

Abstract
Myocardial remodeling is a complex pathological process and its mechanism is unclear. The present study investigated whether epigallocatechin gallate (EGCG) prevents myocardial remodeling by regulating histone acetylation and explored the mechanisms underlying this effect in the heart of a mouse model of transverse aortic constriction (TAC). A TAC mouse model was created by partial thoracic aortic banding (TAB). Subsequently, TAC mice were injected with EGCG at a dose of 50 mg/kg/day for 12 weeks. The hearts of mice were collected for analysis 4, 8 and 12 weeks after TAC. Histopathological changes in the heart were observed by hematoxylin and eosin, Masson trichrome and wheat germ agglutinin staining. Protein expression levels were investigated using western blotting. Cardiac function of mice was detected by echocardiography. The level of histone acetylated lysine 27 on histone H3 (H3K27ac) first increased and then decreased in the hearts of mice at 4, 8 and 12 weeks after TAC. The expression levels of two genes associated with pathological myocardial remodeling, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), also increased initially but then decreased. The expression levels of histone deacetylase 5 (HDAC5) gradually increased in the hearts of mice at 4, 8 and 12 weeks after TAC. Furthermore, EGCG increased acetylation of H3K27ac by inhibiting HDAC5 in the heart of TAC mice treated with EGCG for 12 weeks. EGCG normalized the transcriptional activity of heart nuclear transcription factor myocyte enhancer factor 2A in TAC mice treated for 12 weeks. The low expression levels of myocardial remodeling-associated genes (ANP and BNP) were reversed by EGCG treatment for 12 weeks in TAC mice. In addition, EGCG reversed cardiac enlargement and improved cardiac function and survival in TAC mice when treated with EGCG for 12 weeks. Modification of the HDAC5-mediated imbalance in histone H3K27ac served a key role in pathological myocardial remodeling. The present results show that EGCG prevented and delayed myocardial remodeling in TAC mice by inhibiting HDAC5.
Keywords: EGCG, histone acetylation, myocardial remodeling, mice, HDAC, treatment
Introduction
Cardiovascular disease is the leading cause of mortality worldwide and includes coronary, congenital and rheumatic heart, as well as peripheral arterial disease and chronic heart failure (CHF) (1). Pathological myocardial remodeling is a necessary stage of CHF that refers to the constant adjustment of the structure and function of the heart under the influence of endogenous and/or exogenous factors (2,3). The pathogenesis of pathological myocardial remodeling remains unclear. Therefore, it is important to understand the pathogenesis of myocardial remodeling as this may help to develop more efficient treatment strategies to improve myocardial remodeling. In recent years, epigenetic modification has also been shown to be involved in the occurrence and development of a variety of cardiovascular diseases (4). Epigenetic modification is comprises three processes, namely DNA methylation, histone modification (including methylation, acetylation, phosphorylation, ubiquitination, and SUMOylation), and ncRNAs-based mechanisms (including microRNAs-miRNAs, long non-coding RNAs-lncRNAs, and circular RNAs-circRNAs) (5). It is increasingly recognized that epigenetic gene regulation serves an active role in pathological myocardial remodeling (6,7). Our previous studies confirmed that an imbalance of histone H3K9ac acetylation is involved in the early stages of pathological myocardial remodeling induced by pressure overload and is mediated by histone acetylase (HAT) (8-10). In addition, the association between histone acetylation and both cardiac hypertrophy and heart failure have been extensively studied worldwide (11,12). Histone acetylation modification is regulated by HATs and histone deacetylases (HDACs); different subtypes of HATs and HDACs may serve different roles in pathological myocardial remodeling (13,14).
The natural compound epigallocatechin gallate (EGCG) is an active polyphenolic catechin and accounts for ~59% of total catechins from the leaves of green tea (15). EGCG has been extensively studied as a bioactive dietary component against various types of carcinomas through multiple mechanisms such as antioxidation, induction of apoptosis, inhibition of angiogenesis and metastasis (16). The role of EGCG in cardiovascular disease has been studied, however, the specific mechanism of EGCG remains unknown (17,18). An increasing number of preclinical and clinical studies indicate that EGCG, a green tea extract, regulate histone acetylation modified by HDACs (19,20). The HDAC family comprises 18 members divided into four classes based on their structure and function; HDAC5 is a class II HDAC and is involved in cardiac hypertrophy (21,22). Furthermore, the abnormal expression of HDAC5 has been investigated during pathological myocardial remodeling in a mouse model of transverse aortic constriction (TAC) (23). However, whether EGCG can improve myocardial remodeling in the heart of TAC mice via HDAC5-mediated histone acetylation remains unknown. The present study investigated the effect of HDAC5-mediated histone acetylation on pathological myocardial remodeling in a mouse model of TAC to identify novel strategies to prevent and treat CHF.
Materials and methods
Animal treatment
A total of 120 pathogen-free Kunming male mice (weight, 25-30 g; age, 8-10 weeks) were purchased from the Animal Center of Zunyi Medical University (Guizhou, China). All animal experiments were approved by the Animal Protection and Use Committee of Zunyi Medical University and complied with Directive 2010/63/EU of the European Parliament. Animal studies were performed in the Physiology Research Laboratories of Zunyi Medical University, Guizhou, China. The mice were maintained in individually ventilated cages (25°C and 55-65% humidity) with a 12/12-h light/dark cycle and free access to standard laboratory mouse chow and water. Mice were exposed to pressure-overload by thoracic aortic banding (TAB); this procedure decreased the thoracic aortic diameter by 70%, as previously described (24,25). Prior to TAB surgery, 0.8% sodium pentobarbital (50 mg/kg) was intraperitoneally injected into each mouse for general anesthesia. Anesthesia was confirmed by the disappearance of the righting and pedal withdrawal reflex. Mice received pre- and post-operative analgesia (6 h post-surgery) via subcutaneous injection of 0.02 ml (0.3 mg/ml) buprenorphine hydrochloride solution (Merck KGaA), as previously described (26) Additional analgesia was administered as required. Mice were randomly divided into five groups (n=24/group): Normal, Sham, Vehicle (Veh), TAC and TAC + EGCG. For each animal, the left side of the chest was opened along the median of the sternum to expose the aortic arch. The transverse aortic arch was then ligated between the brachial and left common carotid arteries. Prior to ligation, a curved needle (diameter, ~0.4 mm) was used to narrow the vessel. Finally, the chest was sutured. The postoperative survival rate of TAC mice was 60-70%. After 4, 8 and 12 weeks of TAC, the hearts of experimental mice were collected to evaluate the effect of HDAC5-mediated modification on histone H3K27ac acetylation during myocardial remodeling induced by pressure overload. In addition, the effect of EGCG on myocardial remodeling induced by pressure overload was investigated, and different concentrations (0, 25, 50, 75 and 100 mg/kg) EGCG were used to screen the optimal dose of EGCG. Following the completion of TAB, each mouse received an intraperitoneal injection of EGCG (Selleck Chemicals; PubChem CID 65064) at a dose of 50 mg/kg/day for 12 weeks. Mice in the Sham group received surgery but were not ligated. In the Veh group, normal saline was injected intraperitoneally at the same dose and timepoints beginning 1 week after sham operation for 12 weeks. After 12 weeks, mice were sacrificed using CO2 overdose; death was verified by applying pressure on the mouse nail bed (toe-pinch reflex) and the heart from each animal was collected for analysis.
Stereoscopy and cardiac and lung mass index
The heart and lungs of mice were collected, and cardiac (heart weight/body weight) and lung mass index (lung weight/body weight) were tested. Subsequently, the morphology of hearts and lungs of mice were observed by stereoscope.
