease: 7217 | Chemicals associate with target diseases | 2 686 187 | CTD [19] |
| GD-graph | Gene: 49 776 | Disease: 7078 | Genes associate with target diseases | 26 663 499 | CTD [19] |
To investigate how does the addition of disease entities affects the performance of the BioNet model in CGI prediction, we studied two combo-graphs: ① the CGP graph for relations between chemicals, genes and pathways; ② the CGPD graph for relations between chemicals, genes, pathways and diseases. It is evident that ① is the subgraphs of ②, as illustrated in Figure 2. In the experimental section, we will compare the performance and scalability of our model with different subgraphs. To note, the C-G interactions are featured by many interaction types, while other interactions are considered binary in our study.
Figure 2.

Types of relations are included in each graph.
Graph convolutional encoder
A graph encoder can iteratively aggregate, transform and propagate information across the entire network. The input is a graph structure in which nodes are represented as one-hot vectors and edges are represented as adjacent matrices, as illustrated in Figure 3.
Figure 3.

Graph structure representation.
A GCN module defines the information propagation architecture of each node, so that the node contains its own information and learns the information of all neighbor nodes within k hops. Chebyshev polynomials prove that graph convolutional networks with a depth of two layers usually show better performance [20]. The previous work [2] has been demonstrated that the information encoding by subgraph perspective to aggregate neighbor nodes performs better than the whole graph perspective. Therefore, we set k = 2 and adopted a subgraph perspective to encode the graph in the following steps. Set 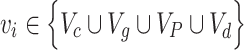 as an example, Figure 4 shows the detailed encoding process.
as an example, Figure 4 shows the detailed encoding process.
Figure 4.

Graph convolutional encoder. Take BioNet-CGPD as an example. Initial embeddings of chemicals 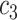 and genes
and genes 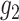 are learned with the binary interaction subgraph.
are learned with the binary interaction subgraph.
The GCN processing in BioNet is a two-step procedure. Firstly, we train the binary association subgraphs (CC-graph, GG-graph, CP-graph, GP-graph, CD-graph and GD-graph), and then we introduce the initial embedding to further train the multi-interaction CG-graph.
In the binary association subgraph 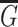 , node embeddings of chemicals are encoded from the neighbor information in the CC-graph, CP-graph and CD-graph, while the node embeddings of genes come from the neighbor nodes in the GG-graph, GP-graph and GD-graph. The binary encoding procedure is performed according to Equation (1). Node features are initialized as one-hot vectors, denoted as
, node embeddings of chemicals are encoded from the neighbor information in the CC-graph, CP-graph and CD-graph, while the node embeddings of genes come from the neighbor nodes in the GG-graph, GP-graph and GD-graph. The binary encoding procedure is performed according to Equation (1). Node features are initialized as one-hot vectors, denoted as 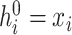 . In the first-layer of GCN, the embedding will aggregate information from
. In the first-layer of GCN, the embedding will aggregate information from 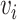 ’s first-order neighbors of different relation types, and get hidden state
’s first-order neighbors of different relation types, and get hidden state 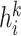 . Stacking one more layer of graph convolutional layers, the embedding will update information from its second-order neighbors explicitly and get hidden state
. Stacking one more layer of graph convolutional layers, the embedding will update information from its second-order neighbors explicitly and get hidden state 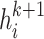 . The formula for the update process is as follows:
. The formula for the update process is as follows:
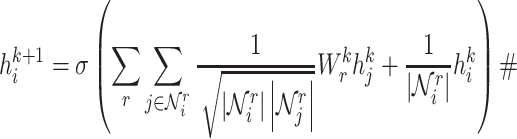 |
(1) |
where 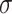 is a non-linear activation function.
is a non-linear activation function. 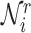 are the neighbors of
are the neighbors of 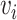 with a link type r.
with a link type r. 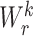 represents a matrix of learnable parameters linked by relation type r.
represents a matrix of learnable parameters linked by relation type r. 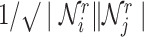 and
and 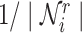 are normalization constants.
are normalization constants.
Then, get the hidden state 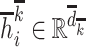 of each hidden layer and output node embedding (
of each hidden layer and output node embedding (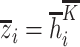 with
with 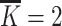 ), and embedded them into the multi-relation subgraph
), and embedded them into the multi-relation subgraph  with the second two-layer graph convolutional network as Equation (2):
with the second two-layer graph convolutional network as Equation (2):
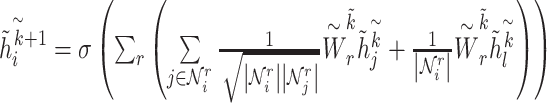 |
(2) |
Similarly, 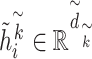 with
with 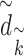 represents the dimension of the
represents the dimension of the 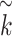 th hidden layer.
th hidden layer. 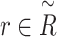 denotes the type of the CG-interaction.
denotes the type of the CG-interaction.
Finally, we assigned 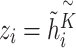 , where
, where 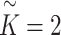 and
and 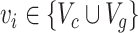 as multiple relational subgraphs to learn high-level node embeddings of chemical and gene nodes.
as multiple relational subgraphs to learn high-level node embeddings of chemical and gene nodes.
Tensor decomposition decoder
Decomposition into directional components (DEDICOM) [21] and RESCAL [21] are used to analysis of social networks with large-scale datasets. They are tensor factorization methods to learn relations between entities. As bilinear models, they capture latent semantics via associating each entity with a vector. Each relation is represented as a matrix that models the paired interactions between potential factors. The core idea of the RESCAL model is to encode the entire knowledge graph into a 3D tensor. This tensor can be factorized to core tensor and a factor matrix, each 2D matrix slice in the core scale representing a relation and each row in the factor matrix representing an entity.
