

Abstract
Non-pathogenic Cellular Prion Protein (PrPC) demonstrates anti-inflammatory activity; however, the responsible mechanisms are incompletely defined. PrPC exists as a glycosylphosphatidylinositol (GPI)-anchored membrane protein in diverse cells; however, PrPC may be released from cells by ADAM proteases or when packaged into extracellular vesicles (EVs). Herein, we show that a soluble PrPC derivative (S-PrP) counteracts inflammatory responses triggered by Pattern Recognition Receptors (PRRs) in macrophages, including Toll-like Receptor (TLR) 2, TLR4, TLR7, TLR9, NOD1, and NOD2. S-PrP also significantly attenuates the toxicity of lipopolysaccharide (LPS) in mice. The response of macrophages to S-PrP is mediated by a receptor assembly that includes the NMDA Receptor (NMDA-R) and LRP1. PrPC was identified in EVs isolated from human plasma. These EVs replicated the activity of S-PrP, inhibiting cytokine expression and IκBα phosphorylation in LPS-treated macrophages. The effects of plasma EVs on LPS-treated macrophages were blocked by PrPC-specific antibody, by antagonists of LRP1 and the NMDA-R, by deleting Lrp1 in macrophages, and by inhibiting Src family kinases. Phosphatidylinositol-specific phospholipase C (PI-PLC) dissociated the LPS-regulatory activity from EVs, rendering the EVs inactive as LPS inhibitors. The LPS-regulatory activity that was lost from PI-PLC-treated EVs was recovered in solution. Collectively, these results demonstrate that GPI-anchored PrPC is the essential EV component required for the observed immune regulatory activity of human plasma EVs. S-PrP and EV-associated PrPC regulate innate immunity by engaging the NMDA-R/LRP1 receptor system in macrophages. The scope of PRRs antagonized by S-PrP suggests that released forms of PrPC may have broad anti-inflammatory activity.
Introduction
Cellular prion protein (PrPC) is a glycosylphosphatidylinositol (GPI)-anchored membrane protein that localizes mainly in lipid rafts (1). Misfolding of PrPC into the β-sheet-rich, scrapie conformation (PrPSc) causes protein aggregation and prion diseases, associated with rapid neurodegeneration (2). The physiological role of non-pathogenic PrPC remains incompletely understood. PrPC is expressed by diverse cell types, inside and outside the nervous system, including T-lymphocytes, natural killer cells, mast cells, and macrophages (3–6). Understanding the function of non-pathogenic PrPC is an important goal.
Non-pathogenic PrPC exists in at least three different states that may be relevant to its function in cell physiology. In addition to GPI-anchored PrPC in cells, derivatives of PrPC may be released from cells into solution by proteases in the ADAM family (7–9). PrPC also may be released from cells after packaging into extracellular vesicles (EVs). EVs are produced by nearly all cells and participate in cell-cell communication (10–12). Both PrPC and its misfolded isoform, PrPSc, have been identified in EVs in various body compartments, including blood (13, 14). PrPC may be enriched in exosomes (15–18), a specific type of EV formed in multivesicular bodies in the endosomal transport pathway (10–12, 19).
There is substantial evidence that PrPC regulates inflammation (3, 20–22). Mice treated with PrPC-specific antibody are protected from the lethal effects of Influenza A virus by a mechanism that apparently involves activation of Src family kinases (SFKs) in macrophages and induction of an M2-like anti-inflammatory state in these cells (23). When the gene encoding PrPC (Prnp) is deleted in mice, susceptibility to the toxic effects of lipopolysaccharide (LPS) is increased (24) and colitis is more severe following treatment with Dextran Sulfate Sodium (DSS) (25).
We demonstrated that a soluble derivative of PrPC (S-PrP), which corresponds closely in sequence to the form of PrPC released by ADAM10 (7), activates cell-signaling and elicits biological responses in PC12 cells and Schwann cells by a mechanism that requires LDL Receptor-related Protein-1 (LRP1) and the N-methyl-D-aspartate receptor (NMDA-R) (26). LRP1 functions as a receptor for over 100 ligands; however, only a subset of these ligands simultaneously engage the NMDA-R to activate cell-signaling (27–29). Tissue-type plasminogen activator (tPA) and α2-macroglobulin (α2M) are examples of LRP1 ligands that activate cell-signaling via the NMDA-R/LRP1 receptor system, similarly to S-PrP (26, 28, 29). In macrophages, tPA blocks the activity of agonists that activate Toll-like Receptors (TLRs), including TLR2, TLR4, and TLR9 (30–33). Thus, the NMDA-R/LRP1 receptor system emerged as an intriguing candidate to explain the anti-inflammatory activity of PrPC.
Prior to our study identifying the NMDA-R and LRP1 as cell-signaling receptors for S-PrP (26), there was already evidence that PrPC interacts with LRP1. Membrane-anchored PrPC physically associates with LRP1 within the plasma membranes of neuron-like cells (34, 35). This interaction apparently controls PrPC trafficking and also may support the function of LRP1 as a cell-signaling receptor for tPA. PrPC has been shown to bind tPA, directly and with high affinity (36). This interaction may be important in the pathway by which membrane-anchored PrPC promotes tPA-initiated cell-signaling via the NMDA-R/LRP1 receptor system (35).
The goal of the current study was to determine whether PrPC, which is released from cells, opposes the activity of Pattern Recognition Receptors (PRRs). We found that S-PrP inhibits responses elicited in macrophages not only by TLR agonists, but also by agents that engage NOD1 and NOD2 (30–33). Regulation of PRRs by S-PrP required the NMDA-R/LRP1 receptor system. We identified PrPC in EVs isolated from human plasma and demonstrated that, like S-PrP, these EVs neutralize the activity of LPS. The anti-LPS/TLR4 activity of plasma EVs was inhibited by PrPC-specific antibody, by antagonists of LRP1 or the NMDA-R, by Lrp1 gene-silencing, by inhibiting Src family kinases (SFKs), and by treating the EVs with phosphatidylinositol-specific phospholipase C (PI-PLC), an enzyme that releases GPI-anchored proteins from plasma membranes (37, 38). The LPS-regulatory activity that was lost from PI-PLC-treated EVs was recovered in solution. Collectively, these results implicate GPI-anchored PrPC as a major EV component responsible for the observed immune regulatory activity of human plasma EVs. Like S-PrP, EV-associated PrPC regulates innate immunity by engaging the NMDA-R/LRP1 receptor system in targeted macrophages. The NMDA-R and LRP1 are not previously identified as receptors for EV-associated membrane proteins. Our results provide a novel mechanistic explanation for the anti-inflammatory activity of PrPC, which is based on the activity of two distinct forms of PrPC released from cells.
Materials and Methods
Proteins and reagents
S-PrP (residues 23–231 from the structure of mouse PrPC) was expressed and purified as previously described (26). In brief, S-PrP was expressed in E. coli BL21 as a His-tagged protein, which was recovered from inclusion bodies, denatured in guanidinium hydrochloride, purified by Ni2+-affinity chromatography, oxidized, and refolded out of denaturant. Thrombin was used to dissociate the N-terminal poly-His tail. The thrombin was then removed by ion exchange chromatography. S-PrP preparations were judged to be >98% pure by SDS-PAGE with silver staining and by LC-MS/MS analysis of tryptic peptides (26). All preparations were processed through high-capacity endotoxin removal columns (Pierce) and determined to be endotoxin-free using an endotoxin detection kit (Thermo Fisher Scientific).
Human enzymatically-inactive tPA (EI-tPA), which carries the S478A mutation and thus lacks catalytic activity and a second mutation (R275E) so the protein remains in single-chain form, was from Molecular Innovations. α2M was purified from human plasma, activated for binding to LRP1 by reaction with methylamine as previously described (39), and determined to be endotoxin-free. LPS serotype 055:B5 from E. coli was from Sigma-Aldrich. Lipoteichoic acid (LTA), ODN 1826, L18-MDP, and C12-iE-DAP were from InvivoGen. Imiquimod (IMQ) was from Frontier Scientific. Endotoxin-free, monomeric Receptor-associated Protein (RAP) was provided by Dr. Travis Stiles (Novoron Biosciences). Dizocilpine (MK801) was from Cayman Chemicals. Dextromethorphan hydrobromide (DXM) and the SFK inhibitor, PP2, were from Abcam. The monoclonal antibodies POM1, POM2, POM3, and POM19, which are directed against different epitopes in PrPC, were purified as previously described (40). PI-PLC from B. cereus was purchased from Thermo Fisher Scientific.
Animals
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of University of California San Diego. Wild-type (WT) C57BL/6J mice were obtained from Jackson Laboratory. To generate mice in which monocytes, macrophages, and neutrophils are LRP1 deficient (mLrp1−/− mice), Lrp1flox/flox mice were bred with mice that express Cre recombinase under the control of the lysozyme-M promoter (LysM-Cre), in the C57BL/6J background, as previously described (41). For experiments with macrophages harvested from mLrp1−/− mice, control cells were harvested from littermates that were LRP1flox/flox but LysM-Cre-negative (mLrp1+/+ mice). Prnp−/− mice were generously provided by Dr. Adriano Aguzzi (University Hospital of Zurich, Zurich, Switzerland).
Cell culture
Bone marrow-derived macrophages (BMDMs) were harvested from 16-week-old male mice, as previously described (30). Briefly, bone marrow cells were flushed from mouse femurs and plated in non-tissue culture-treated dishes. Cells were cultured in DMEM/F-12 medium containing 10% fetal bovine serum (FBS) and 20% L929 cell-conditioned medium for 7 days. Non-adherent cells were eliminated. Adherent cells included >95% BMDMs, as determined by F4/80 and CD11b immunoreactivity.
Quiescent peritoneal macrophages (pMacs) were isolated from 16-week-old male C57BL/6J mice without thioglycollate elicitation and cultured as previously described (42). In brief, 5 ml of PBS (20 mM sodium phosphate, 150 mM NaCl, pH 7.4) with 3% FBS and 1× Gibco Antibiotic-Antimycotic (A/A) (Thermo Fisher Scientific) were injected into the peritoneal space with a 25-gauge needle. The solution was massaged from the abdominal surface and then harvested using the same needle. The procedure was repeated three times. Isolates that contained visible red blood cells were excluded. The remaining isolates were subjected to centrifugation at 800 × g for 5 min, suspended in DMEM/F12 supplemented with 10% FBS and 1x A/A, and then plated at 2 × 106 cells/well in tissue-culture treated 6-well plates. The cells were washed extensively 2 h after plating and maintained in culture for 48 hours before conducting experiments.
Cell viability
BMDMs were transferred to serum-free medium (SFM) for 30 min and then treated with S-PrP (40 or 120 nM) or vehicle for 6 h. The BMDMs were harvested and stained with 7-aminoactinomycin D (7-AAD), using the APC Annexin V Apoptosis Detection Kit (BioLegend), following the manufacturer’s instructions. Apoptotic cells were detected by flow cytometry using a BD FACSCanto II (BD Biosciences). Data were analyzed with FlowJo Software version 10.7.1 (BD Biosciences).
Cell signaling
Cells were transferred to SFM for 30 min and then treated with various proteins and reagents, alone or simultaneously as noted, including: LPS (0.1 μg/ml); LTA (1.0 μg/mL); ODN 1826 (1 μM); IMQ (3 μg/ml); C12-iE-DAP (1 μg/mL); L18-MDP (0.1 μg/mL); S-PrP (20–120 nM); EI-tPA (12 nM); activated α2M (10 nM); human plasma EVs (1.0 – 4.0 μg/mL); POM1, POM2, POM3, POM19, mouse IgG (10 μg/mL); or vehicle (PBS).
Cells were rinsed with ice-cold PBS and proteins were extracted in RIPA buffer (20 mM sodium phosphate, 150 mM NaCl, pH 7.4, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific). Equal amounts of protein, as determined using the detergent-compatible (DC) Protein Assay (Bio-Rad), were subjected to 10% SDS-PAGE and electro-transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% nonfat dried milk and then incubated with primary antibodies from Cell Signaling Technology that recognize: phospho-ERK1/2, total ERK1/2, phospho-IκBα, total IκBα, phospho-Tyr-416 in SFKs (the activation epitope), total SFKs, and β-actin, as a loading control. The membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch). Immunoblots were developed using Radiance, Radiance Q, and Radiance Plus chemiluminescent substrates (Azure Biosystems) and imaged using the Azure Biosystems c300 digital system. The presented results are representative of at least three independent experiments.
RT-qPCR
Cells were transferred to SFM for 30 min and then treated with various proteins and reagents for 6 h. RNA was isolated using the NucleoSpin RNA kit (Macherey-Nagel) and reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad). qPCR was performed using TaqMan gene expression products (Thermo Fisher Scientific). The relative change in mRNA expression was calculated using the 2ΔΔCT method with GAPDH mRNA as an internal normalizer. All results are presented as the fold-increase in mRNA expression relative to a specified control, in which cells were typically not treated with LPS or other reagents.