Hematoxylin and eosin (H&E) and Masson’s staining
Following harvesting, the heart tissue was fixed in 4% paraformaldehyde for 48 h, embedded in paraffin wax and cut into serial sections (4 µm thick). Following dewaxing and dehydration in a series of alcohol concentrations, the sections were washed, soaked with tap water for 5 min and then stained with 0.5% hematoxylin dye for 5 min (to highlight the nucleus), faded in 1% hydrochloric acid with ethanol for 5 min and then placed in 0.5% eosin solution for 2 min (Beijing Solarbio Scince & Technology Co., Ltd.). Additional sections were washed with distilled water and then stained with 0.7% Masson Lichun red acid compound red solution for 5 min (Beijing Solarbio Science & Technology Co., Ltd.). The sections were then stained in 2% glacial acetic acid solution for 3 min. Next, sections were differentiated with 1% phosphomolybdate aqueous solution for 2 min, stained with 0.5% aniline blue for 5 min and then soaked with 0.2% glacial acetic acid aqueous solution for 1 min. H&E- and Masson’s-stained sections were successively treated with 95 and 100% ethanol and xylene I and II, then sealed with neutral resin. Sections were observed under a fluorescence microscope (H&E, magnification, ×4; Masson’s, magnification, ×40). All processes were completed at room temperature.
Echocardiography measurement
Transthoracic echocardiograms were performed on experimental mice using a Vevo 770 High-Resolution echocardiograph (Visual Sonics), as described previously (27,28). Blood flow velocity was measured using p-mode images. The efficacy of TAC was detected by chest Doppler echocardiography 4 weeks after the operation.
Chromatin immunoprecipitation (ChIP)
ChIP trials were performed using a ChIP Assay kit (Merck KGaA). First, a glass homogenizer was used to prepare homogenate from heart tissue. This homogenate was then mixed with formaldehyde (1%) to cross-link DNA/protein complexes at room temperature for 15 min. After cross-linking, 1% SDS Lysis Buffer (cat. no. 20-163, Merck KGaA) was added and then the DNA was fragmented by sonication. Following centrifugation at 100 × g for 2 min for 30 min at 4°C with rotation, pre-cleared solution was used as a DNA input control. A total of 1,800 µl ChIP Dilution Buffer (cat. no. 20-153; Merck KGaA) was added to 200 µl sonicated cell supernatant for a final volume of 2 ml. Next, the protein-DNA complex was precipitated using monoclonal antibodies (anti-MEF2A; 4 µl, 1:500, cat. no. sc-17785; Santa Cruz Biotechnology, Inc.). The antibody-chromatin complexes were precipitated by overnight incubation with Protein A Agarose/Salmon Sperm DNA (60 µl) at 4°C. The agarose pellet was obtained by centrifugation (1,500 × g) at 4°C for 1 min. The immunoprecipitated complexes of Ab-protein-DNA were collected and washed with low-salt, high-salt and LiCl buffer and Tris-EDTA, and then buffered with an elution buffer. The cross-linking of protein-DNA complexes was reversed by incubation with 5 M NaCl at 65°C for 4 h, and the DNA was digested with proteinase K for 1 h at 45°C. A DNA Fragment Purification kit (Merck KGaA) was used to extract the DNA in accordance with the manufacturer’s instructions. All experiments included a positive (precipitated by anti-RNA polymerase II antibody) and a negative control (precipitated by normal mouse IgG) groups. Quantitative (q)PCR was performed after ChIP detection using ChIP Assay kit (Merck KGaA). SYBR-Green Master Mix II (Takara Biotechnology Co., Ltd.) was used to perform qPCR. The thermocycling conditions were as follows: Initial denaturation 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 15 sec and annealing/elongation at 58°C for 30 sec. β-actin was used as an internal reference and the 2−ΔΔCq method was used to determine relative gene expression (29). Lertpa-v1.0 software (Applied Biosystems, Thermo Fisher Scientific, Inc.) was used to analyze the results. The following primers were used for RT-qPCR: ANP forward, 5′-TCC TTG GTG TCT CTC GCT CT-3′ and reverse, 5′-CGC TGG CTT GCT TGT TGT A-3′; BNP forward, 5′-TGC TGT CCC TCT ATG CTT CC-3′ and reverse, 5′-CGC TGG CTT GCT TGT TGT A-3′; β-actin forward, 5′-CCT TTA TCG GTA TGG AGT CTG CG-3′ and reverse, 5′-CCT GAC ATG ACG TTG TTG GCA-3′.
Co-IP
Co-IP trials were performed using a Co-IP Assay kit (Thermo Fisher Scientific, Inc.). A total of 300 µl IP binding Buffer was added to 40 mg heart tissue with 1 mM PMSF protease inhibitor on ice for 15 min. Lysates were centrifuged at 14,000 × g for 10 min at 4°C. Then, ~25% of the supernatant was subjected to input assays and the remaining supernatant was used for the Co-IP assay. IP was performed by combining primary anti-HDAC5 (1 µl, 1:250; cat. no. 16166-1-AP; ProteinTech Group, Inc.), anti-H3K27ac (2.5 µl, 1:100; cat. no. ab4729; Abcam) and anti-MEF2A rabbit polyclonal anti-bodies (1 µl, 1:250; cat. no. sc-17785; Santa Cruz Biotechnology, Inc.), with Dynabeads protein G (1:250; cat. no. 2729S; Invitrogen; Thermo Fisher Scientific, Inc.) and western blotting. All procedures were performed in accordance with the manufacturer’s instructions. First, the primary antibody was combined with protein G magnetic beads. Then, a magnet was used to immunoprecipitate the target antigen (HDAC5) into immunoprecipitation buffer containing 1% Triton X-100, 0.5% NP-40, 20 mmol/l HEPES, 50 mmol/l NaCl and protease inhibitor, at pH 7.4. Next, the samples were washed three times with IP Washing Buffer for 2 min. The immobilized protein complex was eluted at 95°C in 1X SDS-PAGE Loading Buffer (25 µl) for 10 min. The supernatant was collected after magnetic separation. Western blotting was then performed with anti-HDAC5, anti-MEF2A and anti-H3K27ac. IgG was used as a negative control. The HDAC5 IP experiments were performed in triplicate.
Western blotting
Myocardial tissues were dissected. Nucleoproteins were extracted using a Nuclear Extraction kit (Merck KGaA). Protein concentration were tested by BCA method. An equal amount of nucleoprotein extracts (40 µg/lane) were separated by 8/12% SDS-PAGE and then transferred onto polyvinylidene difluoride (PVDF) membranes (Merck KGaA). Next, the PVDF membranes were blocked with 5% non-fat milk in tris-buffered saline containing 0.1% Tween-20 for 2 h at room temperature and incubated at 4°C overnight with monoclonal antibodies [anti-HDAC5, 1:1,000, cat. no. 16166-1-AP; ProteinTech Group Inc.; anti-H3K27ac, 1:200, cat. no. ab4729, Abcam; anti-myocyte enhancer factor (MEF) 2A, 1:1,000, cat. no. sc-17785; Santa Cruz Biotechnology, Inc.; anti-brain natriuretic peptide (BNP), 1:1,000, cat. no. ab239510; Abcam; anti-atrial (A)NP, 1:1,000, cat. no. ab236101; Abcam; anti-H3, 1:2,000, cat. no. ab1791; Abcam and anti-β-actin, 1:1,000; cat. no. TA-09; OriGene Technologies, Inc.]. The next day, membranes were washed in Tris-buffered saline containing 0.1% Tween-20. The membranes were then incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibody (1:2,000, cat. no. Sc-47778; Santa Cruz Biotechnology). Protein bands were visualized by Enhanced Chemiluminescence System (GE Healthcare). Positive bands were quantified using Quantity One (version 4.4) software package (Bio-Rad Laboratories, Inc.).