DEDICOM and RESCAL are appropriate for analyzing inherent asymmetric relations, such as the relations between chemicals and genes. Besides, RESCAL can further simplify the decoding process, especially for modeling multi-type interaction data. Given a chemical 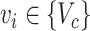 and a gene
and a gene 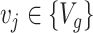 , the decoder will generate the probability
, the decoder will generate the probability 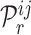 of an edge
of an edge 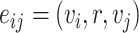 for how likely chemical
for how likely chemical 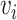 results in an interaction type
results in an interaction type 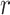 of gene
of gene 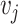 .
.
In DEDICOM (Figure 5A), we characterize CGIs with a tensor (note that the first two modes have the same size) as
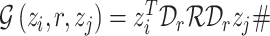 |
(3) |
Figure 5.

Tensor decomposition decoder. The chemical embedding matrix and the gene embedding matrix are learned from the graph convolutional encoder. (A) DELICOM (B) RESCAL. The chemical embedding matrix (Left) and the gene embedding matrix (Right) are learned from the graph convolutional encoder.
Where each slice of  is a
is a 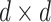 diagonal matrix, giving weights to the columns of the node embeddings
diagonal matrix, giving weights to the columns of the node embeddings 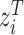 and
and 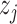 learned by the encoder. Where
learned by the encoder. Where  captures the asymmetric relations, which models to propagate and gather information across different types of interactions.
captures the asymmetric relations, which models to propagate and gather information across different types of interactions.
To reduce the parameters during the training process, The RESCAL (Figure 5B) omitted diagonal matrix, and the decomposition is
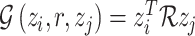 |
(4) |
Parallel optimization
Early BioNet–CGP occupies about 15 GB of memory, which can barely be squeezed into a single NVIDIA v100 GPU (with 16 GB of device memory). Compared with BioNet-CGP, the size of BioNet–CGPD is increased by multiple times due to the addition of tens of millions of disease-related interactions. It composes a computational challenge of model training and prediction. A single GPU can hardly fulfill the computation requirements of our model due to the fact that: (1) the processing time is too long; (2) the data size of the BioNet surpasses the volume of a single device. Therefore, we need to employ parallel processing to optimize the computation process.
The amount of computation in the training process is primarily determined by the number of relations. Therefore, we split the training load into batches, which enables efficient parallel computation across a few GPUs. Gradient all-reduce [22] is an algorithm that aims to efficiently consolidate data from different machines and then distribute the results to individual machines. In each pass, gradient all-reduce is performed in parallel with gradient computation to update the parameters in the BioNet model. The resulting model on each GPU is identical because each GPU starts with an identical copy and weights updates are identical on all GPUs due to the gradient all-reduce operation.
To note, BioNet is ultimately a multi-type interaction model, and each interaction type has a different number of training samples. When splitting the training load across different GPUs, we need to ensure that: (1) the overall workload allocated to each GPU needs to be approximately balanced; (2) the relation type-specific workload allocated to each GPU should also be evenly distributed.
Our training data distribution scheme is depicted in Figure 6. Rn represents different types of relation between chemicals and genes.
Figure 6.

Parallel and distributed computing. The edges of different relation types are randomly sent to different GPU devices in batches. Each GPU device only processes one kind of relation type per step.
Specifically, the input edges (the training data) are divided into mini-batches per relation type, which are sent to GPUs for calculation. The mini-batch size is determined by the number of relations with the least number (70 here according to our data). Firstly, we sort the relations from small to large by number of CG pairs, and set the number of CG pairs containing the fewest CG pairs as batch size 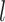 . Secondly, we split other types of CG pairs by
. Secondly, we split other types of CG pairs by 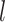 and remove the insufficient
and remove the insufficient 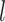 portion. Finally, we distribute the divided data to each GPU. In this way, each GPU maintains a copy of the BioNet model and trains on a sub-minibatch of the training data. For example, R0 represents the relation type “affects^localization,” while R0-0 corresponds to the first 70 edges with an R0 relation.
portion. Finally, we distribute the divided data to each GPU. In this way, each GPU maintains a copy of the BioNet model and trains on a sub-minibatch of the training data. For example, R0 represents the relation type “affects^localization,” while R0-0 corresponds to the first 70 edges with an R0 relation.
In this way, we can achieve load balancing without disrupting the generalizability of our model during the training process.
The introduction of multiples GPUs for parallel training is vital for time efficiency, and it also greatly improves the model capacity of the BioNet, which facilitates incorporating a lot more valuable information vital for the prediction of chemical–gene relations.
Results
Experimental settings
All experiments were conducted on a cluster with NVIDIA Tesla V100 16GB GPUs. The code of the BioNet model was implemented using the Pytorch deep learning framework. Similar to our previous work [2], BioNet uses the hinge loss as the loss function [23], and it is optimized by the Adam optimizer. The parameters used in the model are listed in Table 2.
Table 2.
The parameter used in BioNet
| Parameter | Description | Value |
|---|---|---|
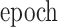
|
The number of training epochs | 20 |
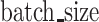
|
The number of samples per training step | 70 |
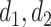
|
The embedding sizes in the total graph perspective | 32, 16 |
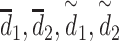
|
The embedding sizes in the subgraph perspective | 128, 64, 32, 16 |
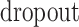
|
The dropout rate | 0.1 |
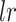
|
The learning rate of the Adam optimizer | 0.001 |
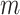
|
The margin value of the hinge loss function | 0.1 |
Performance evaluation
To comprehensively evaluate the performance of BioNet, we employed three classical metrics for performance evaluation, including area under receiver operating characteristic curve (AUROC), area under precision-recall (PR) curve (AUPRC) and average precision of top k recommendations (AP@k). Firstly, all CGI instances were randomly divided into the training set, the validation set and the test set by a ratio of 8:1:1 per interaction type. We evaluated the performance of BioNet along with other state-of-the-art methods on two datasets (CGP and CGPD). The CGP dataset only contains information about chemicals, genes and pathways; the CGPD dataset contains all CGP data plus disease-related information. The size of CGPD is about seven times greater than CGP.
