
Figure 2: FGFR2 EIDs are oncogenic and confer sensitivity to FGFR inhibitors.
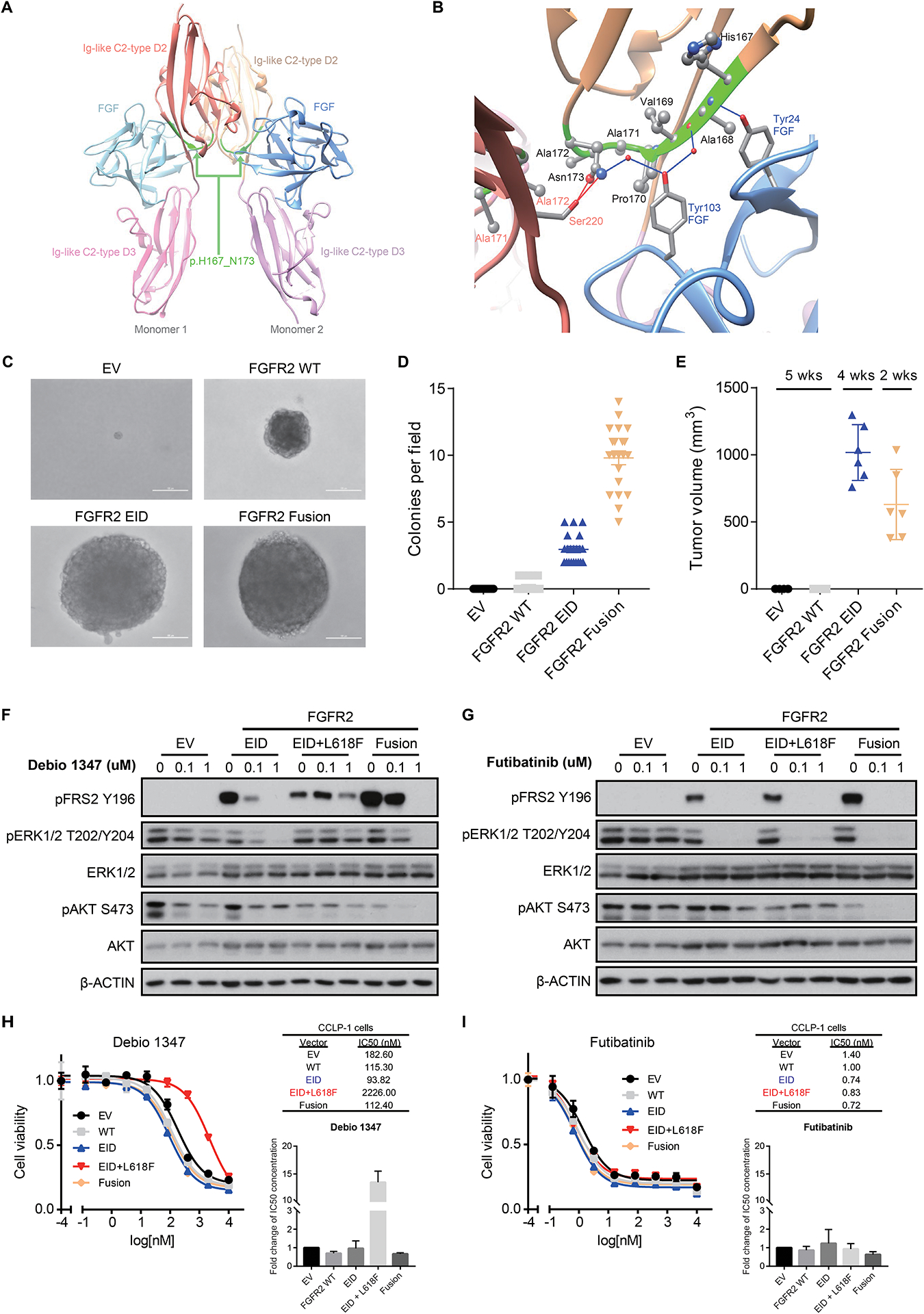
A, B. Modeling alteration p.H167_N173del within the three-dimensional structure of FGFR2. Panel A shows a global view of the p.H167_N173 deletion (ribbon highlighted in green) located in both Ig-like C2-type D2 extracellular domains of FGFR2 dimeric structure; refer to Supplemental Figure 7 (deletion #1) for mapping onto the protein sequence. As shown more closely in panel B, p.H167_N173 (ribbon highlighted in green) is an extended deletion, mostly included in a beta-strand of the C2-type D2 domain (ribbon in beige), and makes several intermolecular interactions with both the FGF ligand (ribbon and residue names in blue) and the C2-type D2 domain of the other monomer (ribbon and residue names in red). Intermolecular hydrogen-bonds (direct or water-mediated) between p.H167_N173 residues and FGF or the other monomer are shown as blue and red lines, respectively. Van der Waals contacts are not displayed explicitly. C, D. Expression of FGFR2 p.H167_N173del in NIH3T3 cells is sufficient for transformation in soft agar colony formation assays. Cells were transduced with retroviral vectors containing empty vector control (EV), wild-type FGFR2 (FGFR2 WT), FGFR2 p.H167_N173del (FGFR2 EID), or FGFR2-OPTN fusion (FGFR2 Fusion). Representative images (C) and quantification of number of colonies per field of view (D) are shown. Line and bars indicate mean with standard error, with 21 fields of view assessed across three biologic replicates. E. Volume of subcutaneous tumors forming in NSG mice following implantation of NIH3T3 cells expressing the indicated constructs (n=6 mice per condition). Both the FGFR2 p.H167_N173del EID and the FGFR2-PHGDH fusion (FGFR2 Fusion) induce tumor formation. Expression of the FGFR2-PHGDH fusion led to slightly faster tumor growth necessitating euthanasia at an earlier time point. Line and bars indicate mean with standard deviation. F, G. Expression of FGFR2 p.H167_N173del (EID), the EID with an L618F mutant kinase domain (EID+L618F), and FGFR2-PHGDH fusions (Fusion) lead to constitutive FGFR signaling (induction of FRS2) in NIH3T3 cells. FGFR2 EIDs and fusions are sensitive to treatment with Debio1347 (reduction in pFRS2 and pERK), whereas the L618F mutation causes resistance (F); all three of these FGFR2 alterations are inhibited by futibatinib (TAS-120) (G). H, I. Cell viability assays in CCLP-1 cells harboring empty vector control (EV), wild-type FGFR2 (WT), FGFR2 p.H167_N173del (EID), the co-occurring FGFR2 kinase mutation L618F (EID+L618F), or FGFR2-PHGDH fusion (Fusion) and treated with Debio 1347 (H) or futibatinib (I). Drug response measurements were performed in three independent experiments with each consisting of two technical replicates. Inset graphs demonstrate the average fold change in IC50 between conditions. Representative dose response curves and IC50 values are shown from a single experiment, with error bars on the curves representing standard deviation from 2 technical replicates.