RNA isolation and reverse transcription (RT)-qPCR
Total RNA was extracted from myocardial tissue using an RNA Extraction kit (BioTeke Corporation) in accordance with the manufacturer’s instructions. Single-stranded complementary DNA (cDNA) was synthesized from total RNA using primers and PrimeScript RT reagent (Takara Biotechnology Co., Ltd.). cDNA was subjected to RT-qPCR using gene-specific primers and SYBR-Green Master Mix II (Takara Biotechnology Co., Ltd.). The thermocycling conditions were conducted as follows: Initial denaturation 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 15 sec and annealing/elongation at 57°C for 30 sec. β-actin was used as an internal reference and the 2−ΔΔCq method was used to determine relative gene expression (29). Lertpa-v1.0 software was used to analyze the results. The following primers were used for RT-qPCR: MEF2A forward, 5′-CAC TTC CTT GGA CTA CTT GTT TCG T-3′ and reverse, 5′-GTT CCT GCT TTT CTA CTG CTC TGT T-3′ and β-actin forward, 5′-AGA AAA TCT GGC ACC ACA CC-3′ and reverse, 5′-CAG AGG CGT ACA GGG ATA GC-3′.
Wheat germ agglutinin (WGA) staining
Paraffin-embedded sections were treated with EDTA antigen repair buffer (pH 8.0; Beijing Solarbio Science & Technology Co., Ltd.) for antigen repair at 37°C for 20 min. After cooling, slides were washed in PBS buffer. Next, WGA dye (1:1,000; GeneTex) was added dropwise, then washed with PBS buffer. After the slides had partially dried, DAPI (5 ng/ml) solution (Beijing Solarbio Science & Technology Co., Ltd.) was added and incubated at room temperature for 3 min in the dark. The slides were then PBS washed and incubated with Autofluorescence Quenching Agent kit (Thermo Fisher Scientific, Inc.). The sections were then shaken dry and sealed with anti-fluorescence quenching sealing tablets. Finally, sections were observed under a fluorescence microscope (magnification, ×20; Olympus Corporation) and images were captured.
Statistical analysis
All data are expressed as the mean ± SD of 3 independent experiments. All statistical analysis was performed using SPSS software version 18.0 (SPSS, Inc.). Differences between multiple groups were analyzed by one-way ANOVA followed by Tukey’s post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Myocardial remodeling in the mouse model of TAC
Echocardiography indicated that the TAC model was successfully generated via TAB (Fig. 1A). Color Doppler imaging showed that blood flow was blocked in the aortic arch (Fig. 1B). The diameter of the transverse aorta decreased following TAC; the diameter of this vessel was similar between the TAC and EGCG + TAC groups (~0.4 mm; Fig. 1C), thus resulting in a similar increase of blood flow in the transverse aorta in both groups (Fig. 1D). In the present study, stereoscopy and H&E staining showed that the hearts of TAC mice at 4, 8 and 12 weeks after TAC were enlarged compared with the Sham group (Fig. 1E and F). Masson’s staining further revealed interstitial fibrosis and collagen accumulation in the myocardial tissue of TAC mice. Myocardial fibrosis and collagen deposition became more notable from 4 to 12 weeks after TAC (Fig. 1G). Echocardiography further showed that the left ventricular (LV) wall in TAC mice thickened between 4 and 8 weeks after TAC compared with the Sham group. At 8-12 weeks after TAC, the LV volume of TAC mice began to gradually increase; this was accompanied by a decrease in myocardial tissue (Fig. 1H). To determine morphological changes of lung in TAC mice, stereoscopy was performed. The results of stereoscopy showed that the lungs at 4, 8 and 12 weeks after TAC were enlarged compared with the Sham group (Fig. 1I). There was a decrease in the LV anterior wall thickness (AWT) in mice at 12 weeks after TAC. The LV ejection fraction (EF) also increased significantly in TAC mice over time. However, 8 weeks after TAC, LVEF showed a significant decrease, indicating that heart failure may have occurred (Table I).
Figure 1.

Color Doppler ultrasound imaging from the aortic arch of a ligated mouse and the structure and volume of the heart in a model of TAC. (A) Echocardiography and (B) Color Doppler imaging in the aortic arch of a ligated mouse (white arrow). (C) Diameter and (D) velocity of blood flow (P-mode ultrasound image) following TAC surgery in the presence or absence of EGCG treatment. (E) Stereoscopy images of complete hearts. (F) Hematoxylin and eosin staining in longitudinal sections of heart tissue (magnification, ×4). (G) Sections of LV treated with Masson’s stain. Scale bar, 100 µm. (H) Echocardiography M-mode imaging from short-axis measurements was used to evaluate LV function. Red, diastolic diameter; green, systolic diameter. (I) Stereoscopy images of complete lungs. *P<0.05 vs. before operation. n=6/group. AAO, ascending aorta; DAO, descending aorta; TAC, transverse aortic constriction; EGCG, epigallocatechin gallate; LV, left ventricle.
Table I.
Cardiac function measurement via echocardiography (n=6).
| Parameter | Sham | Transverse aortic constriction
|
F-value | P-value | ||
|---|---|---|---|---|---|---|
| 4 weeks | 8 weeks | 12 weeks | ||||
| Body weight, g | 38.66±0.22 | 39.15±0.18 | 38.40±0.60 | 38.66±0.60 | 1.500 | 0.2900 |
| Heart rate, bpm | 466.00±4.00 | 466.00±4.00 | 474.00±3.00 | 466.00±6.00 | 2.680 | 0.1200 |
| LV end diastole | ||||||
| AWT, mm | 1.04±0.55 | 1.59±0.11 | 1.32±0.06 | 1.28±0.98a | 47.550 | <0.0001 |
| IVS, mm | 0.89±0.01 | 0.97±0.01 | 0.95±0.25 | 1.02±0.21a | 32.923 | <0.0001 |
| Dimension, mm | 2.74±0.13 | 1.88±0.13 | 1.58±0.29 | 3.37±0.16a | 57.660 | <0.0001 |
| Volume, µl | 75.33±0.74 | 75.81±0.75 | 80.00±1.00 | 77.80±0.12a | 27.270 | <0.0001 |
| LV end systole | ||||||
| AWT, mm | 1.40±0.12 | 1.76±0.05 | 1.67±0.03 | 1.86±0.06a | 13.670 | <0.0001 |
| IVS, mm | 1.05±0.10 | 1.11±0.12 | 1.08±0.10 | 1.12±0.11 | 2.100 | 0.1800 |
| Dimension, mm | 1.14±0.12 | 0.83±0.35 | 0.76±0.02 | 1.42±0.03a | 45.310 | <0.0001 |
| Volume, µl | 22.42±1.27 | 20.08±0.89 | 17.46±0.95 | 25.65±0.77a | 37.500 | <0.0001 |
| EF, % | 82.00±3.00 | 93.33±4.16 | 69.33±1.53 | 58.33±6.03a | 42.630 | <0.0001 |
Data are presented as the mean ± SD.
P<0.0001 vs. Sham. LV, left ventricular; AWT, anterior wall thickness; IVS, interventricular septum; EF, ejection fraction.
HDAC5 may be involved in histone acetylation in the heart of TAC mice
Pathological myocardial remodeling is a key pathophysiological process of heart failure that is regulated by HDACs. Hence, protein expression levels of HDAC5 and H3K27ac were assessed by western blotting. Results showed that expression levels of HDAC5 in the heart of TAC mice increased gradually from 4 to 12 weeks after TAC (Fig. 2A). Western blotting showed that the acetylation levels of histone H3K27ac in TAC mice at 4 and 8 weeks after TAC were significantly higher than at 12 weeks (Fig. 2B). In addition, the expression levels of ANP and BNP at 4 and 8 weeks were significantly higher than those of the Sham group. However, the expression levels of ANP and BNP at 12 weeks after TAC were significantly lower than that at 4 and 8 weeks (Fig. 2C and D).