Table 3 lists the performance of BioNet compared with several baseline algorithms (e.g. Deep walk [24], Node2vec [25], SVD [26], Laplacian [27], GCN-Total [28] and CGINet [2]). As illustrated in Table 3, BioNet consistently outperformed the other approaches on all three metrics, including AUROC, AUPRC and AP@20 either on CGP and CGPD, which confirmed the effectiveness of our BioNet method. To note, previous GCN-based methods including GCN-Total and CGINet cannot efficiently handle the CGPD dataset, so they were only evaluated on the CGP dataset. The experimental results show that the former outperforms the latter with the same subgraph. This is largely due to two reasons: (1) BioNet adopts a parallel strategy to optimize the training process. During the parallel training process, BioNet trains each relation type evenly compared to CGINet, which can alleviate the distribution imbalance of data. (2) Unlike CGINet, BioNet employs RESCAL as the tensor decomposition decoder instead of DEDICOM. RESCAL can further simplify the decoding process, especially for modeling multi-type interaction data.
Table 3.
Performance comparison of our models with baseline approaches
| Model | Dataset | AUROC | AUPRC | AP@20 |
|---|---|---|---|---|
| DeepWalk [24] | CGP | 0.830 | 0.811 | 0.733 |
| CGPD | 0.835 | 0.832 | 0.736 | |
| Node2Vec [25] | CGP | 0.819 | 0.800 | 0.735 |
| CGPD | 0.849 | 0.798 | 0.683 | |
| SVD [26] | CGP | 0.833 | 0.823 | 0.772 |
| CGPD | 0.820 | 0.876 | 0.797 | |
| Laplacian [27] | CGP | 0.839 | 0.841 | 0.765 |
| CGPD | 0.860 | 0.873 | 0.744 | |
| GCN-Total [28] | CGP | 0.823 | 0.768 | 0.571 |
| CGINet [2] | CGP | 0.901 | 0.872 | 0.770 |
| BioNet | CGP | 0.948 | 0.939 | 0.886 |
| CGDP | 0.952 | 0.944 | 0.922 |
Additionally, we can clearly see that the introduction of disease-related data can further improve the performance of BioNet, as demonstrated in Table 3, where BioNet–CGPD is superior to BioNet–CGP.
Specifically, it is inspiring to see that BioNet–CGPD outperforms BioNet–CGP by 5.6% on AP@20, indicating that under the joint embedding of the pathway-related interactions and disease-related interactions, the top-20 instances selected by the BioNet model have higher accuracy. This also indicates that a more comprehensive data collection is essential for relation prediction tasks on a heterogeneous network. However, more data imply a graph with a grander scale, which poses a great challenge for either computation or model robustness. Our previous work [2] can only deal with CGP-graph, whereas BioNet is capable of processing a larger scale graph with a higher precision.
Computational optimization
The device memory of a single NVIDIA TESLA V100 GPU is 16 GB, which is insufficient to fit the scale of our model (the estimated size is about 20 GB). By distributed computing, we use multiple GPUs to enable BioNet to learn all the knowledge provided by all data, and improve the model’s ability to process big data. More importantly, we have accelerated the training time of the model and advanced the training efficiency of the model.
We evaluated the parallel processing performance of BioNet on different numbers of V100 GPUs. Figure 7 shows the time spent with different numbers of GPUs on training BioNet-CGP and BioNet-CGPD. Note that a single GPU cannot fulfill the computation of BioNet-CGPD. As the number of GPUs increases, the time cost decreases significantly. For example, with the same size of datasets (BioNet-CGP), under the parallel algorithm, the calculation time of a single EPOCH is reduced by nearly 7 h. That is, the parallel efficiency when computing with 16 GPUs is:
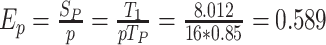 |
(5) |
Figure 7.

The time cost of processing on different numbers of GPUs. Note. A single GPU cannot fulfill the computation of BioNet–CGD and BioNet–CGPD.
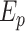 represents parallel efficiency,
represents parallel efficiency,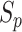 represents speedup,
represents speedup, 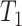 refers to the execution time of the sequential execution algorithm,
refers to the execution time of the sequential execution algorithm,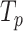 refers to the execution time of the parallel execution algorithm,
refers to the execution time of the parallel execution algorithm,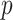 represents the number of GPU.
represents the number of GPU.
Case study
To further exemplify how BioNet can boost relevant biomedical studies, we carried out a few case studies with BioNet to serve as examples.
(1) Identification of targets related to cancer
Because of the various pathogenic mechanisms and changeable pathogenesis sites, the research on cancer has never stopped, and a considerable amount of data have been accumulated. Relevant studies have proved that various therapeutics may have a certain therapeutic effect, such as aspirin, vitamin D, etc.
We retrieved some top genes related to cancer according to CTD. We then used BioNet to obtain all related chemical predictions and exclude existing relation pairs in the source database. The remaining predictions were ranked in a descending order, as listed in Supplementary Table S1 (https://github.com/yangxi1016/BioNet). A higher score indicates a higher predicted probability of the potential interaction. Next, we searched for the entity pairs in search engines (Google Scholar and PubMed) to find supporting evidence in the literature. Table 4 provides the top 3 and partially verifiable CGIs related to cancer predicted by BioNet.
Table 4.