LPS challenge in vivo
Male C57BL/6J mice (16–20 weeks old, ~25 grams) were injected IP with 9 mg/kg LPS. The LD50 for the specific LPS lot was pre-determined in our laboratory, as previously described by us (31) and was 6 mg/kg. One hour later, mice were treated by IV injection with S-PrP (2.5 μg/g body weight) or with PBS. Animals were monitored and scored at 3 h intervals using the Murine Sepsis Score (MSS), as described by Shrum et al. (43). In brief, the following variables were scored from 0–4: appearance, level of consciousness, activity, responses to auditory stimuli, eye function, respiration rate, and respiration quality. Mice were considered moribund and euthanized if the MSS was ≥21. Investigators were blinded to treatment groups. Survival was plotted in Kaplan-Meier curves.
Isolation of plasma EVs by sequential ultracentrifugation
Outdated fresh frozen human plasma (FFP) was obtained from the UCSD Transfusion Medicine service and studied without patient identifiers. The work presented herein was approved by the UCSD IRB for Human Investigation. FFP was subjected to centrifugation at 5,000 × g for 10 min at 4° C to ensure removal of platelets and cellular debris. The supernatant was collected and larger EVs were precipitated by ultracentrifugation (UC) for 2 h at 20,000 × g at 4° C (Avanti J Ultracentrifuge, Beckman Coulter). The supernatant was collected and subjected to UC at 100,000 × g for 18 h at 4° C. The pellet was re-suspended in PBS, sterile-filtered using 0.22 μM syringe filters (EMD Millipore), and washed by UC at 100,000 × g for 2 h at 4° C (Opti-Max E, MLS-50 swinging-bucket rotor, Beckman Coulter). The EV-enriched pellet was re-suspended in PBS for analysis and experiments. The protein content of final EV preparations was determined by DC assay.
Characterization of EVs
NTA was performed using a NanoSight NS300 instrument equipped with a 405 nm laser (Malvern). EV samples were passed through a fluidics flow chamber at a constant flow rate using a syringe pump at room temperature. Each sample was measured in duplicate at a camera setting of 11 with an acquisition time of 30 sec and detection threshold setting of 3. Data were captured and analyzed with NTA software, version 2.3 (Malvern Panalytical).
EV preparations were subjected to immunoblot analysis using antibodies that detect PrPC (Abcam), Flotillin (BD Biosciences), Tsg101 (Abcam), and GM130 (BD Biosciences). For transmission electron microscopy (TEM) studies, isolated EVs were adsorbed to formvar/carbon-coated 100-mesh copper grids for 10 min, washed with water, and negatively stained with 2% uranyl acetate aqueous solution for 1 min. Grids were viewed using a JEOL 1200EX II TEM and photographed using a Gatan digital camera.
Treatment of EVs with PI-PLC
PI-PLC releases GPI-anchored proteins from plasma membranes and thus, may be used to identify proteins that are anchored to plasma membranes by this type of linkage (37, 38). Equal amounts of EVs were treated with PI-PLC (0.1 units/mg EV protein) or with vehicle for 1 h at 4° C with constant agitation. Samples were subjected to UC at 100,000 × g for 2 h at 4° C. Supernatants, containing released proteins, were separated and retained for analysis. EV-containing pellets were washed once, re-suspended in PBS, and also retained for analysis.
Statistics
Statistical analysis was performed using GraphPad Prism 9.0 (GraphPad Software). All results are expressed as the mean ± SEM. When “n” values are reported, each replicate was performed using a different macrophage preparation or, when relevant, an EV preparation isolated from a different human plasma sample. Data were analyzed by one-way ANOVA followed by post-hoc Tukey’s multiple comparison test. Kaplan-Meier survival curves were analyzed using the Mantel-Cox test. P-values of *P<0.05, **P<0.01, ***P<0.001, ****P< 0.0001 were considered statistically significant.
Results
S-PrP neutralizes the activity of LPS in macrophages and in vivo in mice
BMDMs were harvested from WT C57BL/6J mice and treated with 0.1 μg/mL LPS for 6 h in the presence of increasing concentrations of S-PrP. In the absence of S-PrP, LPS significantly increased expression of the mRNAs encoding TNFα and IL-6, as anticipated. S-PrP, at concentrations of 40 nM or higher, blocked the effects of LPS on expression of TNFα and IL-6 (Fig. 1A). In the absence of LPS, 40 nM S-PrP did not significantly regulate expression of TNFα or IL-6. Furthermore, in the absence of LPS, S-PrP (40 nM and 120 nM) did not affect BMDM viability (Supplementary Fig. 1).
FIGURE 1.
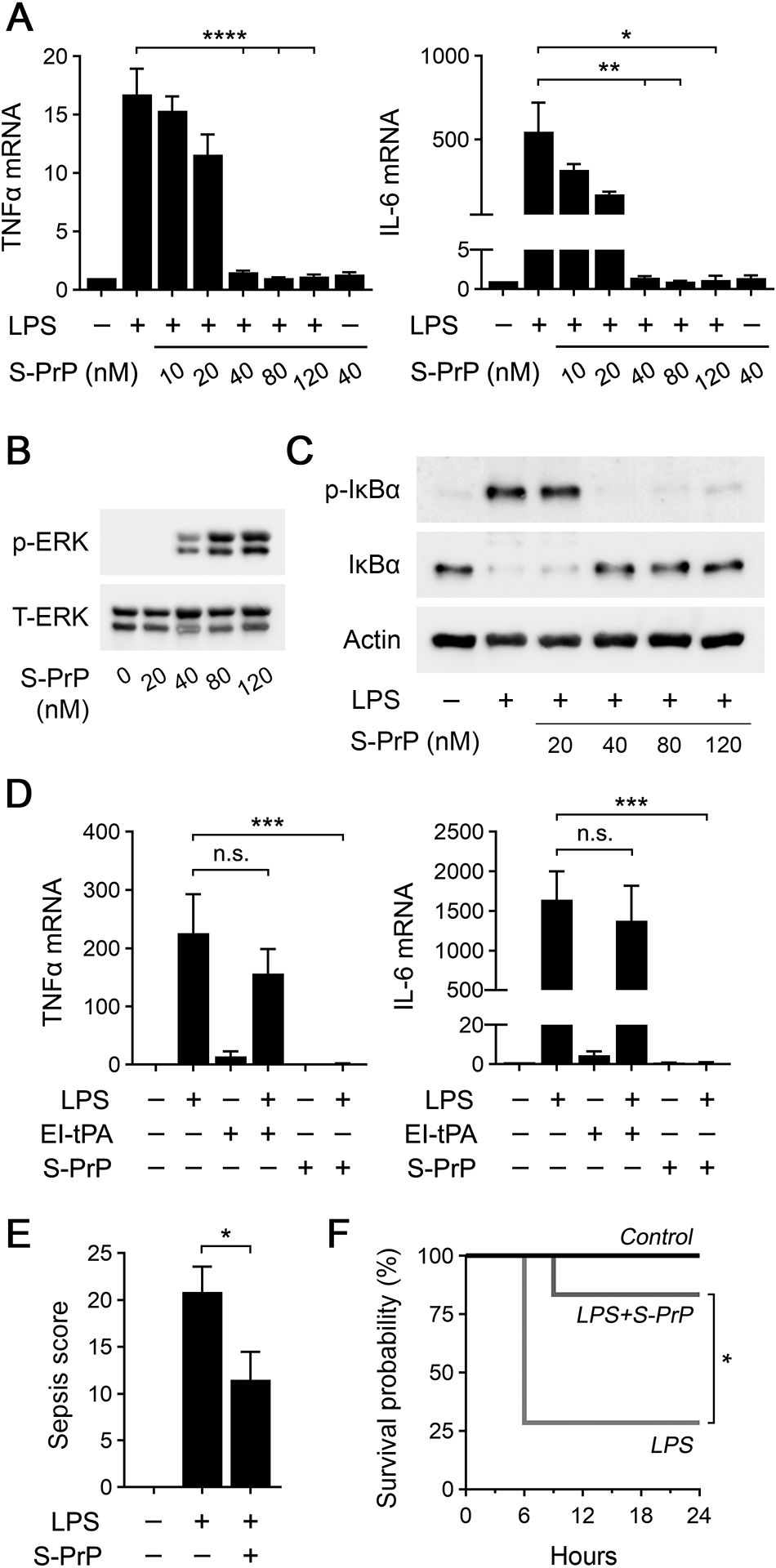
S-PrP counteracts the activity of LPS in vitro and in vivo. (A) BMDMs from C57BL/6J mice were treated for 6 h with LPS (0.1 μg/mL) in presence of increasing concentrations of S-PrP (10–120 nM) or with 40 nM S-PrP in the absence of LPS. RT-qPCR was performed to determine mRNA levels for TNFα and IL-6 (n = 3). (B) BMDMs were stimulated for 1 h with increasing concentrations of S-PrP (20–120 nM). Cell extracts were subjected to immunoblot analysis to detect phospho-ERK1/2 and total ERK1/2. (C) BMDMs were treated for 1 h with LPS (0.1 μg/mL) in presence of increasing concentrations of S-PrP (20–120 nM). Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. (D) pMacs were treated with LPS (0.1 μg/mL), El-tPA (12 nM), S-PrP (40 nM), LPS plus EI-tPA, LPS plus S-PrP, or vehicle for 6 h. RT-qPCR was performed to compare mRNA levels for TNFα and IL-6 (n = 3). (E) Sepsis scores are shown for wild-type C57BL/6J mice treated by IV injection with vehicle (n = 7) or 2.5 μg/g S-PrP (n = 6), 1 h after IP injection of LPS at 1.5 times the LD50 (9 mg/kg). (F) Kaplan-Meier survival curves are shown for mice treated by IV injection with 2.5 μg/g S-PrP (n = 6) or vehicle (n = 7), 1 h after IP injection of LPS at 1.5 times the LD50 (9 mg/kg). Significance was determined by Mantel-Cox test. RT-qPCR and sepsis scoring data are expressed as the mean ± SEM in panels (A), (D) and (E) (one-way ANOVA; *P <0.05, **P <0.01, ***P <0.001, ****P < 0.0001, n.s.: not statistically significant).
To test whether S-PrP directly activates cell-signaling in BMDMs, we treated cells with increasing concentrations of S-PrP for 1 h and then studied ERK1/2 phosphorylation. Fig. 1B shows that S-PrP, at concentrations greater than or equal to 40 nM, activated ERK1/2. The concentrations of S-PrP that activated ERK1/2 matched those that were effective in neutralizing LPS-stimulated cytokine expression (shown in Fig. 1A).
When BMDMs were treated for 1 h with 0.1 μg/mL LPS, in the absence of S-PrP, IκBα was phosphorylated and the abundance of IκBα was decreased (Fig. 1C). These anticipated effects of LPS report activation of NFκB as a transcription factor, which is essential for expression of pro-inflammatory cytokines (44). In BMDMs treated simultaneously with LPS and 40–120 nM S-PrP, the effects of LPS on IκBα phosphorylation and abundance were blocked.
As a second model system to study the activity of S-PrP, we isolated macrophages from the peritoneal space of mice (pMacs) without eliciting or activating agents (33, 42). Compared with BMDMs, pMacs express lower levels of cell-surface NMDA-R and, as a result, are incapable of responding to the NMDA-R/LRP1 receptor system ligand, EI-tPA (33). We examined the ability of S-PrP to neutralize the response to LPS in pMacs and, as a control, we re-examined EI-tPA. Fig. 1D shows that 0.1 μg/mL LPS increased expression of the mRNAs encoding TNFα and IL-6 in pMacs, as anticipated. When pMacs were treated simultaneously with LPS and 12 nM EI-tPA, a concentration of EI-tPA that is fully effective in blocking LPS activity in BMDMs (31), cytokine mRNA expression in pMacs was not inhibited, confirming our earlier results (33). By contrast, 40 nM S-PrP completely blocked LPS-induced cytokine mRNA expression in pMacs. Thus, S-PrP is active as an anti-LPS/TLR4 agent in cells in which the NMDA-R/LRP1 ligand, EI-tPA, does not demonstrate efficacy.
To test the ability of S-PrP to inhibit TLR4 responses in vivo, C57BL/6J mice (~25 gram) were treated by IV injection with 2.5 μg/g body weight S-PrP (n = 6) or vehicle (n = 7), 1 h after injecting LPS at 1.5 × the LD50. Animals were scored for toxicity at 3 h intervals, examining criteria that included level of consciousness, appearance, activity, response to auditory stimuli, respiration quality and rate (43). Mice that entered a moribund state were euthanized immediately. Fig. 1E shows that toxicity scores, determined at 6 h when these scores maximized in animals treated with LPS alone, were significantly decreased in mice treated with S-PrP. Fig. 1F shows that more than half of the mice treated with LPS alone required euthanasia due to the degree of toxicity. A single dose of S-PrP significantly improved survival.