Figure 2.

Modification of histone acetylation in the heart of TAC mice. (A) Protein expression levels of HDAC5. (B) Acetylation levels of histone H3K27ac. Expression levels (C) ANP and (D) BNP. *P<0.05 vs. Sham; #P<0.05 vs. 4 W; ▲P<0.05 vs. 8 W. n=6/group. TAC, transverse aortic constriction; HDAC5, histone deacetylase 5; H3K27ac, histone acetylated lysine 27 on histone H3; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; W, weeks.
EGCG reverses hypoacetylation of H3K27ac by inhibiting HDAC5 and normalizing transcriptional activity of MEF2A
First, the optimal exposure dose for EGCG was determined. According to previous data (30,31), mice were injected intraperitoneally with various concentrations of EGCG (0, 25, 50, 75 and 100 mg/kg). The optimal concentration (50 mg/kg) was selected based on the lowest levels of HDAC5 in the heart of TAC mice (Fig. 3A). Next, the effect of EGCG on myocardial remodeling in TAC mice was investigated. EGCG (50 mg/kg/day) was intraperitoneally injected into mice after TAC for 12 weeks, and the hearts were collected for analysis. Western blotting showed that EGCG reversed the hypoacetylation of H3K27ac in the heart of TAC mice (Fig. 3B). EGCG attenuated the overexpression of HDAC5 in TAC mice (Fig. 3C). Analysis showed that the mRNA and protein expression levels of MEF2A were lower in the TAC group than those in the Sham group, while EGCG improved the mRNA and protein expression levels of MEF2A in TAC mice treated with EGCG (Fig. 3D and E). In addition, Co-IP was performed to verify the formation of a complex between H3K27ac and MEF2A; this demonstrated that HDAC5 may interact with H3K27ac and regulate gene expression levels of MEF2A (Fig. 3F).
Figure 3.

EGCG reverses abnormal expression of ANP and BNP by inhibiting overexpression of HDAC5 and reversing hypoacetylation of H3K27ac in the heart of mice 12 W after TAC. (A) Expression of HDAC5 following treatment with different concentrations of EGCG. (B) Acetylation levels of H3K27ac. (C) Expression of HDAC5. (D) Reverse transcription-quantitative PCR analysis of mRNA levels of MEF2A. (E) Expression of MEF2A. (F) Co-IP using anti-HDAC5-protein G magnetic beads and immunoblotting with anti-H3K27ac, anti-MEF2A or anti-HDAC5 antibody. (G) Chromatin IP-PCR assay was used to identify the regulatory association between MEF2A, ANP and BNP. Input, positive control; IgG, negative control. Western blotting showing expression of (H) ANP and (I) BNP. *P<0.05 vs. Sham; #P<0.05 vs. TAC. n=6/group. EGCG, epigallocatechin gallate; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; TAC, transverse aortic constriction; HDAC5, histone deacetylase 5; H3K27ac, histone acetylated lysine 27 on histone H3; MEF2A, myocyte enhancer factor 2A; IP, immunoprecipitation; IB, immunoblot.
Expression of ANP and BNP is regulated by heart nuclear transcription factor MEF2A
Next, ChIP-PCR was performed to investigate the regulatory association between MEF2A and downstream genes associated with heart development (ANP and BNP). Data showed that MEF2A bound to the promoter of ANP and BNP in the mouse heart (Fig. 3G). Western blotting was performed to investigate the expression levels of ANP and BNP during myocardial remodeling. Data showed that the expression levels of ANP and BNP in the heart of TAC mice were significantly lower compared with the Sham group. Data also showed that EGCG treatment rescued the decreased levels of ANP and BNP in the heart of TAC mice (Fig. 3H and I).
EGCG attenuates myocardial remodeling in TAC mice
Stereoscopy and H&E staining were used to detect the preventive and restorative effects of EGCG in the heart of TAC mice. Stereoscopy showed that the heart of mice in TAC group was larger compared with the Sham group and EGCG reversed cardiac dilatation (Fig. 4A). H&E staining showed that LV volume was higher in the TAC group than in the Sham group. However, EGCG treatment attenuated the thickening of LV in TAC mice (Fig. 4B). Masson’s staining revealed a greater extent of interstitial fibrosis and collagen deposition in the TAC group than in the Sham group. Following treatment with EGCG, the extent of interstitial fibrosis and collagen deposition was notably decreased (Fig. 4C). Furthermore, echocardiography showed that the LV end diastolic dimension (EDD) and end systolic dimension (ESD) in the TAC group were significantly higher than those in the Sham group. However, EGCG treatment attenuated the increased LVEDD and LVESD in the mouse heart. A significant decrease in LVEF was observed in TAC mice; however, EGCG treatment improved LVEF in the heart of TAC mice (Fig. 4D; Table II). In addition, EGCG significantly decreased the cardiac mass index of TAC mice, although the lung mass index remained unchanged (Table III). These data suggested that EGCG attenuated pathological myocardial remodeling in TAC mice.
Figure 4.

EGCG inhibits pressure overload-induced myocardial remodeling in mouse hearts. (A) Stereoscopy images of complete hearts. (B) Hematoxylin and eosin-stained longitudinal sections of heart tissue. (C) Sections of left ventricles stained with Masson’s stain. Scale bar, 100 µm. (D) Echocardiography M-mode imaging. M-mode images obtained from short-axis measurements were used to evaluate left ventricle function. Red, diastolic left ventricular diameter; green, systolic left ventricular diameter. EGCG, epigallocatechin gallate; TAC, transverse aortic constriction.
Table II.
Cardiac function measurement via echocardiography (n=6).
| Parameter | Normal | TAC | Sham | TAC + EGCG | Vehicle | F-value | P-value |
|---|---|---|---|---|---|---|---|
| Body weight, g | 38.28±0.31 | 39.11±0.15 | 38.42±0.38 | 38.43±0.48 | 38.43±0.48 | 2.24 | 0.1400 |
| Heart rate, bpm) | 465.00±4.00 | 468.00±3.00 | 464.00±54.00 | 462.00±3.00 | 461.00±1.00 | 2.47 | 0.1100 |
| LV end diastole | |||||||
| AWT, mm | 1.04±0.04 | 1.23±0.68a | 1.04±0.13 | 1.07±0.05b | 1.03±0.01 | 21.25 | <0.0001 |
| IVS, mm | 0.83±0.15 | 1.02±0.16a | 0.84±0.26 | 0.92±0.15b | 0.85±0.10 | 44.18 | <0.0001 |
| Dimension, mm | 2.92±0.72 | 3.29±0.29a | 2.85±0.50 | 3.01±0.13b | 2.80±0.62 | 15.56 | <0.0001 |
| Volume, µl | 75.33±0.74 | 80.00±1.00a | 75.81±0.75 | 77.80±0.12b | 75.12±0.49 | 27.27 | <0.0001 |
| LV end systole | |||||||
| AWT, mm | 1.43±0.04 | 1.88±0.03a | 1.45±0.06 | 1.63±0.04b | 1.47±0.04 | 65.78 | <0.0001 |
| IVS, mm | 1.08±0.15 | 1.06±0.10 | 1.07±0.10 | 1.09±0.15 | 1.07±0.10 | 1.89 | 0.1900 |
| Dimension, mm | 1.12±0.03 | 1.43±0.03a | 1.17±0.02 | 1.30±0.02b | 1.14±0.02 | 48.72 | <0.0001 |
| Volume, µl | 21.63±1.34 | 29.88±0.96a | 22.81±0.71 | 24.78±0.77b | 23.32±1.02 | 27.56 | <0.0001 |
| EF, % | 82.33±2.52 | 61.00±5.29a | 84.67±4.51 | 76.33±2.52b | 84.33±4.16 | 18.90 | <0.0001 |
TAC, thoracic aorta constriction; EGCG, epigallocatechin gallate; LV, left ventricular; AWT, anterior wall thickness; IVS, interventricular septum; EF, ejection fraction. Data are presented as the mean ± SD.