Partially verifiable CGIs of cancer predicted by BioNet
| Predicted ranking | Relation type | Chemical–gene pairs | Probability score 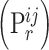
|
Evidence |
|---|---|---|---|---|
| 1 | Increases^abundance | <Succinic Acid,BCL2> | 0.7307 | NA |
| 2 | Increases^abundance | <agathisflavone, BCL2> | 0.7303 | NA |
| 3 | Increases^abundance | <C646 compound, BCL2> | 0.7303 | NA |
| 4 | Increases^glucuronidation | <Calcium, RELA> | 0.7301 | [32] |
| 41 | Increases^glucuronidation | <Navitoclax, AKT1> | 0.7293 | [29] |
| 49 | Increases^glucuronidation | <Icilin, BAX> | 0.7292 | [33] |
| 56 | Decreases^secretion | <Oxytocin, VEGFA> | 0.7292 | [34] |
In Table 4, each row presents a predicted result of chemical C (node 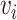 ) and gene G (node
) and gene G (node 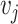 ), with literature evidence listed if applicable. The score
), with literature evidence listed if applicable. The score 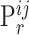 represents the predicted probability of the link
represents the predicted probability of the link 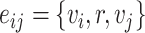 generated by BioNet’s tensor factorization decoder.
generated by BioNet’s tensor factorization decoder.
We easily found direct literature evidence for many predicted CGIs. For instance, the treatment with Akt1 inhibitor and BCL-xL inhibitor (ABT-263/Navitoclax) significantly decreased the cancer cell survival [29].
For predictions without direct literature evidence, we can also find supporting information. For example, the first row in Table 4 indicates a high probability of an “increases^abundance” relation between succinic acid (chemical) and BCL2 (gene). The CTD database confirmed that succinic acid has various interactions with BCL2L1, including promoting product expression, promoting reaction activation, etc., and BCL2L1 is an expression product with a similar structure to BCL2. Moreover, artesunate [30] (dihydrocyanin-10-α- succinate) and 2,3-dimercaptosuccinic acid [31] (2,3-dimercaptosuccinic acid) can promote the expression and reaction of BCL2, and their molecular structures contain the succinic acid. This provides a reasonable explanation and support of the predicted result. For the second predicted result (increases^abundance, agathisflavone, BCL2, 0.73018), we did not find any direct literature evidence, but there are 22 kinds of 3′, 4′-dimrthoxyflavone and chrysin, which belong to the same flavonoids as agathisflavone. The interaction relation with BCL2 can be retrieved, so this prediction result is reasonable. For the third predicted result (increases^abundance, C646 compound, BCL20.72974), compound C646 is a benzoate with a complex structure, and it does have an interaction relation with BCL2’s allotrope BCL2L1 [30], so it is reasonable to assume that this result is explainable.
(Reference for Table 4: [29, 32–34])
The above results demonstrated that our BioNet model can predict correct results not included in curated databases like CTD and provide the potential targets for subsequent experimental verification. In summary, BioNet can help identify CGIs for a given chemical (or a target gene).
(2) COVID-19
The coronavirus pneumonia (COVID-19) caused by the SARS-CoV-2 virus swept the world. In the latest collection of CTD, the genes related to COVID-19 including ACE2, CD9, DPP4, TIPARP, TMPRSS2, etc. To verify the capability that BioNet seeks out novel and credible candidates for COVID-19 (Supplementary Table S2), we obtained the top ten candidates (Table 5) targeted COVID-related genes and sorted the predicted scores in the 65 types of CGIs.
Table 5.
Partially verifiable CGIs of COVID-19 predicted by BioNet
| Predicted ranking | Relation type | Chemical–gene pairs | Probability score | Evidence |
|---|---|---|---|---|
| 1 | Increases^ADP-ribosylation | <Aroclor 1242, DPP4> | 0.727946 | NA |
| 2 | Increases^methylation | <Ethyl methanesulfonate, TMPRSS2> | 0.7279444 | NA |
| 3 | Affects^expression | <Carbamates, TIPARP> | 0.727942 | NA |
| 4 | Affects^reaction | <2-Aminopyrimidine, TIPARP > | 0.7279404 | NA |
| 5 | Increases^glucuronidation | <Lipopolysaccharides, DPP4> | 0.72794 | [35] |
| 11 | Increases^glucuronidation | <Guanylin, DPP4> | 0.72793 | [36] |
| 49 | Increases^glucuronidation | <C-Peptide, DPP4> | 0.72791 | [37] |
(Reference for Table 5: [35–37])
There are 1749 items with a predicted score over 0.7. Among the top 50 predicted relations, we have found four CGIs directly documented in the literature. COVID-19 infection may aggravate nephritis and diabetes. In [35–37], the effects of lipopolysaccharides, guanylin and C-peptide on DDP4 are mentioned, which are used to treat nephropathy and related diabetes.
The first CGI in Table 5 (increases^ADP ribosylation, Aroclor 1242, DPP4, 0.72034). Although this is not curated in the CTD database, we found a polychlorinated biphenyl compound Aroclor 1254 with structural characteristics similar to compound Aroclor 1242, which has a degree^expression effect on DPP4 [38]. More importantly, the relation type mentioned in the prediction results is increases^ADP ribosylation. ADP ribosylation adds one or more ADP riboses to the target protein to affect the protein’s function. Therefore, we have the reason to suspect that Aroclor 1242 can interact with DPP4, and ultimately reduce the expression of DPP4 by promoting ADP ribosylation. The second CGI in Table 5 (increases^methylation, ethyl methanesulfonate, TMPRSS2, 0.71408) is also absent from the curated database. However, according to relevant records in the CTD database, there are nine chemicals containing ethyl, such as atrazine and disopyramide, which have different effects on the expression of TMPRSS2, and the predicted compounds are methanesulfonate, camostat, which belongs to this class of compounds, can reduce the activity of TMPRSS2 [39], so it is predicted that chemicals may affect TMPRSS2 by promoting methylation. The third CGI in Table 5 (affects-expression, carbamates, TIPARP, 0.71146), carbamates represent urethane chemicals. While enterostat and urethane, which also belong to this compound, have the effect of inhibiting and promoting expression with TIPPARP, respectively, and therefore the prediction is reasonable.