S-PrP targets a large continuum of PRRs in innate immunity
LTA is a selective TLR2 agonist, produced by gram-positive bacteria (45, 46). BMDMs treated with 1.0 μg/mL LTA for 6 h demonstrated increased expression of the mRNAs encoding TNFα and IL-6, as anticipated (Fig. 2A). S-PrP (40 nM) neutralized the effects of LTA on expression of TNFα and IL-6.
FIGURE 2.
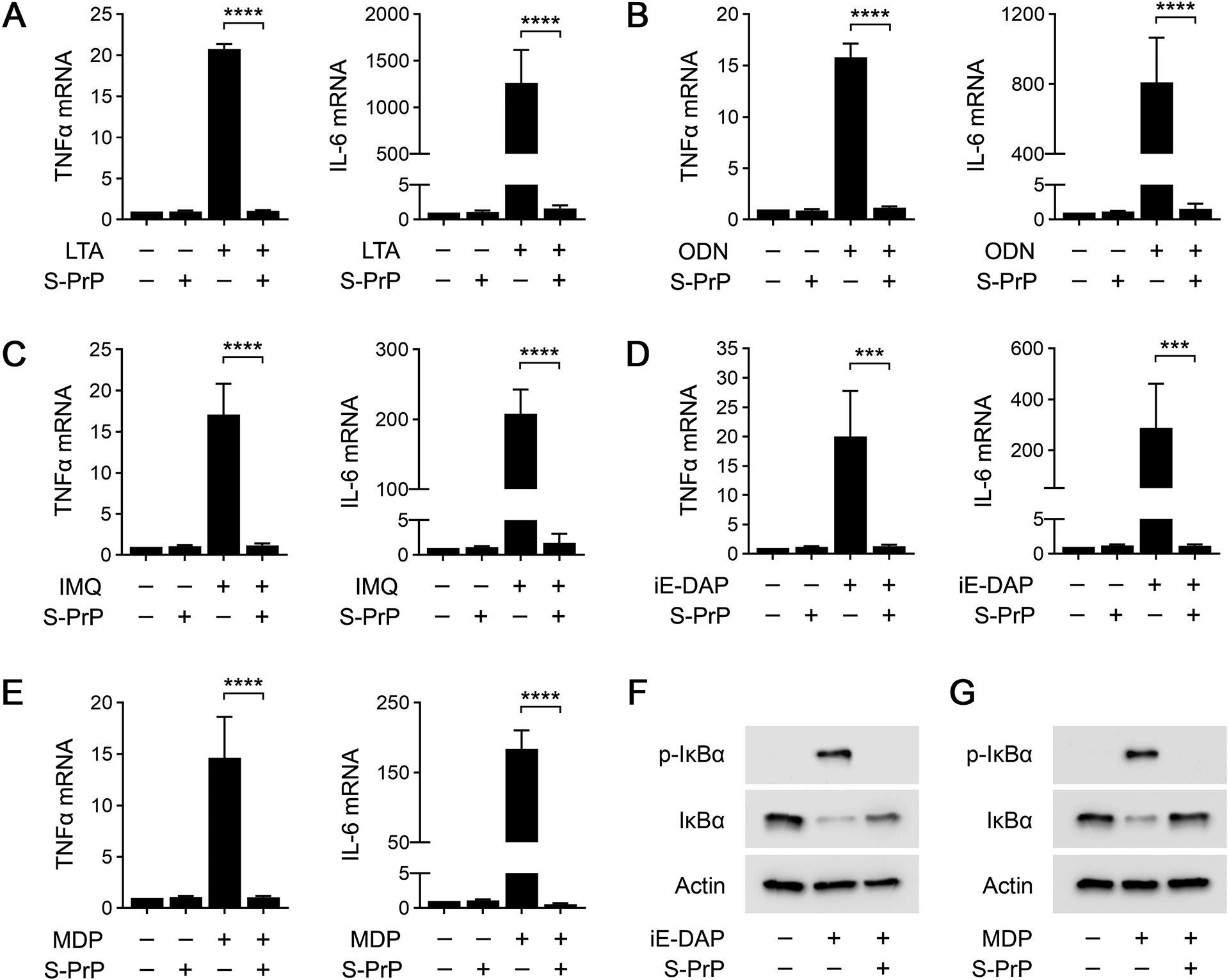
S-PrP inhibits multiple Pattern Recognition Receptors. (A) BMDMs were treated with the TLR2 agonist LTA (1.0 μg/mL), S-PrP (40 nM), or LTA plus S-PrP for 6 h. RT-qPCR was performed to compare mRNA levels of TNFα and IL-6 (n = 4). (B) BMDMs were treated with the TLR9 agonist, ODN 1826 (1.0 μM), S-PrP (40 nM), or ODN 1826 plus S-PrP for 6 h. RT-qPCR was performed to compare mRNA levels of TNFα and IL-6 (n = 3). (C) BMDMs were treated with the TLR7 agonist IMQ (3.0 μg/mL), S-PrP (40 nM), or IMQ plus S-PrP for 6 h. RT-qPCR was performed to compare mRNA levels of TNFα and IL-6 (n = 3). (D) BMDMs were treated with the NOD1 agonist C12-iE-DAP (1 μg/mL), S-PrP (40 nM), or C12-iE-DAP plus S-PrP for 6 h. RT-qPCR was performed to compare mRNA levels of TNFα and IL-6 (n = 3). (E) BMDMs were treated with the NOD2 agonist, L18-MDP (0.1 μg/mL), S-PrP (40 nM), or L18-MDP plus S-PrP for 6 h. RT-qPCR was performed to compare mRNA levels of TNFα and IL-6 (n = 3). Data are expressed as the mean ± SEM (one-way ANOVA; ***P < 0.001, ****P < 0.0001). (F-G) Immunoblot analysis showing total and phosphorylated IκBα in BMDMs treated for 1 h with: (F) C12-iE-DAP (1.0 μg/mL) in presence or absence of S-PrP (40 nM); and (G) L18-MDP (0.1 μg/mL) in presence or absence of S-PrP (40 nM). β-actin levels are shown as a control for load.
ODN 1826 is a TLR9 agonist (47, 48). ODN 1826 (1.0 μM) significantly increased expression of TNFα and IL-6 mRNA in BMDMs (Fig. 2B). S-PrP blocked the effects of ODN 1826 on expression of TNFα and IL-6. Equivalent results were obtained when we studied the TLR7 agonist, IMQ. IMQ (3.0 μg/mL) induced expression of TNFα and IL-6 mRNA in BMDMs and the response was blocked by S-PrP (Fig. 2C). These results support the conclusion that S-PrP is a generalized inhibitor of macrophage responses elicited by TLRs.
NOD1 and NOD2 are intracellular PRRs (49, 50). Unlike TLRs, the responses elicited by NOD1 and NOD2 agonists in BMDMs are not neutralized by EI-tPA and may in fact be amplified (33). We treated BMDMs with the NOD1 agonist, C12-iE-DAP (1 μg/mL), or the NOD2 agonist, L18-muramyl dipeptide (MDP) (0.1 μg/mL), for 6 h. Figs. 2D and 2E show that both agents increased expression of the mRNAs encoding TNFα and IL-6. When BMDMs were treated with C12-iE-DAP and 40 nM S-PrP simultaneously, the effects of C12-iE-DAP on cytokine mRNA expression were neutralized. Similarly, 40 nM S-PrP blocked cytokine mRNA expression in response to MDP.
IκBα was phosphorylated and the abundance of IκBα was decreased in BMDMs treated with C12-iE-DAP (1 μg/mL) for 1 h (Fig. 2F). These effects of C12-iE-DAP on IκBα were blocked by S-PrP (40 nM). S-PrP also inhibited IκBα phosphorylation in response to MDP (0.1 μg/mL) (Fig. 2G). Collectively, these results show that S-PrP neutralizes responses elicited by PRRs in addition to TLRs and thus, may target a broader continuum of PRRs, compared with EI-tPA (33).
S-PrP inhibits PRRs by interacting with the macrophage NMDA-R/LRP1 receptor system
We tested the ability of PrPC-specific monoclonal antibodies with defined epitopes to neutralize the effects of S-PrP on the LPS response in BMDMs. POM1 and POM19 recognize epitopes in the C-terminal globular region of PrPC, whereas POM2 recognizes the tandem octarepeats in the N-terminal unstructured region of PrPC (40). POM3 recognizes an epitope C-terminal to the POM2 epitope, near the center of PrPC (40).
BMDMs were treated with 0.1 μg/mL LPS and 40 nm S-PrP in the presence of each antibody (10 μg/ml) for 1 h. POM2 completely blocked the ability of S-PrP to inhibit LPS-induced IκBα phosphorylation (Fig. 3A). The other antibodies were without effect, as was non-specific IgG. In cytokine expression studies, POM2 blocked the ability of S-PrP to inhibit expression of TNFα and IL-6 in BMDMs treated with 0.1 μg/mL LPS (Fig. 3B). POM2 did not significantly affect cytokine expression in BMDMs in the absence of S-PrP. These results implicate a site in the N-terminal unstructured region of S-PrP as critical for the anti-LPS/TLR4 activity of S-PrP.
FIGURE 3.
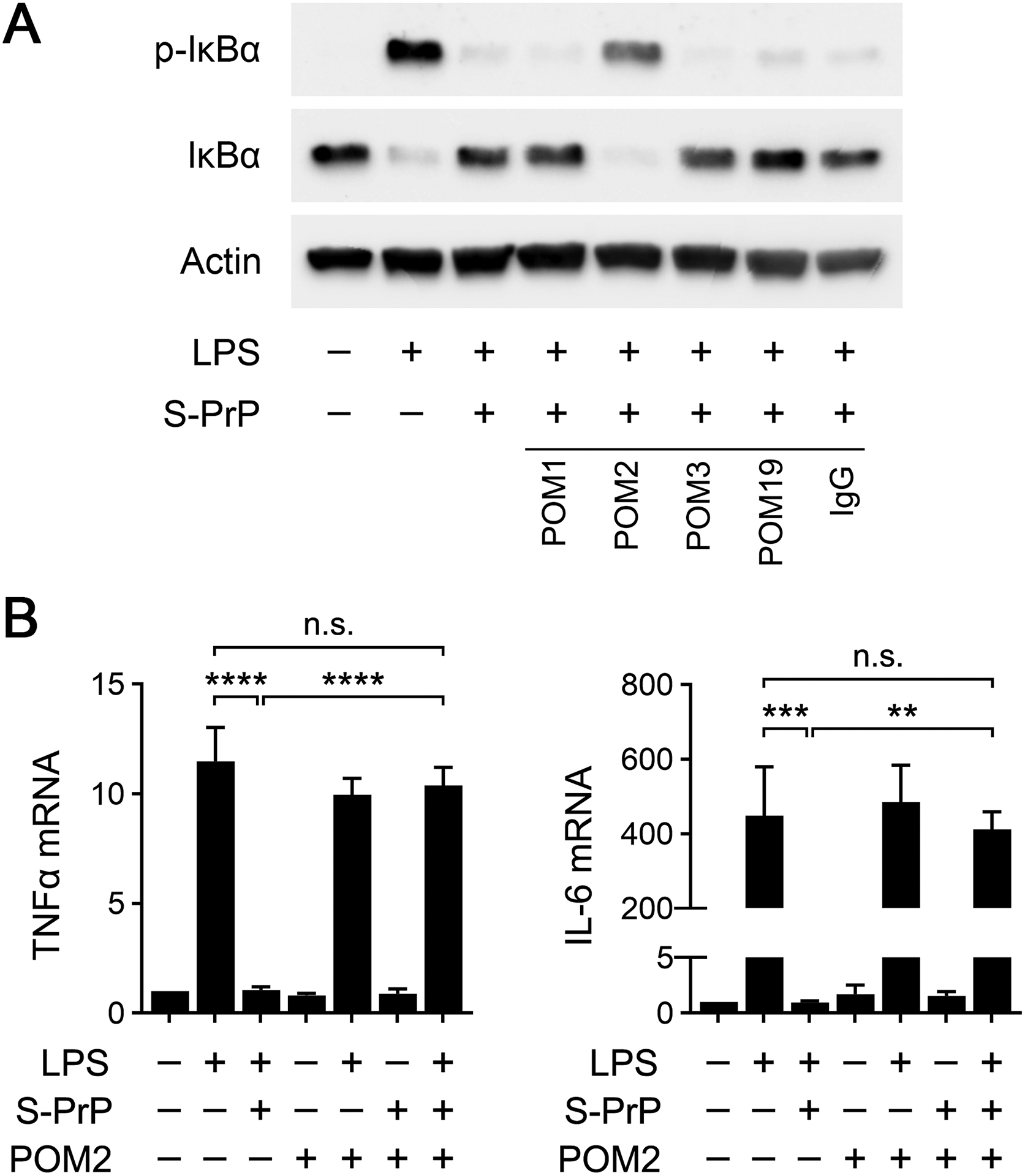
Effects of PrPC-specific monoclonal antibodies on the activity of S-PrP. (A) BMDMs were treated for 1 h with LPS (0.1 μg/mL), LPS plus S-PrP (40 nM), or vehicle in the presence of POM1, POM2, POM3, POM19, or nonspecific IgG (10 μg/ml). Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. (B) BMDMs were treated for 6 h with LPS (0.1 μg/mL), LPS plus S-PrP (40 nM), or with vehicle in presence or absence of POM2 antibody (10 μg/ml). RT-qPCR was performed to compare mRNA levels for TNFα and IL-6 (n = 3). RT-qPCR data are expressed as the mean ± SEM (one-way ANOVA; **P <0.01, ***P <0.001, ****P < 0.0001, n.s.: not statistically significant).