P<0.05 vs. normal;
P<0.05 vs. TAC.
Table III.
Cardiac and lung MI in mice (n=6).
| Parameter | Normal | TAC | Sham | TAC + EGCG | Vehicle | F-value | P-value |
|---|---|---|---|---|---|---|---|
| BW, g | 36.37±1.50 | 42.22±1.11a | 37.35±0.78 | 41.23±1.24 | 38.80±1.57 | 21.17 | <0.0001 |
| HW, mg | 173.48±9.12 | 282.87±20.52a | 171.06±12.78 | 223.46±15.73b | 183.912±14.33 | 23.12 | <0.0001 |
| LW, mg | 333.87±23.56 | 398.13±21.89a | 341.38±23.34 | 360.55±18.95b | 348.03±24.74 | 6.86 | 0.0400 |
| HW/BW, mg/g | 4.77±0.88 | 6.70±0.15a | 4.58±0.36 | 5.42±0.16b | 4.74±0.24 | 78.48 | <0.0001 |
| LW/BW, mg/g | 9.18±0.39 | 9.43±0.23 | 9.14±0.31 | 9.23±0.37 | 8.97±0.54 | 1.83 | 0.1500 |
HW, heart weight; BW, body weight; LW, lung weight; MI, mass index; TAC, thoracic aorta constriction; EGCG, epigallocatechin gallate.
P<0.05 vs. Sham;
P<0.05 vs. TAC.
EGCG attenuates myocardial cell hypertrophy and improves survival rate in TAC mice
Next, wheat germ agglutinin experiments were performed to evaluate the surface area of cardiomyocytes in TAC mice. Data showed that the surface area of cardiomyocytes in the TAC group increased significantly compared with the Sham group. However, EGCG treatment attenuated the increased surface area of cardiomyocytes in the heart of TAC mice (Fig. 5A and B). For the HDAC inhibitor EGCG to be used clinically, it is important to evaluate survival rate following administration. To investigate this issue, mice were subjected to TAC or sham operation and treated with EGCG (50 mg/kg/day) for 8 weeks, a period that corresponds to 6-8 years in humans (8). The data showed that EGCG treatment improved the survival rate of TAC mice [Sham + Veh, 90; TAC, 51; TAC + Veh, 51% and TAC + EGCG, 67% (n=35/group); Fig. 5C].
Figure 5.

Effect of EGCG on cardiomyocyte hypertrophy and survival rate in TAC mice. (A) Wheat germ agglutinin staining was performed to detect (B) cardiomyocyte area. Scale bar, 50 µm. Data are presented as the mean ± SD. *P<0.01 vs. Sham; #P<0.01 vs. TAC. n=6/group. (C) Survival rate of TAC mice treated with EGCG or Veh. EGCG, epigallocatechin gallate; TAC, transverse aortic constriction; Veh, vehicle.
Discussion
Studies have confirmed that one of the basic mechanisms underlying the occurrence and development of chronic heart failure is ventricular remodeling (32-34). Over past years, epigenetic regulation has become a novel target for intervention in modern medical research, especially with regards to the reversible regulatory effect of histone acetylation modification, which makes it possible to switch gene expression on or off (35). Histone acetylation includes numerous subtypes and acetylation sites, such as H3K4ac, H3K9ac, H3K14ac, H3K18ac, H3K27ac, H3K36ac and H3K56ac (36). Previous studies confirmed that multiple histone acetylation sites (H3K9ac, H3K14ac and H3K27ac) are involved in pathological cardiac hypertrophy (8-11,37,38). However, the mechanism by which histone acetylation is modified by HDAC5 in pathological myocardial remodeling remains unclear. Specifically, expression levels of HDAC5 remain at low levels during the CHF compensatory period (39-41). However, during the decompensated stage of this pathological process, HDAC5 is expressed at high levels. In addition, genes associated with cardiac development (ANP and BNP) are highly expressed during the early stages of pathological myocardial remodeling (cardiac hypertrophy stage) and expressed at low levels during the subsequent CHF decompensated stage (42,43). It was hypothesized that the decreased expression of ANP and BNP, markers of cardiac hypertrophy, during the decompensated stage of myocardial remodeling may be due to the inability to synthesize ANP and BNP under cardiac decompensation (due to a decreased number of myocardial cells and severe myocardial fibrosis in the heart of model mice). Therefore, it was hypothesized that HDAC5-mediated histone hypoacetylation is a key regulatory factor for pathological myocardial remodeling in response to pressure overload.
HDACs are considered effective interventional targets for the treatment of numerous types of human disease (44,45). Studies have confirmed that HDACs are involved in the occurrence and development of cardiac hypertrophy in mice (46), and that HDAC-specific inhibitors may improve heart failure and maintain normal systolic function of the heart (47). It has also been reported that deer antler in traditional Chinese medicine improves cardiac hypertrophy and CHF by regulating histone acetylation modification (48). Further studies have revealed that an imblance in HDAC-mediated histone acetylation serves an important role in pathological myocardial remodeling (49,50). Furthermore, HDAC inhibitors exert cardiac and vascular protective effects in rats with cardiac hypertrophy caused by pressure overload (51,52). In addition, Liu et al (30), and Bagchi and Weeks (53) proposed that HDAC inhibitors possess clinical value for the treatment of myocardial fibrosis, cardiac hypertrophy and heart failure by inhibiting the levels of histone acetylation modification. Studies have also confirmed that EGCG specifically inhibits activity of HDAC1/3/8 subtypes in tumors and other types of disease (54), and that EGCG downregulates activity of HDAC1 in mouse myocardial tissue, thus regulating expression of the cardiac structural gene cardiac troponin I (31). Other researchers reported that HDAC5 may be involved in regulating the occurrence and development of cardiac hypertrophy (22,55). Therefore, it was hypothesized that HDAC5 may be a key regulator of EGCG in the prevention of pathological myocardial remodeling in mice. ChIP-PCR data indicated that MEF2A bound to the promoter of ANP and BNP; this implied that there may be a regulatory association between these factors. These results suggested that an imbalance in HDAC5-mediated histone acetylation is involved in abnormal expression of genes associated with myocardial remodeling. However, the potential role of upstream signaling pathways, and whether histone modification other than deacetylation and acetylation are involved in myocardial remodeling, remain unclear. Further studies are needed in the field of myocardial remodeling to address this.
EGCG is the primary polyphenolic compound in green tea and exhibits cardiovascular health-promoting activity by regulating various pathways (56,57). Because EGCG is extracted from green tea, it has almost no side effects (56,58,59), and this compound exhibits cardioprotection, neuroprotection, renal protection, osteoprotection and anticancer properties, as well as the ability to manage obesity, metabolic syndrome and type 2 diabetes (60), thus making it an ideal safe and effective drug for the treatment of myocardial remodeling (61). In certain epidemiological studies, drinking green tea has been shown to decrease the risk of cardiovascular disease (62,63). In recent years, studies have found that EGCG serves an important role in epigenetic regulation (20,64,65). For example, EGCG inhibits DNA methyltransferase from turning on genes that have been silenced by methylation and inhibits the ability of HDACs to regulate histone acetylation modification in vivo (31,66). EGCG improves congestive heart failure caused by knockout of the manganese superoxide gene in a mouse model of dilated cardiomyopathy and significantly improves survival rate (67). Furthermore, EGCG may inhibit cardiac hypertrophy, fibrosis and apoptosis caused by aging (68). These data suggest that EGCG serves a key role in cardiac protection. However, it remains unclear whether EGCG improves cardiac function in patients with CHF. In the present study, low expression levels of MEF2A mRNA were detected in TAC mice. MEF2A is a transcriptional regulator associated with cardiac hypertrophy and leads to low expression levels of genes associated with cardiac hypertrophy, thus inducing cardiac remodeling and heart failure. Inhibiting HDAC activity by drug action could either increase or decrease the expression of associated genes (69). Expression levels of MEF2A protein in the heart of TAC mice were significantly decreased. However, administration of EGCG reversed the low acetylation levels of H3K27ac by inhibiting the action of HDAC5, thus increasing MEF2A transcription activity. EGCG also normalized the abnormal expression of two genes associated with cardiac development, ANP and BNP, in the cardiac muscle tissue of TAC mice. Echocardiographic data demonstrated that EGCG improved cardiac function in TAC mice. However, more preclinical studies are needed to confirm that this novel HDAC inhibitor can be used to prevent or reverse cardiac remodeling and CHF before novel drugs can be developed.