In addition, the spike protein of the new coronavirus is located on the surface of the virus and mediates the binding of the virus to the ACE2 receptor [40] of the host cell, thereby helping the virus invade and infect the host, and therefore become the target of many vaccines and antibody drugs.
We choose ACE2 as the target and construct a dataset that includes all chemicals-ACE2 pairs as test data. Then, we used BioNet to make predictions. The full result set is publicly available on our GitHub repository (https://github.com/yangxi1016/BioNet).
We listed the top 10 predicted CGIs related to ACE2 in Table 6. We found direct literature evidence for some CGI predictions. It is particularly noteworthy that according to the latest research outcomes in 2021, defibrotide can be used to treat endothelins that can complicate COVID-19 [41], L-carnitine tartrate downregulates the ACE2 receptor to limits SARS-CoV-2 infection [42] and fluoxetine [43] may interact with ACE2 receptors and can be used to treat COVID19.
Table 6.
Chemicals–ACE2 interaction prediction probability score Top-10
| Predicted ranking | Relation type | Chemicals | Probability score | Evidence |
|---|---|---|---|---|
| 1 | Affects^activity | Defibrotide | 0.72686 | [41] |
| 2 | Increases^stability | Carnitine | 0.722646 | [42] |
| 3 | Affects^chemical synthesis | Sodium | 0.711425 | NA |
| 4 | Affects^chemical synthesis | Alectinib | 0.719485 | NA |
| 5 | Increases^sumoylation | Moxonidine | 0.710244 | NA |
| 6 | Affects^cotreatment | Thiamylal | 0.709306 | NA |
| 7 | Increases^uptake | Pirprofen | 0.704563 | NA |
| 8 | Increases^sumoylation | Huangqin-Tang | 0.697992 | NA |
| 9 | Increases^transport | Thiobarbituric Acid Reactive Substances | 0.687303 | NA |
| 10 | Increases^uptake | Fluoxetine | 0.676407 | [43] |
Other predictions without direct literature evidence provide valuable possibilities for further studies.
In a recent study, Zhe et al. [44] reported a virtual screening method with accelerated free energy-perturbation-based absolute binding free energy (FEP-ABFE) predictions and confirmed 15 chemicals targeting the SARS-CoV-2 main protease (Mpro) with wet experiments, including candesartan cilexetil, dipyridamole, atazanavir, indinavir and omeprazole, etc. Thus, we retrieve all related genes (provided by CTD) and chemicals from the source data set obtained a validation data set 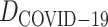 , and predict relation probabilities with BioNet. The prediction scores are mostly over 0.7, which indicates the chemical screening by Mpro has high interaction on genes related to COVID-19 (Supplementary Table S3). Most importantly, the commonly used antithrombus drug dipyridamole has been proved as an effective ancillary drug in the therapy of severely ill patients with COVID-19 [45], which is also supported by the BioNet model. Figure 8 shows partial interaction prediction in
, and predict relation probabilities with BioNet. The prediction scores are mostly over 0.7, which indicates the chemical screening by Mpro has high interaction on genes related to COVID-19 (Supplementary Table S3). Most importantly, the commonly used antithrombus drug dipyridamole has been proved as an effective ancillary drug in the therapy of severely ill patients with COVID-19 [45], which is also supported by the BioNet model. Figure 8 shows partial interaction prediction in 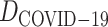 . This further proved that our model can provide new theoretical support for drug screening.
. This further proved that our model can provide new theoretical support for drug screening.
Figure 8.

Partial relation prediction diagram in 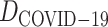 .
.
Based on a high-performance computer, the researchers screened out potential anti-coronavirus emergency 33 drugs based on FEP-ABFE. We found 19 FDA-proved chemicals were contained in BioNet. BioNet predicts that some drugs are likely to act on COVID-19’s target genes, as shown in Table 7, and 10 associations have a predicted probability of more than 0.6.
Table 7.
High-probability prediction of FDA-proved chemicals interaction with COVID-19 targets gene
| Predicted ranking | Relation type | Chemicals | Probability score |
|---|---|---|---|
| 1 | Increases^ADP-ribosylation | <Hesperetin, TMPRSS4> | 0.717661 |
| 2 | Increases^hydrolysis | <Riociguat, TMPRSS11A> | 0.697253 |
| 3 | Decreases^acetylation | <Proanthocyanidins, CD9> | 0.691185 |
| 4 | Decreases^activity | <Doxazosin, CD9> | 0.672306 |
| 5 | Increases^sumoylation | <Nisoldipine, CD9> | 0.663409 |
| 6 | Increases^mutagenesis | <Demeclocycline, TMPRSS4> | 0.644651 |
| 7 | Increases^oxidation | <Riociguat, ACE2> | 0.642546 |
| 8 | Increases^hydroxylation | <Doxazosin, TMPRSS4> | 0.633889 |
| 9 | Increases^reaction | <Hesperetin, TMPRSS2> | 0.627183 |
| 10 | Decreases^transport | <Proanthocyanidins, ACE2> | 0.618641 |
The FEP-based method consumes an astonishing amount of computing resources and computing time. Although BioNet, a smart drug screening method, cannot completely replace the traditional FEP-based method, its effective calculation results and efficient screening speed, on the one hand, can pre-screen drugs before using traditional methods, reducing the need for traditional methods The data range of the calculation, thereby improving the calculation efficiency and reducing the consumption of computing resources. On the other hand, it can mutually confirm the results of traditional methods and provide a certain degree of mechanism explanation.