The NMDA-R is expressed by macrophages and essential for the anti-inflammatory activity of EI-tPA and activated α2M (31). Because the NMDA-R mediates S-PrP cell-signaling responses in neuron-like cells and Schwann cells (26), we tested whether the NMDA-R is required for the anti-inflammatory activity of S-PrP in macrophages. BMDMs were treated with the non-competitive NMDA-R antagonists, MK801 (1.0 μM) or DXM (10 μM). Both reagents completely blocked the ability of 40 nM S-PrP to neutralize inflammatory cytokine expression in BMDMs treated with 0.1 μg/mL LPS (Fig. 4A). MK801 and DXM also blocked the ability of 40 nM S-PrP to inhibit LPS-induced IκBα phosphorylation (Fig. 4B). The effects of the NMDA-R antagonists were not overcome by increasing the concentration of S-PrP to 120 nM. In control experiments, MK801 and DXM did not alter the effects of LPS on IκBα phosphorylation or total abundance in the absence of S-PrP, as anticipated (Supplementary Fig. 2).
FIGURE 4.
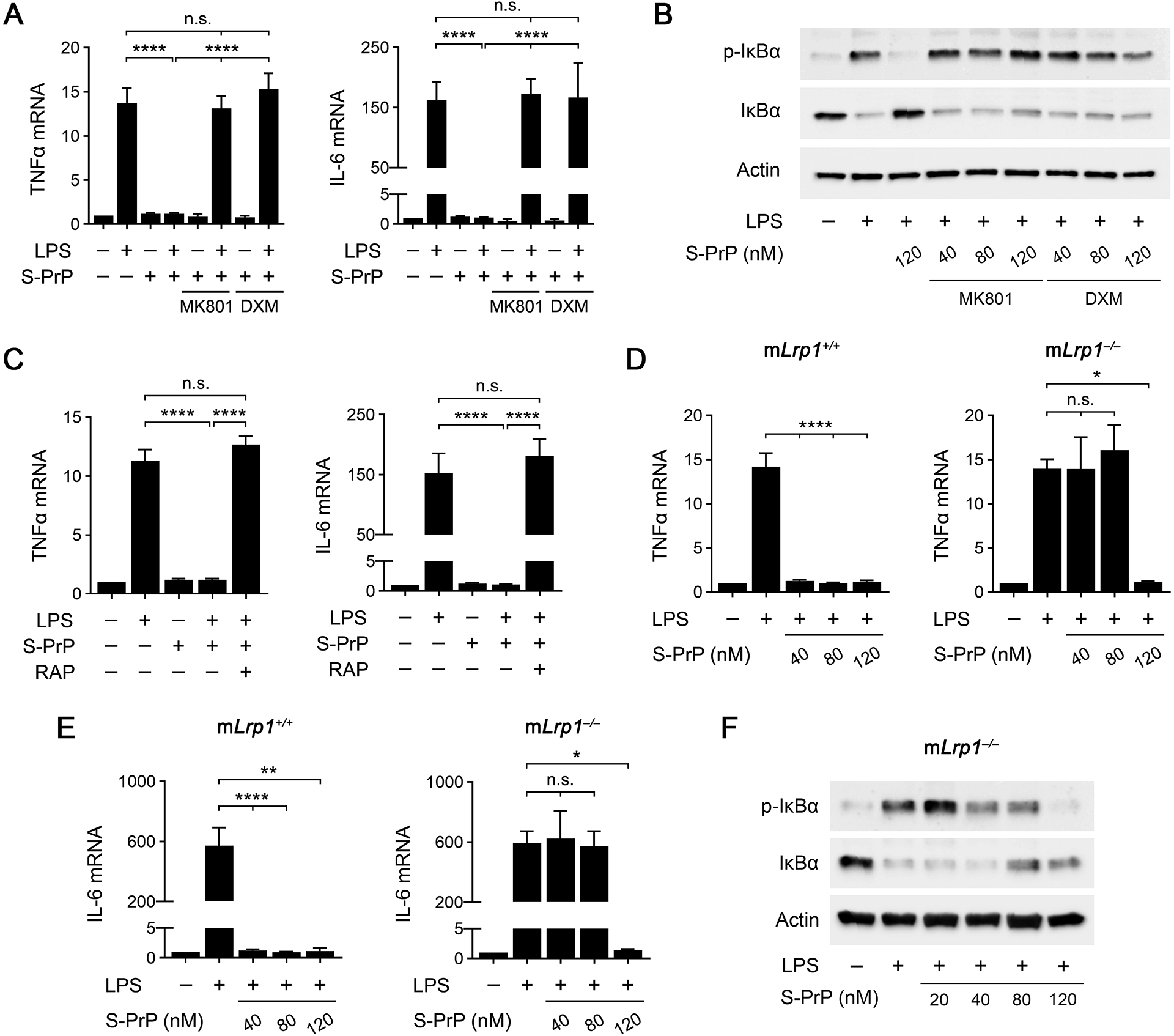
The NMDA-R/LRP1 system mediates the anti-LPS/TLR4 activity of S-PrP. (A) BMDMs were pre-incubated with MK801 (1.0 μM), DXM (10 μM), or vehicle for 30 min. The cells were then treated with LPS (0.1 μg/mL), S-PrP (40 nM), LPS plus S-PrP, or with vehicle for 6 h. RT-qPCR was performed to compare mRNA levels for TNFα and IL-6 (n = 3). (B) BMDMs were pre-treated with MK801 (1.0 μM), DXM (10 μM), or vehicle for 30 min, and then with LPS (0.1 μg/mL) and increasing concentrations of S-PrP (40–120 nM) for 1 h, as indicated. Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. (C) BMDMs were pre-treated with RAP (150 nM) or vehicle for 30 min, and then with LPS (0.1 μg/mL) and S-PrP (40 nM) for 6 h, as indicated. RT-qPCR was performed to compare mRNA levels for TNFα and IL-6 (n = 3). (D) BMDMs from mLrp1+/+ and mLrp1−/− mice were treated for 6 h with LPS (0.1 μg/mL) in presence of increasing concentrations of S-PrP (40–120 nM). RT-qPCR was performed to determine mRNA levels for TNFα (n = 3). (E) BMDMs from mLrp1+/+ and mLrp1−/− mice were treated for 6 h with LPS (0.1 μg/mL) in presence of increasing concentrations of S-PrP (40–120 nM). RT-qPCR was performed to determine mRNA levels for IL-6 (n = 3). (F) BMDMs from mLrp1−/− mice were treated for 1 h with LPS (0.1 μg/mL) in the presence of increasing concentrations of S-PrP (20–120 nM). Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. RT-qPCR data are expressed as the mean ± SEM (one-way ANOVA; *P <0.05, ****P < 0.0001, n.s.: not statistically significant).
Next, we studied the activity of LRP1 as a mediator of the response to S-PrP in BMDMs. BMDMs were treated with 0.1 μg/mL LPS and 40 nM S-PrP, in the presence and absence of RAP (150 nM), a protein antagonist of ligand-binding to LRP1 and other members of the LDL receptor family (27, 51). RAP blocked the ability of 40 nM S-PrP to neutralize expression of TNFα and IL-6 in LPS-treated BMDMs (Fig. 4C), suggesting a role for LRP1.
To confirm the role of LRP1, we isolated BMDMs from mLrp1−/− mice. LRP1 protein is undetectable in macrophages harvested from these mice (30, 41). Control LRP1-expressing BMDMs were harvested from mLrp1+/+ mice. LPS (0.1 μg/mL) increased expression of the mRNAs encoding TNFα and IL-6 in LRP1-expressing and –deficient BMDMs similarly (Fig. 4D, E). S-PrP (40–120 nM) blocked LPS-induced cytokine mRNA expression in LRP1-expressing BMDMs isolated from mLrp1+/+ mice, as anticipated. However, S-PrP (40–80 nM) was ineffective at inhibiting LPS-induced cytokine expression in LRP1-deficient BMDMs. The activity of S-PrP was restored in LRP1-deficient BMDMs when the S-PrP concentration was increased to 120 nM.
Fig. 4F shows that the concentration of S-PrP required to neutralize the effects of LPS on IκBα phosphorylation was increased from 40 nM, in LRP1-expressing BMDMs (see Fig. 1C), to 120 nM in LRP1-deficient BMDMs from mLRP1−/− mice. These results suggest that LRP1 deficiency in BMDMs does not eliminate the anti-LPS/TLR4 activity of S-PrP, but instead, increases the concentration of S-PrP required for efficacy.
Mattei et al (35) reported that membrane-anchored PrPC is required to support NMDA-R/LRP1-dependent, tPA-activated cell-signaling in neurons. To test whether membrane-anchored PrPC is required for macrophages to respond to S-PrP, we studied BMDMs isolated from mice with global deletion of Prnp, the gene encoding PrPC (52). S-PrP (40 nM) blocked the effects of LPS on expression of TNFα and IL-6 equivalently in WT BMDMs (Fig. 5A) and PrPC-deficient BMDMs (Fig. 5B). Thus, macrophages do not appear to require membrane-anchored PrPC to respond to S-PrP.
FIGURE 5.
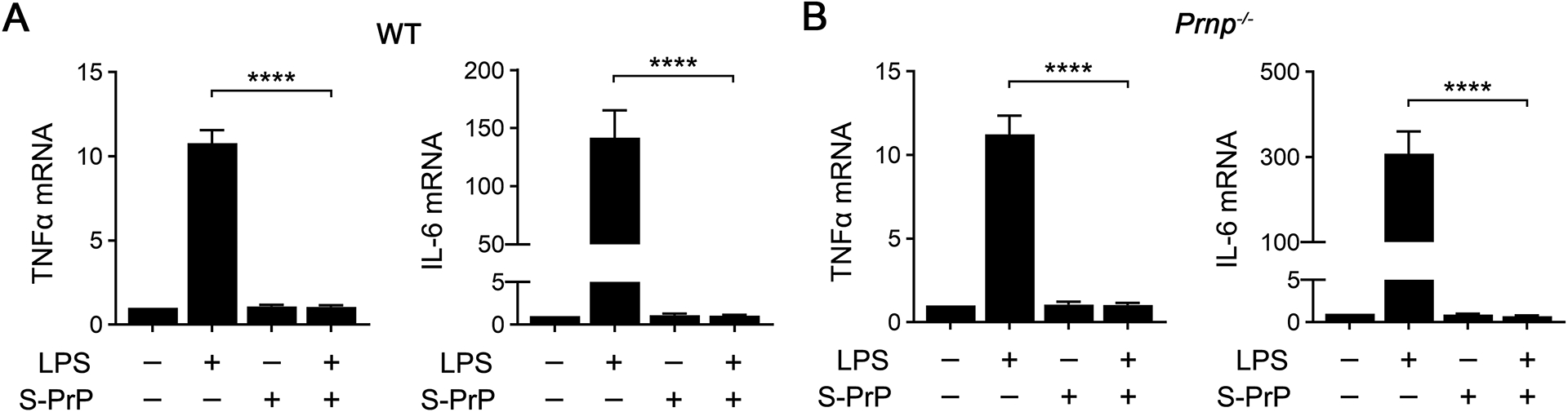
Macrophage PrPC is not necessary for the anti-LPS/TLR4 activity of S-PrP. (A-B) BMDMs isolated from WT mice (A) and from Prnp−/− mice (B) were treated with LPS (0.1 μg/mL), S-PrP (40 nM), LPS plus S-PrP, or vehicle for 6 h. RT-qPCR was performed to compare mRNA levels for TNFα and IL-6 (n = 3). Data are expressed as the mean ± SEM (one-way ANOVA; ****P <0.0001).
Human plasma EVs demonstrate anti-inflammatory activity by engaging the NMDA-R/LRP1 receptor system
We isolated EVs from human blood bank plasma by sequential ultracentrifugation (UC), as previously described (13, 53). Nanoparticle Tracking Analysis (NTA) demonstrated particles of variable size, ranging from 50–400 nm (Fig. 6A). The NTA results indicate that the isolated EVs were heterogeneous, as anticipated.
FIGURE 6.