In conclusion, myocardial remodeling is a complex process and is associated with gene transcription and modification. In the present study, EGCG downregulated histone H3K27ac acetylation mediated by HDAC5 to attenuate myocardial remodeling induced by pressure overload in mice (Fig. 6). Meanwhile, the present results provided evidence that HDAC inhibition is a potential treatment strategy for treatment of cardiac dysfunction caused by pressure overload.
Figure 6.
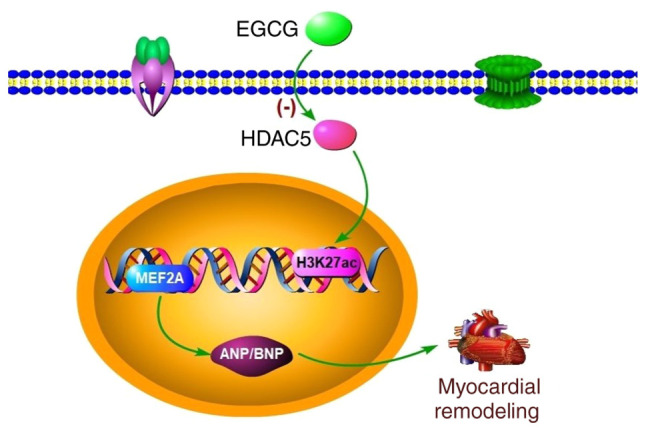
Schematic representation of the potential mechanism by which EGCG attenuates pressure overload-induced myocardial remodeling. EGCG may ameliorate myocardial remodeling by regulating the overexpression of HDAC5. ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; EGCG, epigallocatechin gallate; H3K27ac, histone acetylated lysine 27 on histone H3; HDAC5, histone deacetylase 5; MEF2A, myocyte enhancer factor 2A.
Acknowledgments
Not applicable.
Abbreviations
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
- CHF
chronic heart failure
- ChIP
chromatin immunoprecipitation
- Co-IP
co-immunoprecipitation
- EGCG
epigallocatechin gallate
- HAT
histone acetylase
- H3K27ac
histone acetylated lysine 27 on histone H3
- HDAC5
histone deacetylase 5
- LVAWT
left ventricular anterior wall thickness
- LVEDD
left ventricular end diastolic dimension
- LVEF
left ventricular ejection fraction
- LVESD
left ventricular end systolic dimension
- TAB
thoracic aortic banding
- TAC
transverse aortic constriction
Funding Statement
The present study was supported by the National Natural Science Foundation of China (grant nos. 82060046 and 81560040).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
XH and CP conceived and designed the experiments. XH, XL and LH performed the experiments. CP, QM and SW analyzed the data and confirm the authenticity of all the raw data. XH and HZ wrote the paper. CP revised the paper. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The present study was approved by the Institutional Animal Care and Use Committee of Zunyi Medical University (approval no. SYXK 2017-0012).
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
References
- 1.Khan N, Mukhtar H. Tea polyphenols in promotion of human health. Nutrients. 2018;11:39. doi: 10.3390/nu11010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao L, Cheng G, Choksi K, Samanta A, Girgis M, Soder R, Vincent RJ, Wulser M, De Ruyter M, McEnulty P, et al. Transplantation of human umbilical cord blood-derived cellular fraction improves left ventricular function and remodeling after myocardial ischemia/reperfusion. Circ Res. 2019;125:759–772. doi: 10.1161/CIRCRESAHA.119.315216. [DOI] [PubMed] [Google Scholar]
- 3.Grobe JL, Der Sarkissian S, Stewart JM, Meszaros JG, Raizada MK, Katovich MJ. ACE2 overexpression inhibits hypoxia-induced collagen production by cardiac fibroblasts. Clin Sci (Lond) 2007;113:357–364. doi: 10.1042/CS20070160. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Du W, Zhao R, Hu J, Li H, Han R, Yue Q, Wu R, Li W, Zhao J. New insights into epigenetic modifications in heart failure. Front Biosci (Landmark Ed) 2017;22:230–247. doi: 10.2741/4483. [DOI] [PubMed] [Google Scholar]
- 5.Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci. 2009;66:596–612. doi: 10.1007/s00018-008-8432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh AK, Rai R, Flevaris P, Vaughan DE. Epigenetics in reactive and reparative cardiac fibrogenesis: The promise of epigenetic therapy. J Cell Physiol. 2017;232:1941–1956. doi: 10.1002/jcp.25699. [DOI] [PubMed] [Google Scholar]
- 7.Segers VFM, Gevaert AB, Boen JRA, Van Craenenbroeck EM, De Keulenaer GW. Epigenetic regulation of intercellular communication in the heart. Am J Physiol Heart Circ Physiol. 2019;316:H1417–H1425. doi: 10.1152/ajpheart.00038.2019. [DOI] [PubMed] [Google Scholar]
- 8.Li S, Peng B, Luo X, Sun H, Peng C. Anacardic acid attenuates pressure-overload cardiac hypertrophy through inhibiting histone acetylases. J Cell Mol Med. 2019;23:2744–2752. doi: 10.1111/jcmm.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng C, Zhang W, Zhao W, Zhu J, Huang X, Tian J. Alcohol-induced histone H3K9 hyperacetylation and cardiac hypertrophy are reversed by a histone acetylases inhibitor anacardic acid in developing murine hearts. Biochimie. 2015;113:1–9. doi: 10.1016/j.biochi.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Peng C, Luo X, Li S, Sun H. Phenylephrine-induced cardiac hypertrophy is attenuated by a histone acetylase inhibitor anacardic acid in mice. Mol Biosyst. 2017;13:714–724. doi: 10.1039/C6MB00692B. [DOI] [PubMed] [Google Scholar]
- 11.Ooi JY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ, El-Osta A. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics. 2015;10:418–430. doi: 10.1080/15592294.2015.1024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorski PA, Jang SP, Jeong D, Lee A, Lee P, Oh JG, Chepurko V, Yang DK, Kwak TH, Eom SH, et al. Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca2+-ATPase in heart failure. Circ Res. 2019;124:e63–e80. doi: 10.1161/CIRCRESAHA.118.313865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J, Cai L. Dysregulation of histone acetyltransferases and deacetylases in cardiovascular diseases. Oxid Med Cell Longev. 2014;2014;641979 doi: 10.1155/2014/641979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang M, Zhang Y, Ren J. Acetylation in cardiovascular diseases: Molecular mechanisms and clinical implications. Biochim Biophys Acta Mol Basis Dis. 1866(165836):2020. doi: 10.1016/j.bbadis.2020.165836. [DOI] [PubMed] [Google Scholar]
- 15.Steinmann J, Buer J, Pietschmann T, Steinmann E. Anti-infective properties of epigallocatechin-3-gallate (EGCG), a component of green tea. Br J Pharmacol. 2013;168:1059–1073. doi: 10.1111/bph.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang CS, Landau JM, Huang MT, Newmark HL. Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu Rev Nutr. 2001;21:381–406. doi: 10.1146/annurev.nutr.21.1.381. [DOI] [PubMed] [Google Scholar]
- 17.Khurana S, Venkataraman K, Hollingsworth A, Piche M, Tai TC. Polyphenols: Benefits to the cardiovascular system in health and in aging. Nutrients. 2013;5:3779–3827. doi: 10.3390/nu5103779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li K, Teng C, Min Q. Advanced nanovehicles-enabled delivery systems of epigallocatechin gallate for cancer therapy. Front Chem. 8(573297):2020. doi: 10.3389/fchem.2020.573297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Y, McIntosh GH, Le Leu RK, Somashekar R, Meng XQ, Gopalsamy G, Bambaca L, McKinnon RA, Young GP. Supplementation with Brazil nuts and green tea extract regulates targeted biomarkers related to colorectal cancer risk in humans. Br J Nutr. 2016;116:1901–1911. doi: 10.1017/S0007114516003937. [DOI] [PubMed] [Google Scholar]