Discussion
The abundance of drug-related data offers tremendous opportunities to generate new insights and develop better approaches for drug discovery. The heterogeneous information fusion of biomedical data from different sources can systematically understand the mechanisms of biopharmaceuticals, provide a more comprehensive and effective support for drug repurposing, and increase the accuracy of predictions.
Besides CGI, other types of relations are also important for drug repurposing, according to recent studies that have attracted a lot of attention. Zhao et al. [46] first used a graph convolutional network to learn the features for each drug–protein pairs (DPP), and presented (GCN)-DTI to the identification of new drug–target interactions (DTIs). By evaluation, (GCN)–DTI outperforms superior to state-of-the-art DTI prediction methods. Liu et al. [47] presented DeepCDR, a hybrid graph convolutional network (UGCN) for exploring intrinsic chemical structures of drugs for predicting cancer drug response (CDR), and the successful use of the synergy of multi-omics profiles significantly improves the performance of CDR prediction. Kumar et al. [48] comprehensively analyzed the works and data tackling the COVID-19 pandemic and integrated heterogeneous COVID-19 data sources by various data processing methods, provided biomedical research and drug/vaccine designers with available systematic datasets, and computational biology and bioinformatics approaches.
The above models provide important foundation and evidence to support our study. In addition, Wang et al. [49] introduced a bipartite GCN model named BiFusion, which presents a better method of extracting and fusing information from the protein–protein interaction (PPI) network for discovering novel drug–disease association. The results provide inspiration that the addition of PPI network as an extra dimension of information that could be potentially valuable to enhance of our model in the future work. Based on the subgraph segmentation strategy, our model shows strong expansibility for different types of relation information. Therefore, it is feasible to integrate extra information like protein–protein interactions using the framework provided in this paper. We will further expand the type of entities and interactions in our future work.
Conclusion
In this study, we proposed BioNet, a graph neural network model that integrates interaction information of biomedical entities including chemicals, genes, pathways and diseases to predict CGIs. BioNet adopts the graph encoder–decoder architecture. In the encoder part, initial node embeddings are first learnt from binary subgraphs and then transferred to the whole graph for a second round of encoding. This scheme greatly reduces the complexity of the model. The decoder is implemented as a tensor decomposition task based on the RESCAL algorithm, which significantly reduce the number of parameters compared with the canonical DEDECOM method. Evaluation results indicate that our model outperformed existing methods, which can be attributed to the fact that we employed more curated data in the context and we developed a more suitable architecture for multi-type relation prediction. To note, the introduction of more curated information leads to a massive graph, which is computationally challenging and goes beyond the capability of previous models. Therefore, our parallel processing strategy is also key to enable scalable computation in the training and the prediction process. Finally, to further manifest the reliability and high quality of the prediction results of BioNet, we performed case studies related to cancer and COVID-19. The cases demonstrate how BioNet can be applied to prioritize drug candidate given the complex relations related to the disease of interest.
In the future work, we would like to further expand the entity types in BioNet. Currently, the network used in this article consists of only four types of entities: chemicals, genes, pathways and diseases. As we have solved the scalability and computing capacity problem, we can add more types of entities like symptoms and enrich the prediction of various biomedical relations to give more research inspiration to biomedical studies.
- Abbreviation
- GCN
graph convolution network
- CGI
chemical–gene interaction
- CTD
comparative toxicogenomics database
Key Points
We constructed a comprehensive and large-scale heterogeneous biological interaction network by integrating curated datasets related to chemicals, genes, pathways and diseases.
We proposed a deep graph neural network model named BioNet based on an encoder–decoder architecture, which utilizes a graph convolution encoder to learn entity embeddings from subgraphs and employs a tensor decomposition decoder to predict chemical–gene interactions (CGIs).
We developed a parallel strategy to boost the learning process and improved the model’s ability to handle large-scale data.
We exemplified the value of BioNet by evaluating the CGIs of cancer and COVID-19, which prioritizes chemicals with higher potential for effective therapeutics.
Xi Yang is currently a Ph.D. student in the College of Computer, National University of Defense Technology, China. Her researches focus on biomedical text mining, knowledge graph and high-performance computing.
Wei Wang is currently a software engineer in the National Supercomputer Center in Tianjin, China. His researches focus on biomedical text mining, knowledge graph and high-performance computing.
Jing-Lun Ma is currently a postgraduate student in the College of Computer, National University of Defense Technology, China. His researches focus on the biomedical knowledge graph.
Yan-Long Qiu is currently a Ph.D. student in the College of Computer, National University of Defense Technology, China. His researches focus on biomedical text mining and knowledge graph.
Kai Lu is currently the Dean of the College of Computer, National University of Defense Technology. His researches focus on operating system, computer architecture, high-performance computing and quantum computing.
Dong-Sheng Cao is currently a professor in the Xiangya School of Pharmaceutical Sciences, Central South University, China. His research interests include chemo-informatics, bioinformatics, drug design, chemo- and geo-informatics, web server and database, machine learning. Further information about Dong-Sheng Cao can be found at the website of his group: http://www.scbdd.com.
Cheng-Kun Wu is currently an associate professor Institute for Quantum Information & State Key Laboratory of High Performance Computing, College of Computer Science and Technology, National University of Defense Technology, Changsha 410073, China. His research focus on high-performance computing, computational systems biology and biomedical data mining.
Contributor Information
Xi Yang, College of Computer, National University of Defense Technology, China.
Wei Wang, National Supercomputer Center in Tianjin, China.
Jing-Lun Ma, College of Computer, National University of Defense Technology, China.
Yan-Long Qiu, College of Computer, National University of Defense Technology, China.