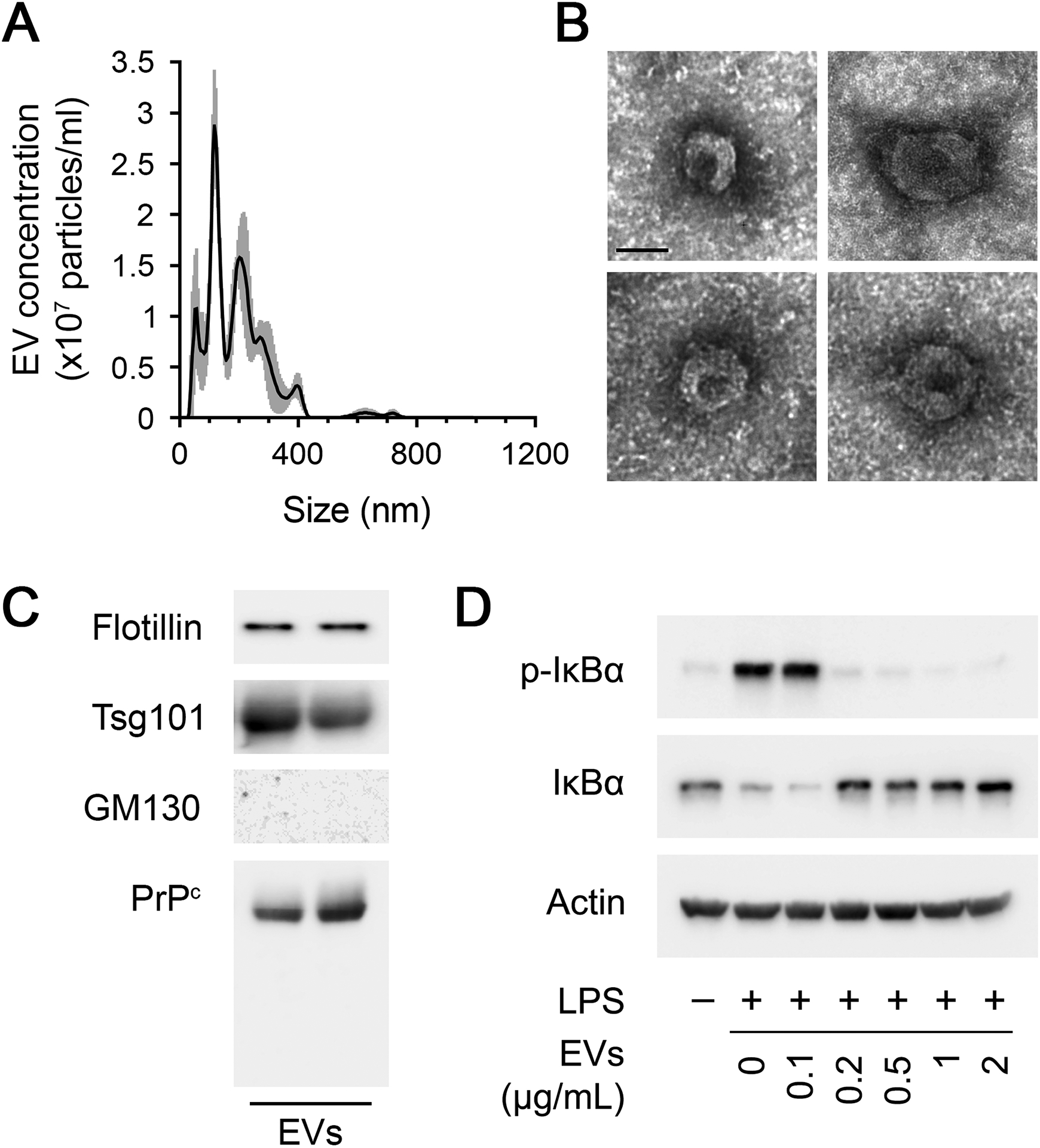
Human plasma EVs inhibit the response of macrophages to LPS. (A) NTA of a representative human plasma EV preparation obtained by sequential UC. (B) Representative TEM images of EVs isolated from human plasma (Scale bar = 50 nm). (C) Characterization of two representative human plasma EV UC preparations. Immunoblot analysis was performed to detect the exosome biomarkers, flotillin and Tsg101. The same blots were probed for PrPC and GM130. (D) BMDMs were treated for 1 h with LPS (0.1 μg/mL) in presence of increasing concentrations of human plasma EVs (0.1–2.0 μg/mL). Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin.
Human plasma EVs were negatively stained and examined by TEM. Fig. 6B shows representative examples of EVs present in the UC EV preparations from human plasma. The exosome biomarker, flotillin, a lipid raft associated protein, and the cytosolic marker, Tumor Susceptibility 101 (Tsg101), were identified in two representative plasma UC EV preparations, together with PrPC (Fig. 6C). The golgi matrix protein, GM130, was absent from human plasma EVs, as anticipated.
Because human plasma EVs carry PrPC, we tested whether these EVs replicate the activity of S-PrP in experiments with cultured macrophages. BMDMs were treated with LPS (0.1 μg/mL) and increasing amounts of human plasma EVs for 1 h. IκBα phosphorylation and abundance were examined. Human plasma EVs blocked the effects of LPS on IκBα phosphorylation and abundance in an EV concentration-dependent manner (Fig. 6D). Complete inhibition of the LPS response was observed when the amount of EV-associated protein added to the cultures was equal to or exceeded 0.2 μg/mL.
Next, we tested whether human plasma EVs regulate cytokine mRNA expression in BMDMs treated with LPS. Fig. 7A shows that EVs (1.0 μg/mL) blocked expression of TNFα and IL-6 in BMDMs treated with 0.1 μg/mL LPS for 6 h. Plasma EVs did not regulate expression of TNFα or IL-6 in the absence of LPS. The effects of the EVs on LPS-induced cytokine expression were neutralized by the NMDA-R antagonist, MK801, suggesting an essential role for the NMDA-R, and by RAP, suggesting a role for LRP1. MK801 and RAP also blocked the ability of human plasma EVs to inhibit IκBα phosphorylation and the accompanying decrease in IκBα abundance in LPS-treated BMDMs (Fig. 7B).
FIGURE 7.
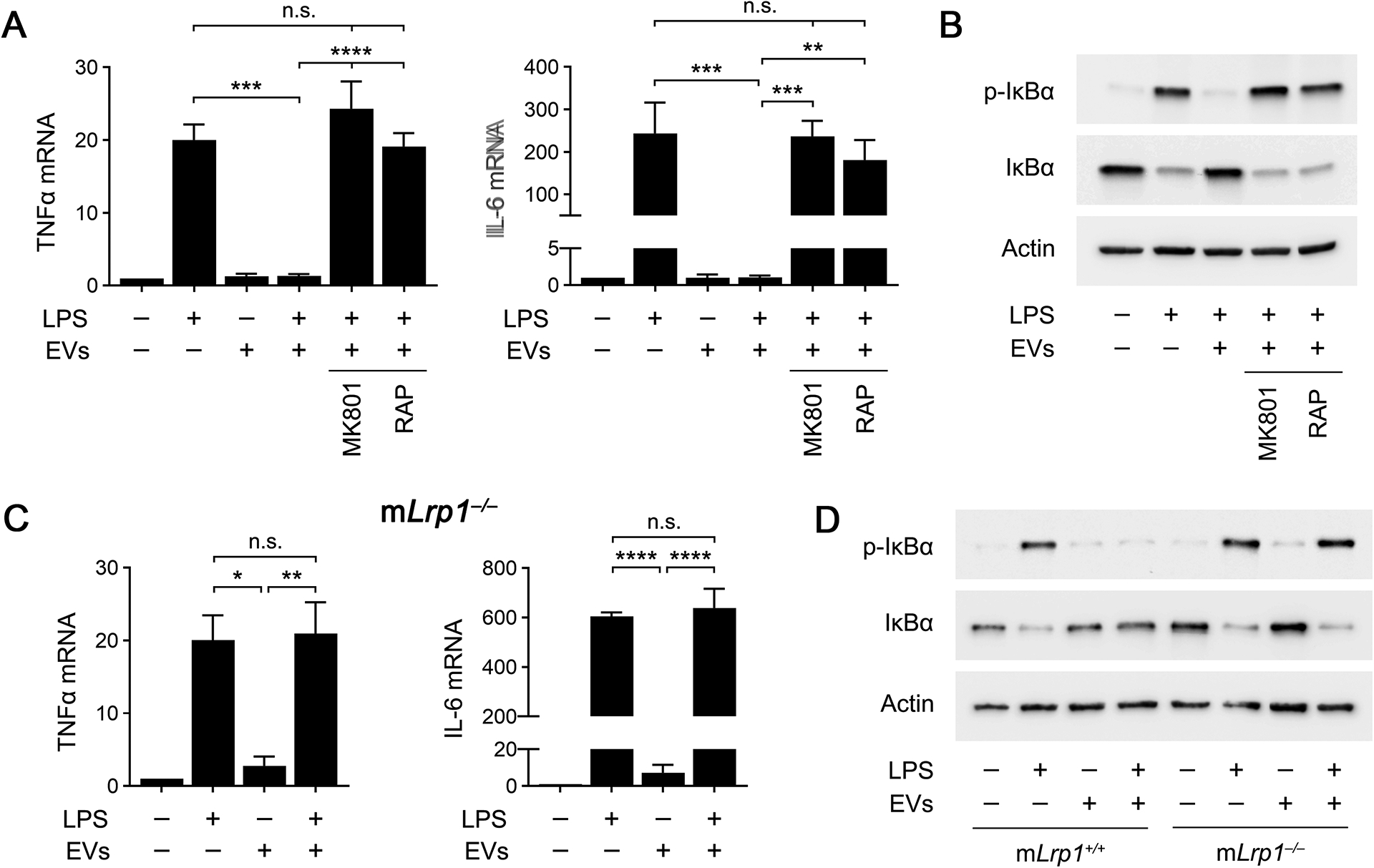
The NMDA-R/LRP1 receptor complex is required for the anti-LPS/TLR4 activity of human plasma EVs. (A) BMDMs were pre-incubated with MK801 (1.0 μM), RAP (150 nM), or vehicle for 30 min. The cells were then treated with LPS (0.1 μg/mL), human plasma EVs (1.0 μg/mL), LPS plus EVs, or with vehicle for 6 h. RT-qPCR was performed to compare mRNA levels for TNFα and IL-6 (n = 3). (B) BMDMs were pre-treated with MK801 (1.0 μM), RAP (150 nM), or vehicle for 30 min, and then with LPS (0.1 μg/mL) and/or human plasma EVs (1.0 μg/mL) for 1 h, as indicated. Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. (C) BMDMs from mLrp1−/− mice were treated for 6 h with LPS (0.1 μg/mL), human plasma EVs (1.0 μg/mL), or LPS plus EVs. RT-qPCR was performed to determine mRNA levels for TNFα and IL-6 (n = 3). (D) BMDMs from mLrp1−/− mice were treated with LPS (0.1 μg/mL), human EVs (1.0 μg/mL), LPS plus EVs, or with vehicle for 1 h. Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. RT-qPCR data are expressed as the mean ± SEM (one-way ANOVA; *P <0.05, **P <0.01, ***P <0.001, ****P < 0.0001, n.s.: not statistically significant).
To confirm that macrophage LRP1 mediates the anti-LPS/TLR4 activity of human plasma EVs, we studied LRP1-deficient BMDMs isolated from mLrp1−/− mice. LPS (0.1 μg/mL) increased expression of the mRNAs encoding TNFα and IL-6 in LRP1-deficient BMDMs, as anticipated (Fig. 7C); however, human plasma EVs (1.0 μg/mL) failed to inhibit the LPS response in these cells. Similarly, human plasma EVs failed to inhibit the effects of LPS on IκBα phosphorylation in LRP1-deficient BMDMs (Fig. 7D). These results confirm that macrophage LRP1 mediates the anti-LPS/TLR4 activity of human plasma EVs.
The anti-LPS/TLR4 activity of human plasma EVs requires GPI-anchored PrPC
To test whether EV-associated PrPC is responsible for the effects of human plasma EVs on LPS-induced TNFα expression, LPS (0.1 μg/mL) and EVs (1.0 μg/mL) were added to BMDM cultures in the presence of the PrPC-specific antibodies: POM1, POM2, POM3, or POM19 (each at 10 μg/ml). POM2 completely neutralized the anti-LPS activity of the EVs, restoring TNFα expression to the level observed in cells treated with LPS alone (Fig. 8A). The other antibodies were entirely ineffective. This is an important result because POM2 is the only PrPC-targeting antibody that blocked the anti-LPS/TLR4 activity of recombinant S-PrP. In control studies, the antibodies examined did not regulate TNFα expression in the absence of LPS.
FIGURE 8.