- 20.Evans LW, Athukorala M, Martinez-Guryn K, Ferguson BS. The role of histone acetylation and the microbiome in phytochemical efficacy for cardiovascular diseases. Int J Mol Sci. 21(4006):2020. doi: 10.3390/ijms21114006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Deng M, Lu A, Chen Y, Chen Y, Wu C, Tan Z, Boini KM, Yang T, Zhu Q, Wang L. Sodium butyrate attenuates angiotensin II-induced cardiac hypertrophy by inhibiting COX2/PGE2 pathway via a HDAC5/HDAC6-dependent mechanism. J Cell Mol Med. 2019;23:8139–8150. doi: 10.1111/jcmm.14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandrasekaran S, Peterson RE, Mani SK, Addy B, Buchholz AL, Xu L, Thiyagarajan T, Kasiganesan H, Kern CB, Menick DR. Histone deacetylases facilitate sodium/calcium exchanger up-regulation in adult cardiomyocytes. FASEB J. 2009;23:3851–3864. doi: 10.1096/fj.09-132415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melleby AO, Romaine A, Aronsen JM, Veras I, Zhang L, Sjaastad I, Lunde IG, Christensen G. A novel method for high precision aortic constriction that allows for generation of specific cardiac phenotypes in mice. Cardiovasc Res. 2018;114:1680–1690. doi: 10.1093/cvr/cvy141. [DOI] [PubMed] [Google Scholar]
- 25.Riehle C, Bauersachs J. Small animal models of heart failure. Cardiovasc Res. 2019;115:1838–1849. doi: 10.1093/cvr/cvz161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mecklenburg J, Patil MJ, Koek W, Akopian AN. Effects of local and spinal administrations of mu-opioids on postoperative pain in aged versus adult mice. Pain Rep. 2017;2:e584. doi: 10.1097/PR9.0000000000000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaw AM, Williams CM, Law HK, Chow BK. Minimally invasive transverse aortic constriction in mice. J Vis Exp: 2017;55293 doi: 10.3791/55293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Wang C, Hong X, Miao J, Liao Y, Hou FF, Zhou L, Liu Y. Wnt/β-catenin signaling mediates both heart and kidney injury in type 2 cardiorenal syndrome. Kidney Int. 2019;95:815–829. doi: 10.1016/j.kint.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Liu L, Zhao W, Liu J, Gan Y, Liu L, Tian J. Epigallocatechin-3 gallate prevents pressure overload-induced heart failure by up-regulating SERCA2a via histone acetylation modification in mice. PLoS One. 2018;13:e0205123. doi: 10.1371/journal.pone.0205123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan B, Quan J, Liu L, Xu Z, Zhu J, Huang X, Tian J. Epigallocatechin gallate reverses cTnI-low expression-induced age-related heart diastolic dysfunction through histone acetylation modification. J Cell Mol Med. 2017;21:2481–2490. doi: 10.1111/jcmm.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uriel N, Sayer G, Annamalai S, Kapur NK, Burkhoff D. Mechanical unloading in heart failure. J Am Coll Cardiol. 2018;72:569–580. doi: 10.1016/j.jacc.2018.05.038. [DOI] [PubMed] [Google Scholar]
- 33.Bouwens E, Brankovic M, Mouthaan H, Baart S, Rizopoulos D, van Boven N, Caliskan K, Manintveld O, Germans T, van Ramshorst J, et al. Temporal patterns of 14 blood biomarker candidates of cardiac remodeling in relation to prognosis of patients with chronic heart failure-the Bio-SH i FT study. J Am Heart Assoc. 2019;8:e009555. doi: 10.1161/JAHA.118.009555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X, Ferrara F, Contaldi C, Bossone E. Right ventricular size and function in chronic heart failure: Not to be forgotten. Heart Fail Clin. 2019;15:205–217. doi: 10.1016/j.hfc.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Sabia C, Picascia A, Grimaldi V, Amarelli C, Maiello C, Napoli C. The epigenetic promise to improve prognosis of heart failure and heart transplantation. Transplant Rev (Orlando) 2017;31:249–256. doi: 10.1016/j.trre.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Barnes CE, English DM, Cowley SM. Acetylation & Co: An expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019;63:97–107. doi: 10.1042/EBC20180061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu X, Pan B, Liu L, Zhao W, Zhu J, Huang X, Tian J. In utero exposure to PM2.5 during gestation caused adult cardiac hypertrophy through histone acetylation modification. J Cell Biochem. 2019;120:4375–4384. doi: 10.1002/jcb.27723. [DOI] [PubMed] [Google Scholar]
- 38.Chen K, Jian D, Zhao L, Zang X, Song W, Ma J, Jia Z, Wang X, Gao C. Protective effect of histone methyltransferase NSD3 on ISO-induced cardiac hypertrophy. FEBS Lett. 2019;593:2556–2565. doi: 10.1002/1873-3468.13515. [DOI] [PubMed] [Google Scholar]
- 39.Xu WP, Yao TQ, Jiang YB, Zhang MZ, Wang YP, Yu Y, Li JX, Li YG. Effect of the angiotensin II receptor blocker valsartan on cardiac hypertrophy and myocardial histone deacetylase expression in rats with aortic constriction. Exp Ther Med. 2015;9:2225–2228. doi: 10.3892/etm.2015.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parra M. Class IIa HDACs-new insights into their functions in physiology and pathology. FEBS J. 2015;282:1736–1744. doi: 10.1111/febs.13061. [DOI] [PubMed] [Google Scholar]
- 41.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo Y, Xu Y, Liang C, Xing W, Zhang T. The mechanism of myocardial hypertrophy regulated by the interaction between mhrt and myocardin. Cell Signal. 2018;43:11–20. doi: 10.1016/j.cellsig.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 43.Gaggin HK, Januzzi JL., Jr Biomarkers and diagnostics in heart failure. Biochim Biophys Acta. 18322013:2442–2450. doi: 10.1016/j.bbadis.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 44.Scholz B, Schulte JS, Hamer S, Himmler K, Pluteanu F, Seidl MD, Stein J, Wardelmann E, Hammer E, Völker U, Müller FU. HDAC (histone deacetylase) inhibitor valproic acid attenuates atrial remodeling and delays the onset of atrial fibrillation in mice. Circ Arrhythm Electrophysiol. 2019;12:e007071. doi: 10.1161/CIRCEP.118.007071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou G, Zhong W, Wu F, Wang X, Liu L. Catalpol attenuates cardiomyocyte apoptosis in diabetic cardiomyopathy via Neat1/miR-140-5p/HDAC4 axis. Biochimie. 2019;165:90–99. doi: 10.1016/j.biochi.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 46.Yoon S, Kim M, Min HK, Lee YU, Kwon DH, Lee M, Lee S, Kook T, Joung H, Nam KI, et al. Inhibition of heat shock protein 70 blocks the development of cardiac hypertrophy by modulating the phosphorylation of histone deacetylase 2. Cardiovasc Res. 2019;115:1850–1860. doi: 10.1093/cvr/cvy317. [DOI] [PubMed] [Google Scholar]