Kai Lu, College of Computer, National University of Defense Technology, China.
Dong-Sheng Cao, Xiangya School of Pharmaceutical Sciences, Central South University, China.
Cheng-Kun Wu, Institute for Quantum Information & State Key Laboratory of High Performance Computing, College of Computer Science and Technology, National University of Defense Technology, Changsha 410073, China.
Authors’ contributions
C.W. and D.C. set up the general idea of this project and revised the whole manuscript. X.Y. and W.W. developed the algorithms and drafted the manuscript. They developed the codes, prepared the datasets for testing. J.M. and Y.L. designed a test experiment. Drafted the discussion and revised the whole manuscript together with C.W. and K.L.. All authors have read and approved the manuscript.
Funding
Computing resources are supported by Tianhe Supercomputer Project 2018YFB0204301. Publication costs are funded by the National Key R&D project by the Ministry of Science and Technology of China (2018YFB1003203), the open fund from the State Key Laboratory of High-Performance Computing (No. 201901-11) the National Science Foundation of China (U1811462,22173118), and Hunan Provincial Science Fund for Distinguished Young Scholars (2021JJ10068). The funder C.W. and K.L. took part in the formulation and development of methodology and provided financial support for this study.
Data availability
All datasets and codes used in this study are available at GitHub: https://github.com/yangxi1016/BioNet.
References
- 1. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 2019;18:41–58. [DOI] [PubMed] [Google Scholar]
- 2. Wang W, Yang X, Wu C, et al. CGINet: graph convolutional network-based model for identifying chemical-gene interaction in an integrated multi-relational graph. BMC Bioinformatics 2020;21(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ge L, Wu K, Zeng Y, et al. Multi-scale spatiotemporal graph convolution network for air quality prediction. Appl Intell 2021;51(6):3491–505. [Google Scholar]
- 4. Abu-El-Haija S, Kapoor A, Perozzi B, et al. N-GCN: multi-scale graph convolution for semi-supervised node classification, Proceedings of The 35th Uncertainty in Artificial Intelligence Conference, Israel: PMLR. Tel Aviv, 2020;841–851.
- 5. Koreneva M, Visheratin AA, Nasonov D. Decoupling graph convolutional networks for large-scale supervised classification. Proc Comput Sci 2020;178:337–44. [Google Scholar]
- 6. Hopkins AL. Network pharmacology. Nat Biotechnol 2007;25:1110–1. [DOI] [PubMed] [Google Scholar]
- 7. Kun-Yi H, Yukiko M, Yoshiyuki A, et al. SystemsDock: a web server for network pharmacology-based prediction and analysis. Nucleic Acids Res 2016;44(W1):W507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sosa DN, Derry A, Guo M, et al. A literature-based knowledge graph embedding method for identifying drug repurposing opportunities in rare diseases, In PACIFIC SYMPOSIUM ON BIOCOMPUTING. the Big Island of Hawaii. 2020, 463–474. [PMC free article] [PubMed]
- 9. Wang R, Li S, Cheng L, et al. Predicting associations among drugs, targets and diseases by tensor decomposition for drug repositioning. BMC Bioinformatics 2019;20(S26):628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen H, Li J. Learning data-driven drug-target-disease interaction via neural tensor network. Yokohama, Japan: International Joint Conferences on Artificial Intelligence IJCAI. Yokohama, Japan. 2020, 3452–3458.
- 11. Capuzzi SJ, Thornton TE, Liu K, et al. Chemotext: a publicly available web server for mining drug–target–disease relationships in PubMed. J Chem Inf Model 2018;58:212–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao P, Zhang J, Sun Y, et al. Accurate predictions of aqueous solubility of drug molecules via the multilevel graph convolutional network (MGCN) and SchNet architectures. J Mach Learn Res 2020;22:23766–72. [DOI] [PubMed] [Google Scholar]
- 13. Yu Z, Huang F, Zhao X, et al. Predicting drug-disease associations through layer attention graph convolutional network. Brief Bioinform 2020;22(4). [DOI] [PubMed] [Google Scholar]
- 14. Ioannidis VN, Zheng D, Karypis G. Few-shot link prediction via graph neural networks for covid-19 drug-repurposing . arXiv preprint arXiv:2007.10261. 2020.
- 15. Das D, Arber N, Jankowski JA. Chemoprevention of colorectal cancer. Digestion 2007;76:51–67. [DOI] [PubMed] [Google Scholar]
- 16. Redka DS, Mackinnon SS, Landon M, et al. PolypharmDB, a deep learning-based resource quickly identifies repurposed drug candidates for COVID-19. ChemRxiv 2020. doi: 10.26434/chemrxiv.12071271.v1. [DOI]
- 17. Kuhn M, Mering CV, Campillos M, et al. STITCH: interaction networks of chemicals and proteins. Nucleic Acids Res 2007;36:D684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marinka Z, Monica A, Jure L. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018;34(13):i457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis AP, Grondin CJ, Johnson RJ, et al. The comparative toxicogenomics database: update 2019. Nucleic Acids Res 2018;47(D1):D948–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Z, Li M, Lin X, et al. Network-wide traffic flow estimation with insufficient volume detection and crowdsourcing data. Transport Res Part C Emerg Technol 2020;121:102870. [Google Scholar]
- 21. Papalexakis Evangelos E, Christos Faloutsos, Nicholas D. Sidiropoulos. Tensors for data mining and data fusion: Models, applications, and scalable algorithms. ACM Trans Intell Syst Technol 2016;8(2):1–44.28344853 [Google Scholar]
- 22. Technologies behind distributed deep learning . 2018. https://tech.preferred.jp/en/blog/technologies-behind-distributed-deep-learning-allreduce/.