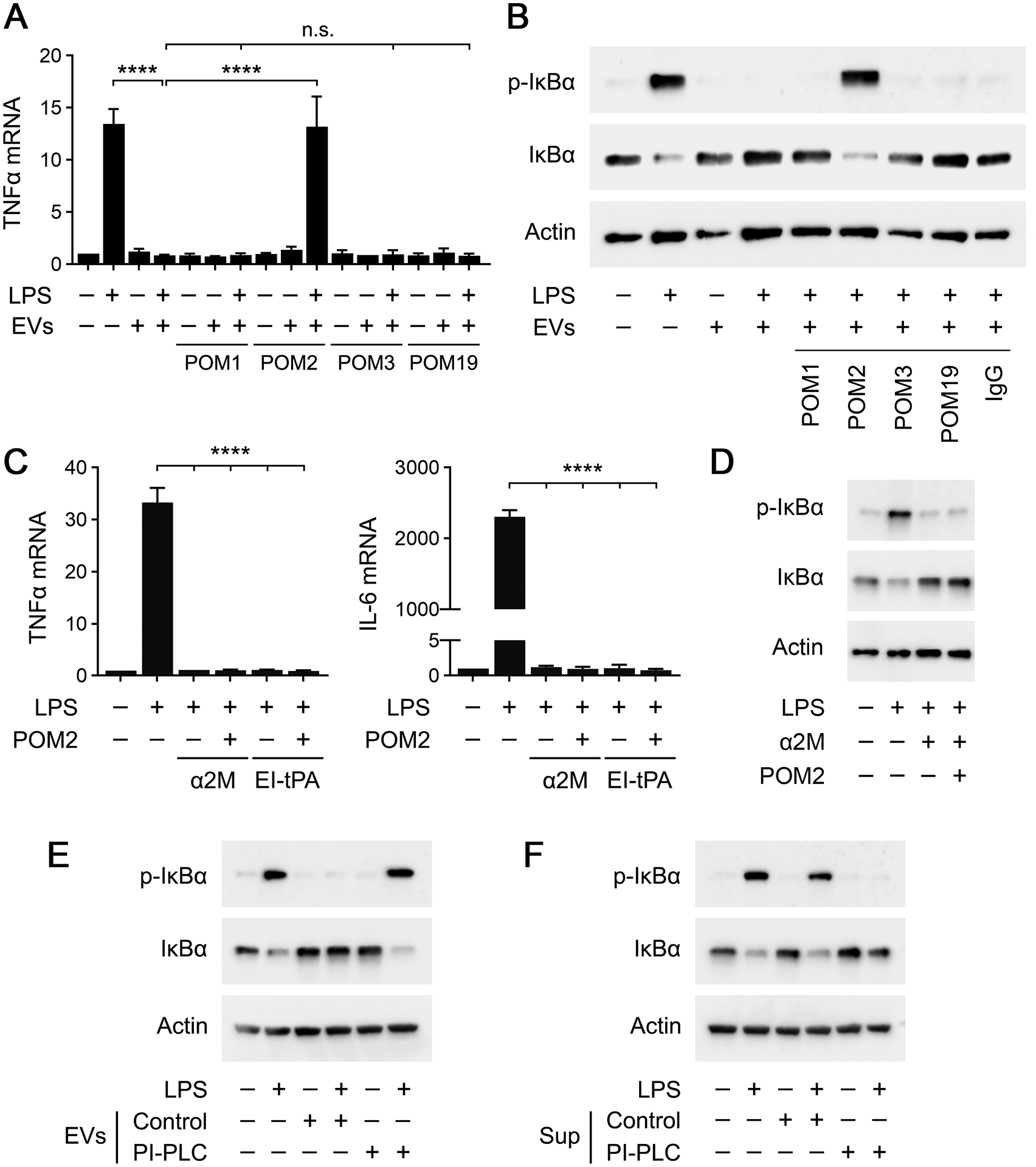
GPI-anchored PrPC is responsible for the anti-LPS/TLR4 activity of human plasma EVs. (A) BMDMs were treated for 6 h with LPS (0.1 μg/mL), human EVs (1.0 μg/mL), LPS plus EVs, or vehicle in the presence of POM1, POM2, POM3, or POM19 (10 μg/ml). RT-qPCR was performed to determine TNFα mRNA expression (n = 3). (B) BMDMs were treated for 1 h with LPS (0.1 μg/mL), human plasma EVs (1.0 μg/mL), LPS plus EVs, or vehicle in the presence of nonspecific IgG, POM1, POM2, POM3, or POM19 (10 μg/ml). Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. (C) BMDMs were treated with LPS (0.1 μg/mL) in the presence or absence of α2M (10 nM), EI-tPA (12 nM), and POM2 (10 μg/mL) for 6 h, as indicated. RT-qPCR was performed to determine TNFα and IL-6 mRNA expression (n = 3). (D) BMDMs were treated with LPS (0.1 μg/mL), α2M (10 nM), and POM2 (10 μg/mL) for 1 h, as indicated. Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. (E) Human plasma EVs were treated with PI-PLC or with vehicle and subjected to UC to separate the EVs from soluble proteins in the supernatants. BMDMs were treated for 1 h with LPS (0.1 μg/mL), 2.0 μg/mL of EVs that were unmodified (Control), LPS + unmodified EVs, 2.0 μg/mL of PI-PLC-treated EVs (PI-PLC), or LPS + PI-PLC-treated EVs, as indicated. Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. (F) Human plasma EVs were treated with PI-PLC or with vehicle. The supernatants were separated from the EVs by UC and studied. BMDMs were treated for 1 h with LPS (0.1 μg/mL), supernatants harvested from 2.0 μg of control EVs (Control), LPS + supernatants from control EVs, supernatants from 2.0 μg of PI-PLC-treated EVs (PI-PLC), or LPS + supernatants from PI-PLC-treated EVs, as indicated. Immunoblot analysis was performed to detect phospho-IκBα, total IκBα, and β-actin. RT-qPCR data are expressed as the mean ± SEM (one-way ANOVA; ****P <0.0001, n.s.: not statistically significant).
Next, we examined the activity of the POM antibodies in IκBα phosphorylation experiments. Fig. 8B shows that POM2 completely blocked the ability of human plasma EVs to inhibit IκBα phosphorylation and the accompanying decrease in the IκBα abundance in BMDMs treated with LPS (0.1 μg/mL). The PrPC-specific antibodies, POM1, POM3, and POM19, and non-specific IgG were without effect.
Our results with BMDMs isolated from Prnp−/− mice suggested that macrophage-associated PrPC is not required to mediate the anti-LPS/TLR4 activity of S-PrP. To confirm that POM2 blocks the anti-inflammatory activity of human plasma EVs by targeting EV-associated PrPC and not macrophage-associated PrPC, we studied the activity of POM2 in experiments with two distinct proteins that block LPS/TLR4 responses in macrophages by engaging the NMDA-R/LRP1 receptor system: α2M and EI-tPA. Fig. 8C shows that 10 μg/mL POM2 had no effect on the ability of 12 nM EI-tPA or 10 nM α2M to inhibit expression of the mRNAs encoding TNFα or IL6 in LPS-treated BMDMs. Similarly, POM2 did not interfere with the ability of 10 nM α2M to inhibit LPS-induced IκBα phosphorylation (Fig. 8D). These results support our conclusion that POM2 inhibits the anti-LPS/TLR4 activity of human plasma EVs by targeting EV-associated PrPC and not macrophage PrPC.
To confirm the role EV-associated PrPC as the principal factor responsible for the anti-LPS/TLR4 activity of human plasma EVs and to determine the state of PrPC in plasma EVs, we treated human plasma EVs with PI-PLC, which cleaves and releases GPI-anchored proteins from plasma membranes (37, 38). Following PI-PLC treatment, EVs were washed by UC at 100,000 × g to separate the EVs from solution-phase components. Control EVs were not PI-PLC-treated but still subjected to the same washing protocol. Fig. 8E shows that, unlike control EVs, PI-PLC-treated EVs were totally inactive as inhibitors of LPS-induced IκBα phosphorylation in BMDMs. In control experiments, PI-PLC-treated EVs did not independently induce IκBα phosphorylation.
Next, human plasma EVs were treated with PI-PLC or vehicle and subjected to washing by UC. Instead of studying the EV-containing pellet, we studied the supernatants. Supernatants that were harvested from control EVs, which were not PI-PLC-treated, were inactive at inhibiting LPS-induced IκBα phosphorylation (Fig. 8F). By contrast, supernatants that were harvested from PI-PLC-treated EVs were active, blocking LPS-induced IκBα phosphorylation and the accompanying decrease in abundance of IκBα. Thus, PI-PLC treatment dissociates the anti-LPS/TLR4 activity from human plasma EVs without destroying this activity. Collectively, the POM2 and PI-PLC experiments strongly suggest that EV-associated PrPC is responsible for the anti-LPS/TLR4 activity of human plasma EVs and that the bioactive form of PrPC in EVs is GPI-anchored.
SFKs are required for the anti-LPS/TLR4 activity of human plasma EVs
In neuron-like cells, SFKs are important upstream mediators of cell-signaling responses elicited by S-PrP and other ligands that engage the NMDA-R/LRP1 receptor system (26, 54). SFKs also have been implicated in anti-inflammatory responses mediated by membrane-anchored PrPC (23). Thus, we performed studies to determine whether SFKs function in the pathway by which human plasma EV-associated PrPC regulates macrophage physiology.
Fig. 9A shows that human plasma EVs rapidly activated SFKs in BMDMs. Phospho-SFK was observed within 5 min of adding 4 μg/mL EVs. Fig. 9B shows that ERK1/2 was activated in BMDMs treated with human plasma EVs, in the absence of LPS, for 1 h. This response was entirely blocked by MK801, as anticipated, and by the SFK inhibitor, PP2 (1 μM). Similarly, PP2 decreased the ability of human plasma EVs to inhibit IκBα phosphorylation in BMDMs treated with LPS (Fig. 9C). PP2 had no effect on IκBα phosphorylation when added to BMDM cultures alone or together with LPS, in the absence of plasma EVs.
FIGURE 9.
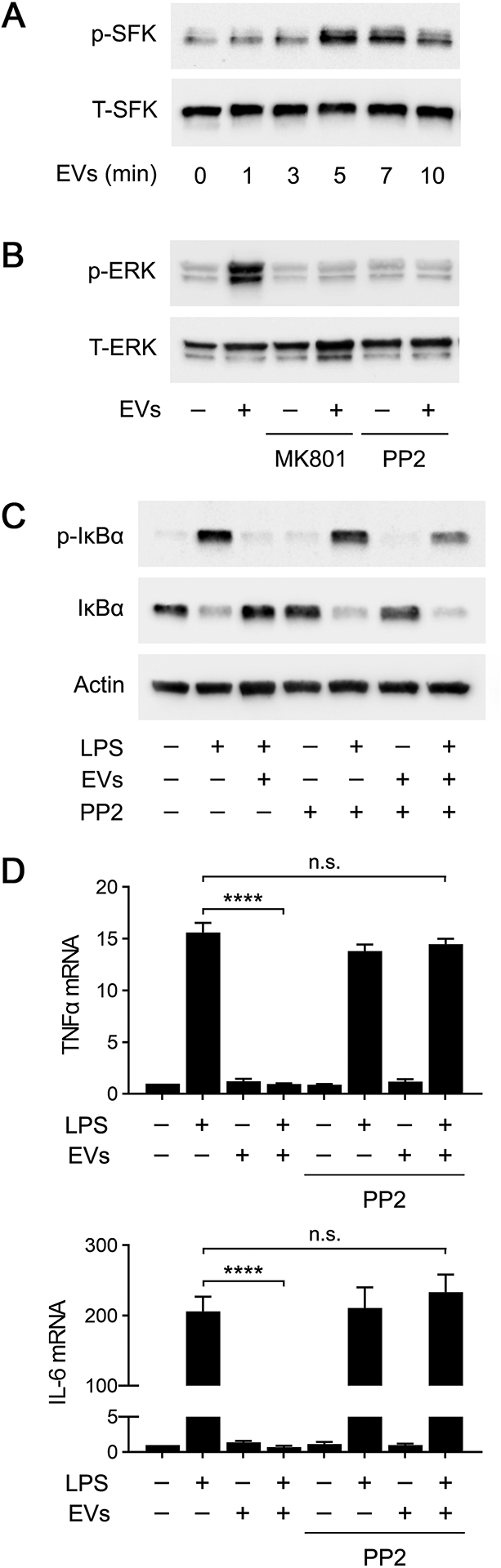
Inhibiting SFKs blocks the anti-LPS/TLR4 activity of human plasma EVs. (A) BMDMs from wild-type mice were treated with human plasma EVs (4 μg/mL), for the indicated times. Phospho-SFKs and total SFKs were determined by immunoblot analysis. (B) BMDMs were treated with human plasma EVs (4 μg/mL), MK801 (1.0 μM), PP2 (1.0 μM), EVs plus MK801, or EVs plus PP2 for 1 h, as indicated. Phospho-ERK1/2 and total ERK1/2 were determined by immunoblot analysis. (C) BMDMs were treated with LPS (0.1 μg/mL), EVs, and/or PP2 (1.0 μM) for 1 h, as indicated. Immunoblot analysis was performed to detect phospho-IκBα and total IκBα. (D) BMDMs were treated for 6 h with LPS, EVs, and/or PP2, as indicated. RT-qPCR was performed to determine mRNA levels for TNFα and IL-6. RT-qPCR data are expressed as the mean ± SEM (n = 3; one-way ANOVA; ****P <0.0001).