- 47.Lee E, Lee HA, Kim M, Do GY, Cho HM, Kim GJ, Jung H, Song JH, Cho JM, Kim I. Upregulation of C/EBPβ and TSC2 by an HDAC inhibitor CG200745 protects heart from DOCA-induced hypertrophy. Clin Exp Pharmacol Physiol. 2019;46:226–236. doi: 10.1111/1440-1681.13022. [DOI] [PubMed] [Google Scholar]
- 48.Zhao W, Hu W, Wang X, Xia N, Hu Q, Zhou H. A traditional Chinese medicine, Lujiao prescription, as a potential therapy for hypertrophic cardiomyocytes by acting on histone acetylation. J Chin Med Assoc. 2015;78:486–493. doi: 10.1016/j.jcma.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 49.Habibian J, Ferguson BS. The crosstalk between acetylation and phosphorylation: Emerging new roles for hDAC inhibitors in the heart. Int J Mol Sci. 2018;20:102. doi: 10.3390/ijms20010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, Wu J, Jeong MY, Lin YH, Borghetti G, et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci Transl Med. 2020;12:eaay7205. doi: 10.1126/scitranslmed.aay7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung H, Lee E, Kim I, Song JH, Kim GJ. Histone deacetylase inhibition has cardiac and vascular protective effects in rats with pressure overload cardiac hypertrophy. Physiol Res. 2019;68:727–737. doi: 10.33549/physiolres.934110. [DOI] [PubMed] [Google Scholar]
- 52.Yan H, Yi S, Zhuang H, Wu L, Wang DW, Jiang J. Sphingosine-1-phosphate ameliorates the cardiac hypertrophic response through inhibiting the activity of histone deacetylase-2. Int J Mol Med. 2018;41:1704–1714. doi: 10.3892/ijmm.2017.3325. [DOI] [PubMed] [Google Scholar]
- 53.Bagchi RA, Weeks KL. Histone deacetylases in cardiovascular and metabolic diseases. J Mol Cell Cardiol. 2019;130:151–159. doi: 10.1016/j.yjmcc.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 54.Kim DJ, Dunleavey JM, Xiao L, Ollila DW, Troester MA, Otey CA, Li W, Barker TH, Dudley AC. Suppression of TGFβ-mediated conversion of endothelial cells and fibroblasts into cancer associated (myo)fibroblasts via HDAC inhibition. Br J Cancer. 2018;118:1359–1368. doi: 10.1038/s41416-018-0072-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu T, Schreiter FC, Bagchi RA, Tatman PD, Hannink M, McKinsey TA. HDAC5 catalytic activity suppresses cardiomyocyte oxidative stress and NRF2 target gene expression. J Biol Chem. 2019;294:8640–8652. doi: 10.1074/jbc.RA118.007006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eng QY, Thanikachalam PV, Ramamurthy S. Molecular understanding of epigallocatechin gallate (EGCG) in cardiovascular and metabolic diseases. J Ethnopharmacol. 2018;210:296–310. doi: 10.1016/j.jep.2017.08.035. [DOI] [PubMed] [Google Scholar]
- 57.Papadaki M, Vikhorev PG, Marston SB, Messer AE. Uncoupling of myofilament Ca2+ sensitivity from troponin I phosphorylation by mutations can be reversed by epigallocatechin-3-gallate. Cardiovasc Res. 2015;108:99–110. doi: 10.1093/cvr/cvv181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oliveira MR, Nabavi SF, Daglia M, Rastrelli L, Nabavi SM. Epigallocatechin gallate and mitochondria-a story of life and death. Pharmacol Res. 2016;104:70–85. doi: 10.1016/j.phrs.2015.12.027. [DOI] [PubMed] [Google Scholar]
- 59.Isbrucker RA, Edwards JA, Wolz E, Davidovich A, Bausch J. Safety studies on epigallocatechin gallate (EGCG) preparations. Part 3: Teratogenicity and reproductive toxicity studies in rats. Food Chem Toxicol. 2006;44:651–661. doi: 10.1016/j.fct.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 60.Afzal M, Safer AM, Menon M. Green tea polyphenols and their potential role in health and disease. Inflammopharmacology. 2015;23:151–161. doi: 10.1007/s10787-015-0236-1. [DOI] [PubMed] [Google Scholar]
- 61.Qin S, Chen MH, Fang W, Tan XF, Xie L, Yang YG, Qin T, Li N. Cerebral protection of epigallocatechin gallate (EGCG) via preservation of mitochondrial function and ERK inhibition in a rat resuscitation model. Drug Des Devel Ther. 2019;13:2759–2768. doi: 10.2147/DDDT.S215358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hertog MG, Feskens EJ, Hollman PC, Katan MB, Kromhout D. Dietary antioxidant flavonoids and risk of coronary heart disease: The zutphen elderly study. Lancet. 1993;342:1007–1011. doi: 10.1016/0140-6736(93)92876-U. [DOI] [PubMed] [Google Scholar]
- 63.Nakachi K, Matsuyama S, Miyake S, Suganuma M, Imai K. Preventive effects of drinking green tea on cancer and cardiovascular disease: Epidemiological evidence for multiple targeting prevention. Biofactors. 2000;13:49–54. doi: 10.1002/biof.5520130109. [DOI] [PubMed] [Google Scholar]
- 64.Potenza MA, Iacobazzi D, Sgarra L, Montagnani M. The intrinsic virtues of EGCG, an extremely good cell guardian, on prevention and treatment of diabesity complications. Molecules. 25(3061):2020. doi: 10.3390/molecules25133061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang L, Zhang W, Chopra S, Kaur D, Wang H, Li M, Chen P, Zhang W. The epigenetic modification of epigallocatechin gallate (EGCG) on cancer. Curr Drug Targets. 21:1099–1104. 2020. doi: 10.2174/1389450121666200504080112. [DOI] [PubMed] [Google Scholar]
- 66.Sheng J, Shi W, Guo H, Long W, Wang Y, Qi J, Liu J, Xu Y. The inhibitory effect of (-)-epigallocatechin-3-gallate on breast cancer progression via reducing SCUBE2 methylation and DNMT activity. Molecules. 2019;24:2899. doi: 10.3390/molecules24162899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oyama JI, Shiraki A, Nishikido T, Maeda T, Komoda H, Shimizu T, Makino N, Node K. EGCG, a green tea catechin, attenuates the progression of heart failure induced by the heart/muscle-specific deletion of MnSOD in mice. J Cardiol. 2017;69:417–427. doi: 10.1016/j.jjcc.2016.05.019. [DOI] [PubMed] [Google Scholar]
- 68.Muhammed I, Sankar S, Govindaraj S. Ameliorative effect of epigallocatechin gallate on cardiac hypertrophy and fibrosis in aged rats. J Cardiovasc Pharmacol. 2018;71:65–75. doi: 10.1097/FJC.0000000000000545. [DOI] [PubMed] [Google Scholar]
- 69.Li C, Sun XN, Chen BY, Zeng MR, Du LJ, Liu T, Gu HH, Liu Y, Li YL, Zhou LJ, et al. Nuclear receptor corepressor 1 represses cardiac hypertrophy. EMBO Mol Med. 2019;11:e9127. doi: 10.15252/emmm.201809127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.