- 23. Wu Y, Liu Y. Robust truncated hinge loss support vector machines. J Am Stat Assoc 2007;102:974–83. [Google Scholar]
- 24. Perozzi B, Al-Rfou R, Skiena S. DeepWalk: online learning of social representations. Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. New York, New York, USA: Association for Computing Machinery, 2014, 701–710. [Google Scholar]
- 25. Grover A, Leskovec J. node2vec: scalable feature learning for networks. Proceedings of the 22nd ACM SIGKDD international conference on Knowledge discovery and data mining San Francisco, CA, USA, 2016, 855–864. [DOI] [PMC free article] [PubMed]
- 26. Golub GH. Singular value decomposition and least squares solutions. Numer Math 1970;14:403–20. [Google Scholar]
- 27. Deng C, He X, Han J, et al. Graph regularized non-negative matrix factorization for data representation. IEEE Trans Pattern Anal Mach Intell 2011;33:1548–60. [DOI] [PubMed] [Google Scholar]
- 28. Kip FTN, Welling M. Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907, 2016.
- 29. Kim H, Prasain N, Vemula S, et al. Human platelet lysate improves human cord blood derived ECFC survival and vasculogenesis in three dimensional (3D) collagen matrices. Microvasc Res 2015;101:72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou X, Chen Y, Wang F, et al. Artesunate induces autophagy dependent apoptosis through upregulating ROS and activating AMPK-mTOR-ULK1 axis in human bladder cancer cells. Chem Biol Interact 2020;331:109273. [DOI] [PubMed] [Google Scholar]
- 31. Pachauri V, Mehta A, Mishra D, et al. Arsenic induced neuronal apoptosis in guinea pigs is Ca2+ dependent and abrogated by chelation therapy: role of voltage gated calcium channels. Neurotoxicology 2013;35:137–45. [DOI] [PubMed] [Google Scholar]
- 32. Xia W, Bacus S, Husain I, et al. Resistance to ErbB2 tyrosine kinase inhibitors in breast cancer is mediated by calcium-dependent activation of RelA. Mol Cancer Ther 2010;9:292. [DOI] [PubMed] [Google Scholar]
- 33. Cheng QY, Yang MC, Wu J, et al. Reduced cardiac ischemia/reperfusion injury by hypothermic reperfusion via activation of transient receptor potential M8 channel. Life Sci 2019;232:116658. [DOI] [PubMed] [Google Scholar]
- 34. Zhong M, Boseman ML, Millena AC, et al. Oxytocin as a potential autocrine regulator of prostate cancer metastasis. ENDOCRINE REVIEWS 2010;31(3):20815–5817. [Google Scholar]
- 35. Xingyun H, Yan L, Shanying L, et al. Effect of DPP4/CD26 inhibitor on LPS-induced inflammation in islet β cells. Chin J Clin (Electronic Edition) 2014;8:102–105. [Google Scholar]
- 36. Vallon V, Docherty NG. Intestinal regulation of urinary sodium excretion and the pathophysiology of diabetic kidney disease: a focus on glucagon-like peptide 1 and dipeptidyl peptidase 4. Experimental physiology 2014;99(9):1140–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alsalim W, Persson M, Ahrén B. Different glucagon effects during DPP-4 inhibition versus SGLT-2 inhibition in metformin-treated type 2 diabetes patients. Diabetes Obes Metab 2018;20(7):1652–58. [DOI] [PubMed] [Google Scholar]
- 38. Cai J, Wang C, Huang L, et al. A novel effect of polychlorinated biphenyls: impairment of the tight junctions in the mouse epididymis. Toxicol Sci Off J Soc Toxicol 2013;134:382–90. [DOI] [PubMed] [Google Scholar]
- 39. Qiao Y, Wang XM, Mannan R, et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc Natl Acad Sci 2021;118:e2021450118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moraleda JM, Carlo-Stella C, García-Bernal D, et al. D3Targets-2019-nCoV: a webserver for predicting drug targets and for multi-target and multi-site based virtual screening against COVID-19. Acta Pharm Sin B 2020;10(7):1239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moraleda JM, Carlo-Stella C, García-Bernal D, et al. Defibrotide for the treatment of endotheliitis complicating SARS-CoV-2 infection: rationale and ongoing studies as part of the international DEFACOVID Study Group. Blood 2020;136:6–8.32614958 [Google Scholar]
- 42. Bellamine A, Pham TNQ, Jain J, et al. L-carnitine tartrate downregulates the ACE2 receptor and limits SARS-CoV-2 infection. Nutrients 2021;13(4):1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheikhpour M. The current recommended drugs and strategies for the treatment of coronavirus disease (COVID-19). Ther Clin Risk Manag 2020;16:933–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Z, Li X, Huang Y, et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc Natl Acad Sci 2020;117:27381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu X, Li Z, Liu S, et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm Sin B 2020;10(7):1205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao T, Hu Y, Valsdottir LR, et al. Identifying drug–target interactions based on graph convolutional network and deep neural network. Brief Bioinform 2021;22:2141–50. [DOI] [PubMed] [Google Scholar]
- 47. Liu Q, Hu Z, Jiang R, et al. DeepCDR: a hybrid graph convolutional network for predicting cancer drug response. Bioinformatics 2020;36:i911–8. [DOI] [PubMed] [Google Scholar]
- 48. Kumar Das J, Tradigo G, Veltri P, et al. Data science in unveiling COVID-19 pathogenesis and diagnosis: evolutionary origin to drug repurposing. Brief Bioinform 2021;22:855–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Z, Zhou M, Arnold C. Toward heterogeneous information fusion: bipartite graph convolutional networks for in silico drug repurposing. Bioinformatics 2020;36:i525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets and codes used in this study are available at GitHub: https://github.com/yangxi1016/BioNet.