In cytokine expression studies, PP2 blocked the ability of plasma EVs to inhibit expression of TNFα and IL-6 in LPS-treated BMDMs (Fig. 9D). PP2 did not independently regulate cytokine expression or affect the ability of LPS to induce cytokine expression in the absence of EVs. These results implicate SFKs as essential upstream mediators of the response to EV-associated PrPC, which neutralizes the activity of LPS in macrophages.
Discussion
This study defines a novel pathway by which PrPC may express anti-inflammatory activity when it is released from cells as a soluble derivative or as an EV-associated protein. The NMDA-R and LRP1 are central components of this pathway. These receptors were previously shown to be responsible for the anti-inflammatory activity of tPA in macrophages (30, 31, 33). We considered the NMDA-R and LRP1 as candidates for mediating the response to S-PrP in macrophages because of their known role in S-PrP-initiated cell-signaling in PC12 cells and Schwann cells (26). The activity of the NMDA-R and LRP1, as receptors for EV-associated PrPC, was unanticipated because EV-associated proteins are not previously described as ligands for LRP1 or the NMDA-R. Inflammatory cells in addition to macrophages express the NMDA-R and LRP1 (55). Thus, the NMDA-R/LRP1 receptor system may mediate the anti-inflammatory activity of shed and EV-associated PrPC derivatives, in vivo, in target cells in addition to macrophages.
S-PrP blocked inflammatory cytokine mRNA expression in macrophages in response to ligands that activate diverse TLRs and also inhibited TLR-induced IκBα phosphorylation. S-PrP inhibited the toxicity of LPS in vivo. Furthermore, S-PrP directly activated ERK1/2 in cultured macrophages, in the absence of TLR agonists. These S-PrP activities were similar to those demonstrated previously for EI-tPA, which also engages the NMDA-R/LRP1 receptor complex (31, 33); however, there are important differences. First, S-PrP neutralized the response of macrophages to agonists that activate NOD1 and NOD2, whereas EI-tPA amplified these responses (33). Furthermore, EI-tPA was ineffective at neutralizing the effects of LPS on quiescent pMacs (33), whereas S-PrP was effective, suggesting that S-PrP may require a lower cell-surface abundance of NMDA-R in target cells. We conclude that S-PrP is capable of targeting an expanded continuum of PRRs compared with previously studied anti-inflammatory proteins that engage the NMDA-R/LRP1 system. Understanding the unique qualities of S-PrP, which allow it to function as an inhibitor of NOD1 and NOD2, in addition to TLRs, is an important future goal. The activity of S-PrP as an inhibitor of NOD2 is particularly intriguing because mutations in NOD2 have been associated with susceptibility to Crohn’s Disease (56, 57).
Results obtained with BMDMs harvested from Prnp−/− mice demonstrated that the NMDA-R/LRP1 complex does not require membrane-anchored PrPC as a co-receptor to trigger cell-signaling in response to S-PrP. Experiments with the LRP1 antagonist, RAP, and with LRP1-deficient BMDMs from mLrp1−/− mice suggested that LRP1 is important but not essential for mediating the anti-inflammatory activity of S-PrP. In the absence of LRP1, S-PrP was still active as an inhibitor of LPS-induced cytokine expression and IκBα phosphorylation; however, higher concentrations of S-PrP were required. These results are consistent with a model in which LRP1 sequesters S-PrP and delivers it to the NMDA-R to trigger cell-signaling. In the absence of LRP1, other macrophage cell surface macromolecules may sequester S-PrP for delivery to the NMDA-R. Alternatively, S-PrP may bind directly to the NMDA-R, albeit with lower avidity in the absence of LRP1, as has previously been described for tPA (29, 58, 59).
S-PrP is a recombinant protein; however, its structure is similar to that of a PrPC derivative released from cells by ADAM10 (7). PrPC serves as a substrate for other ADAMs, which generate solubilized PrPC products differing in size and structure (7, 8). Our results with POM2 suggest that the critical motif that interacts with the NMDA-R/LRP1 system to mediate anti-inflammatory cell-signaling is localized in the N-terminal unstructured region of PrPC (26, 40). Other soluble PrPC products that retain this epitope also may be active in regulating PPR activity. In a general sense, our studies demonstrate that solubilized PrPC derivatives may be responsible for, or at least contribute to previously described anti-inflammatory activities of PrPC (3, 20–22). Importantly, in many inflammatory cells, activators of innate immunity increase ADAM activity (60). Thus, release of soluble PrPC derivatives may represent a feedback pathway by which innate immunity pathways are controlled.
By examining human blood bank plasma, we confirmed work by others (13, 14) demonstrating that EVs from blood carry PrPC. EVs are known to function in cell-cell communication, mainly through their ability to transfer biologically active cargo, including mRNAs, microRNAs, and proteins from a cell of origin to a target cell (10–12). Our results demonstrate that human plasma EVs may regulate innate immunity; however, the identified mechanism does not involve cargo transfer. Instead, the key event is the ability of a GPI-anchored membrane protein in EVs to function as a ligand and engage the NMDA-R/LRP1 receptor system in macrophages, which has known anti-inflammatory activity (30–33).
By performing experiments with PI-PLC, we demonstrated that the immune regulatory factor in EVs is GPI-anchored. PI-PLC completely eliminated the LPS-regulatory activity of human plasma EVs and this activity was recovered in the supernatant, as would be anticipated for a GPI-anchored protein that is known to be active in soluble form. We determined that the GPI-anchored protein in human plasma EVs is PrPC by neutralizing its activity with POM2. It is noteworthy that, from a battery of POM-specific monoclonal antibodies with defined epitopes (40), POM2 was the only antibody that blocked the activity of both S-PrP and EV-associated PrPC. This result supports a model in which S-PrP and membrane-anchored PrPC in EVs regulate PRRs by the equivalent mechanism.
Our studies implicating the macrophage NMDA-R/LRP1 receptor complex as responsible for the anti-LPS/TLR4 activity of EV-associated PrPC are novel because, although LRP1 functions as a receptor for numerous soluble ligands (27, 61, 62), LRP1 is not recognized as a receptor for EV-associated membrane proteins. The ability of EV-associated PrPC to activate NMDA-R/LRP1-dependent cell-signaling in macrophages represents an entirely novel mechanism by which EVs may regulate immunity. The ability of LRP1 to function as an EV receptor merits further consideration. Because our results suggest that the NMDA-R/LRP1 receptor complex triggers cell-signaling in response to a membrane-anchored EV protein, it will be important to test whether the NMDA-R/LRP1 receptor system triggers cell-signaling in response to plasma membrane proteins on neighboring cells.
Our results demonstrating that PI-PLC completely inactivates human plasma EVs as regulators of LPS suggest that PrPC alone may be responsible for this EV activity. Although PI-PLC targets other GPI-anchored proteins, the conclusion that PrPC is exclusively responsible for the anti-LPS/TLR4 activity of plasma EVs was supported by our results with POM2. Given the known heterogeneity in plasma EVs (10–12, 63–65), it is likely that the activities demonstrated here reflect a sub-population of human plasma EVs. It will be important to determine whether the total abundance of plasma EV-associated PrPC varies in diseases with chronic inflammatory components. Furthermore, more work will be necessary to determine whether EV-associated PrPC is active in regulating innate immunity in vivo.
The ability of LRP1 to function as a receptor for EV-associated PrPC is not entirely unanticipated because LRP1 is known to function in phagocytosis of large particles (66) and in efferocytosis, as a receptor for apoptotic cells (67, 68). Based on these prior studies, it is reasonable to hypothesize that LRP1 may function in EV targeting, binding, and cargo internalization, specifically for EVs with abundant PrPC. If this is correct, then the anti-inflammatory activity of EV PrPC may reflect a more complicated set of mechanisms, beyond the ability to trigger NMDA-R/LRP1 receptor system-dependent cell-signaling.
SFKs were rapidly activated in macrophages treated with human plasma EVs. Furthermore, the effects of human plasma EVs on ERK1/2 phosphorylation, LPS-induced IκBα phosphorylation, and LPS-induced pro-inflammatory cytokine expression, in macrophages, were inhibited by PP2, suggesting an essential role for SFKs. This is the first study implicating SFKs as critical upstream activators of the cell-signaling pathway triggered by any anti-inflammatory ligand for the NMDA-R/LRP1 receptor system in macrophages. SFKs have been implicated NMDA-R/LRP1 cell-signaling in neurons and neuron-like cells (26, 54). Similarly, SFKs are activated downstream of the NMDA-R in neurons treated with NMDA (69). Thus, although the outcome of NMDA-R-activated cell-signaling may depend on the cell type in which the NMDA-R is expressed, the upstream cell-signaling pathway may be at least partially conserved.
In conclusion, we have identified two states of PrPC that are active in the regulation of innate immunity, shed PrPC and PrPC that is incorporated into EVs as a GPI-anchored protein. Both forms of PrPC may contribute to the known anti-inflammatory activity of this gene product. The NMDA-R/LRP1 receptor system in macrophages serves as a receptor for both S-PrP and EV-associated PrPC. These receptor-ligand interactions constitute a novel innate immunity regulatory system in macrophages.
Supplementary Material
KEY POINTS.
Shed PrPC derivatives and PrPC in extracellular vesicles regulate innate immunity
The macrophage NMDA-R/LRP1 complex mediates the activity of PrPC released by cells
NMDA-R/LRP1 complex functions as an extracellular vesicle receptor in target cells
Acknowledgments
The authors acknowledge Mr. Cory B. Gunner for technical assistance.
This work was supported by NIH grant R01 HL136395 (to S.L.G.)
References
- 1.Taylor DR, and Hooper NM. 2006. The prion protein and lipid rafts. Mol. Membr. Biol 23: 89–99. [DOI] [PubMed] [Google Scholar]
- 2.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, and Prusiner SB. 1993. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U. S. A 90: 10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakkebø MK, Mouillet-Richard S, Espenes A, Goldmann W, Tatzelt J, and Tranulis MA. 2015. The cellular prion protein: A player in immunological quiescence. Front. Immunol 6: 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almeida C. J. G. de, Chiarini LB, da Silva JP, e Silva PMR, Martins MA, Linden R, de Almeida CJG, Chiarini LB, da Silva JP, e Silva PMR, Martins MA, and Linden R. 2005. The cellular prion protein modulates phagocytosis and inflammatory response. J. Leukoc. Biol 77: 238–246. [DOI] [PubMed] [Google Scholar]
- 5.Dürig J, Giese A, Schulz-Schaeffer W, Rosenthal C, Schmücker U, Bieschke J, Dührsen U, and Kretzschmar HA. 2000. Differential constitutive and activation-dependent expression of prion protein in human peripheral blood leucocytes. Br. J. Haematol 108: 488–495. [DOI] [PubMed] [Google Scholar]
- 6.James Haddon D, Hughes MR, Antignano F, Westaway D, Cashman NR, and McNagny KM. 2009. Prion protein expression and release by mast cells after activation. J. Infect. Dis 200: 827–831. [DOI] [PubMed] [Google Scholar]
- 7.Taylor DR, Parkin ET, Cocklin SL, Ault JR, Aschcroft AE, Turner AJ, and Hooper NM. 2009. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J. Biol. Chem 284: 22590–22600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald AJ, Dibble JP, Evans EGB, and Millhauser GL. 2014. A new paradigm for enzymatic control of α-cleavage and β-cleavage of the prion protein. J. Biol. Chem 289: 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang J, Wang W, Sorensen D, Medina S, Ilchenko S, Kiselar J, Surewicz WK, Booth SA, and Kong Q. 2012. Cellular prion protein regulates its own α-cleavage through ADAM8 in skeletal muscle. J. Biol. Chem 287: 16510–16520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Théry C, Zitvogel L, and Amigorena S. 2002. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol 2: 569–579. [DOI] [PubMed] [Google Scholar]
- 11.Raposo G, and Stoorvogel W. 2013. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol 200: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maas SLN, Breakefield XO, and Weaver AM. 2017. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 27: 172–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ritchie AJ, Crawford DM, Ferguson DJP, Burthem J, and Roberts DJ. 2013. Normal prion protein is expressed on exosomes isolated from human plasma. Br. J. Haematol 163: 678–680. [DOI] [PubMed] [Google Scholar]
- 14.Robertson C, Booth SA, Beniac DR, Coulthart MB, Booth TF, and McNicol A. 2006. Cellular prion protein is released on exosomes from activated platelets. Blood 107: 3907–3911. [DOI] [PubMed] [Google Scholar]
- 15.Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, and Raposo G. 2004. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U. S. A 101: 9683–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vella LJ, Greenwood DLV, Cappai R, Scheerlinck JPY, and Hill AF. 2008. Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet. Immunol. Immunopathol 124: 385–393. [DOI] [PubMed] [Google Scholar]
- 17.Falker C, Hartmann A, Guett I, Dohler F, Altmeppen H, Betzel C, Schubert R, Thurm D, Wegwitz F, Joshi P, Verderio C, Krasemann S, and Glatzel M. 2016. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J. Neurochem 137: 88–100. [DOI] [PubMed] [Google Scholar]
- 18.Hartmann A, Muth C, Dabrowski O, Krasemann S, and Glatzel M. 2017. Exosomes and the prion protein: More than one truth. Front. Neurosci 11: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abels ER, and Breakefield XO. 2016. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol 36: 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onodera T, Sakudo A, Tsubone H, and Itohara S. 2014. Review of studies that have used knockout mice to assess normal function of prion protein under immunological or pathophysiological stress. Microbiol. Immunol 58: 361–374. [DOI] [PubMed] [Google Scholar]
- 21.Tsutsui S, Hahn JN, Johnson TA, Ali Z, and Jirik FR. 2008. Absence of the cellular prion protein exacerbates and prolongs neuroinflammation in experimental autoimmune encephalomyelitis. Am. J. Pathol 173: 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gourdain P, Ballerini C, Nicot AB, and Carnaud C. 2012. Exacerbation of experimental autoimmune encephalomyelitis in prion protein (PrPc)-null mice: Evidence for a critical role of the central nervous system. J. Neuroinflammation 9: 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chida J, Hara H, Uchiyama K, Takahashi E, Miyata H, Kosako H, Tomioka Y, Ito T, Horiuchi H, Matsuda H, Kido H, and Sakaguchi S. 2020. Prion protein signaling induces M2 macrophage polarization and protects from lethal influenza infection in mice. PLOS Pathog. 16: e1008823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Zhao D, Liu C, Ding T, Yang L, Yin X, and Zhou X. 2015. Prion Protein Participates in the Protection of Mice from Lipopolysaccharide Infection by Regulating the Inflammatory Process. J. Mol. Neurosci 55: 279–287. [DOI] [PubMed] [Google Scholar]
- 25.Martin GR, Keenan CM, Sharkey KA, and Jirik FR. 2011. Endogenous prion protein attenuates experimentally induced colitis. Am. J. Pathol 179: 2290–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mantuano E, Azmoon P, Banki MA, Lam MS, Sigurdson CJ, and Gonias SL. 2020. A soluble derivative of PrPC activates cell-signaling and regulates cell physiology through LRP1 and the NMDA receptor. J. Biol. Chem 295: 14178–14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strickland DK, Gonias SL, and Argraves WS. 2002. Diverse roles for the LDL receptor family. Trends Endocrinol. Metab 13: 66–74. [DOI] [PubMed] [Google Scholar]
- 28.Martin AM, Kuhlmann C, Trossbach S, Jaeger S, Waldron E, Roebroek A, Luhmann HJ, Laatsch A, Weggen S, Lessmann V, and Pietrzik CU. 2008. The functional role of the second NPXY motif of the LRP1 β-chain in tissue-type plasminogen activator-mediated activation of N-methyl-D-aspartate receptors. J. Biol. Chem 283: 12004–12013. [DOI] [PubMed] [Google Scholar]
- 29.Mantuano E, Lam MS, and Gonias SL. 2013. LRP1 assembles unique co-receptor systems to initiate cell signaling in response to tissue-type plasminogen activator and myelin-associated glycoprotein. J. Biol. Chem 288: 34009–34018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mantuano E, Brifault C, Lam MS, Azmoon P, Gilder AS, and Gonias SL. 2016. LDL receptor-related protein-1 regulates NFκB and microRNA-155 in macrophages to control the inflammatory response. Proc. Natl. Acad. Sci 113: 1369–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mantuano E, Azmoon P, Brifault C, Banki MA, Gilder AS, Campana WM, and Gonias SL. 2017. Tissue-type Plasminogen Activator Regulates Macrophage Activation and Innate Immunity. Blood 130: 1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zalfa C, Azmoon P, Mantuano E, and Gonias SL. 2019. Tissue-type plasminogen activator neutralizes LPS but not protease-activated receptor-mediated inflammatory responses to plasmin. J. Leukoc. Biol 105: 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das L, Azmoon P, Banki MA, Mantuano E, and Gonias SL. 2019. Tissue-type plasminogen activator selectively inhibits multiple toll-like receptors in CSF-1differentiated macrophages. PLoS One 14: e0224738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parkyn CJ, Vermeulen EGM, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A, and Morris RJ. 2008. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell Sci 121: 773–783. [DOI] [PubMed] [Google Scholar]
- 35.Mattei V, Manganelli V, Martellucci S, Capozzi A, Mantuano E, Longo A, Ferri A, Garofalo T, Sorice M, and Misasi R. 2020. A multimolecular signaling complex including PrPC and LRP1 is strictly dependent on lipid rafts and is essential for the function of tissue plasminogen activator. J. Neurochem 152: 468–481. [DOI] [PubMed] [Google Scholar]
- 36.Ellis V, Daniels M, Misra R, and Brown DR. 2002. Plasminogen activation is stimulated by prion protein and regulated in a copper-dependent manner. Biochemistry 41: 6891–6896. [DOI] [PubMed] [Google Scholar]
- 37.Hirose S, Knez JJ, and Edward Medof M. 1995. Mammalian Glycosylphosphatidylinositol-Anchored Proteins and Intracellular Precursors. Methods Enzymol. 250: 582–614. [DOI] [PubMed] [Google Scholar]
- 38.Griffith OH, and Ryan M. 1999. Bacterial phosphatidylinositol-specific phospholipase C: Structure, function, and interaction with lipids. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1441: 237–254. [DOI] [PubMed] [Google Scholar]
- 39.Imber MJ, and V Pizzo S. 1981. Clearance and binding of two electrophoretic “fast” forms of human alpha 2-macroglobulin. J Biol Chem 256: 8134–8139. [PubMed] [Google Scholar]
- 40.Polymenidou M, Moos R, Scott M, Sigurdson C, Shi Y, Yajima B, Hafner-Bratkovič I, Jerala R, Hornemann S, Wuthrich K, Bellon A, Vey M, Garen G, James MNG, Kav N, and Aguzzi A. 2008. The POM Monoclonals: A Comprehensive Set of Antibodies to Non-Overlapping Prion Protein Epitopes. PLoS One 3: e3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Staudt ND, Jo M, Hu J, Bristow JM, Pizzo DP, Gaultier A, VandenBerg SR, and Gonias SL. 2013. Myeloid cell receptor LRP1/CD91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res. 73: 3902–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Goncalves R, and Mosser DM. 2008. The isolation and characterization of murine macrophages. Curr Protoc Immunol Chapter 14: Unit 14.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shrum B, V Anantha R, Xu SX, Donnelly M, Haeryfar SMM, McCormick JK, and Mele T. 2014. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res. Notes 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tak PP, and Firestein GS. 2001. NF-κB: A key role in inflammatory diseases. J. Clin. Invest 107: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwandner R, Dziarski R, Wesche H, Rothe M, and Kirschning CJ. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J. Biol. Chem 274: 17406–17409. [DOI] [PubMed] [Google Scholar]
- 46.Schneewind O, and Missiakas D. 2014. Lipoteichoic acids, phosphate-containing polymers in the envelope of gram-positive bacteria. J Bacteriol 196: 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, Wader T, Tluk S, Liu M, Davis HL, and Krieg AM. 2004. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol 34: 251–262. [DOI] [PubMed] [Google Scholar]
- 48.Ballas ZK, Krieg AM, Warren T, Rasmussen W, Davis HL, Waldschmidt M, and Weiner GJ. 2001. Divergent Therapeutic and Immunologic Effects of Oligodeoxynucleotides with Distinct CpG Motifs. J. Immunol 167: 4878–4886. [DOI] [PubMed] [Google Scholar]
- 49.Strober W, Murray PJ, Kitani A, and Watanabe T. 2006. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 6: 9–20. [DOI] [PubMed] [Google Scholar]
- 50.Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, and Núñez G. 2008. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity 28: 246–257. [DOI] [PubMed] [Google Scholar]
- 51.Williams SE, Ashcom JD, Argraves WS, and Strickland DK. 1992. A novel mechanism for controlling the activity of alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein. Multiple regulatory sites for 39-kDa receptor-associated protein. J. Biol. Chem 267: 9035–9040. [PubMed] [Google Scholar]
- 52.Nuvolone M, Hermann M, Sorce S, Russo G, Tiberi C, Schwarz P, Minikel E, Sanoudou D, Pelczar P, and Aguzzi A. 2016. Strictly co-isogenic C57BL/6J-Prnp−/− mice: A rigorous resource for prion science. J. Exp. Med 213: 313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das S, The Extracellular RNA Communication Consortium, Ansel KM, Bitzer M, Breakefield XO, Charest A, Galas DJ, Gerstein MB, Gupta M, Milosavljevic A, McManus MT, Patel T, Raffai RL, Rozowsky J, Roth ME, Saugstad JA, Van Keuren-Jensen K, A. M. Weaver, and L. C. Laurent. 2019. The Extracellular RNA Communication Consortium: Establishing Foundational Knowledge and Technologies for Extracellular RNA Research. Cell 177: 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi Y, Mantuano E, Inoue G, Campana WM, and Gonias SL. 2009. Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci Signal 2: ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Das L, Banki MA, Azmoon P, Pizzo D, and Gonias SL. 2021. Enzymatically Inactive Tissue-Type Plasminogen Activator Reverses Disease Progression in the Dextran Sulfate Sodium Mouse Model of Inflammatory Bowel Disease. Am. J. Pathol 191: 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nuñez G, and Cho JH. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411: 603–606. [DOI] [PubMed] [Google Scholar]
- 57.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, and Thomas G. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411: 599–603. [DOI] [PubMed] [Google Scholar]
- 58.Lesept F, Chevilley A, Jezequel J, Ladépêche L, Macrez R, Aimable M, Lenoir S, Bertrand T, Rubrecht L, Galea P, Lebouvier L, Petersen KU, Hommet Y, Maubert E, Ali C, Groc L, and Vivien D. 2016. Tissue-type plasminogen activator controls neuronal death by raising surface dynamics of extrasynaptic NMDA receptors. Cell Death Dis. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mantuano E, Lam MS, Shibayama M, Campana WM, and Gonias SL. 2015. The NMDA receptor functions independently and as an LRP1 co-receptor to promote Schwann cell survival and migration. J. Cell Sci 128: 3478–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lambrecht BN, Vanderkerken M, and Hammad H. 2018. The emerging role of ADAM metalloproteinases in immunity. Nat. Rev. Immunol 18: 745–758. [DOI] [PubMed] [Google Scholar]
- 61.Gonias SL, and Campana WM. 2014. LDL receptor-related protein-1: A regulator of inflammation in atherosclerosis, cancer, and injury to the nervous system. Am. J. Pathol 184: 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fernandez-Castaneda A, Arandjelovic S, Stiles TL, Schlobach RK, Mowen KA, Gonias SL, and Gaultier A. 2013. Identification of the low density lipoprotein (LDL) receptor-related protein-1 interactome in central nervous system myelin suggests a role in the clearance of necrotic cell debris. J. Biol. Chem 288: 4538–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnstone RM 2006. Exosomes biological significance: A concise review. Blood Cells, Mol. Dis 36: 315–321. [DOI] [PubMed] [Google Scholar]
- 64.Jan A, Rahman S, Khan S, Tasduq S, and Choi I. 2019. Biology, Pathophysiological Role, and Clinical Implications of Exosomes: A Critical Appraisal. Cells 8: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu L, Wu L-F, and Deng F-Y. 2019. Exosome: An Emerging Source of Biomarkers for Human Diseases. Curr. Mol. Med 19: 387–394. [DOI] [PubMed] [Google Scholar]
- 66.Gaultier A, Wu X, Le Moan N, Takimoto S, Mukandala G, Akassoglou K, Campana WM, and Gonias SL. 2009. Low-density lipoprotein receptor-related protein 1 is an essential receptor for myelin phagocytosis. J. Cell Sci 122: 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ogden CA, DeCathelineau A, Hoffmann PR, Bratton D, Fadok B, Ghebrehiwet VA, and Henson PM. 2001. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med 194: 781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, Walport MJ, Fisher JH, Henson PM, and Greene KE. 2002. Role of Surfactant Proteins A, D, and C1q in the Clearance of Apoptotic Cells In Vivo and In Vitro: Calreticulin and CD91 as a Common Collectin Receptor Complex. J. Immunol 169: 3978–3986. [DOI] [PubMed] [Google Scholar]
- 69.Head BP, Patel HH, Tsutsumi YM, Hu Y, Mejia T, Mora RC, Insel PA, Roth DM, Drummond JC, and Patel PM. 2008. Caveolin-1 expression is essential for N-methyl-D-aspartate receptor-mediated Src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 22: 828–840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.