
Figure 3. Functional effects of patient mutations in SCN1A, SCN2A and SCN3A.
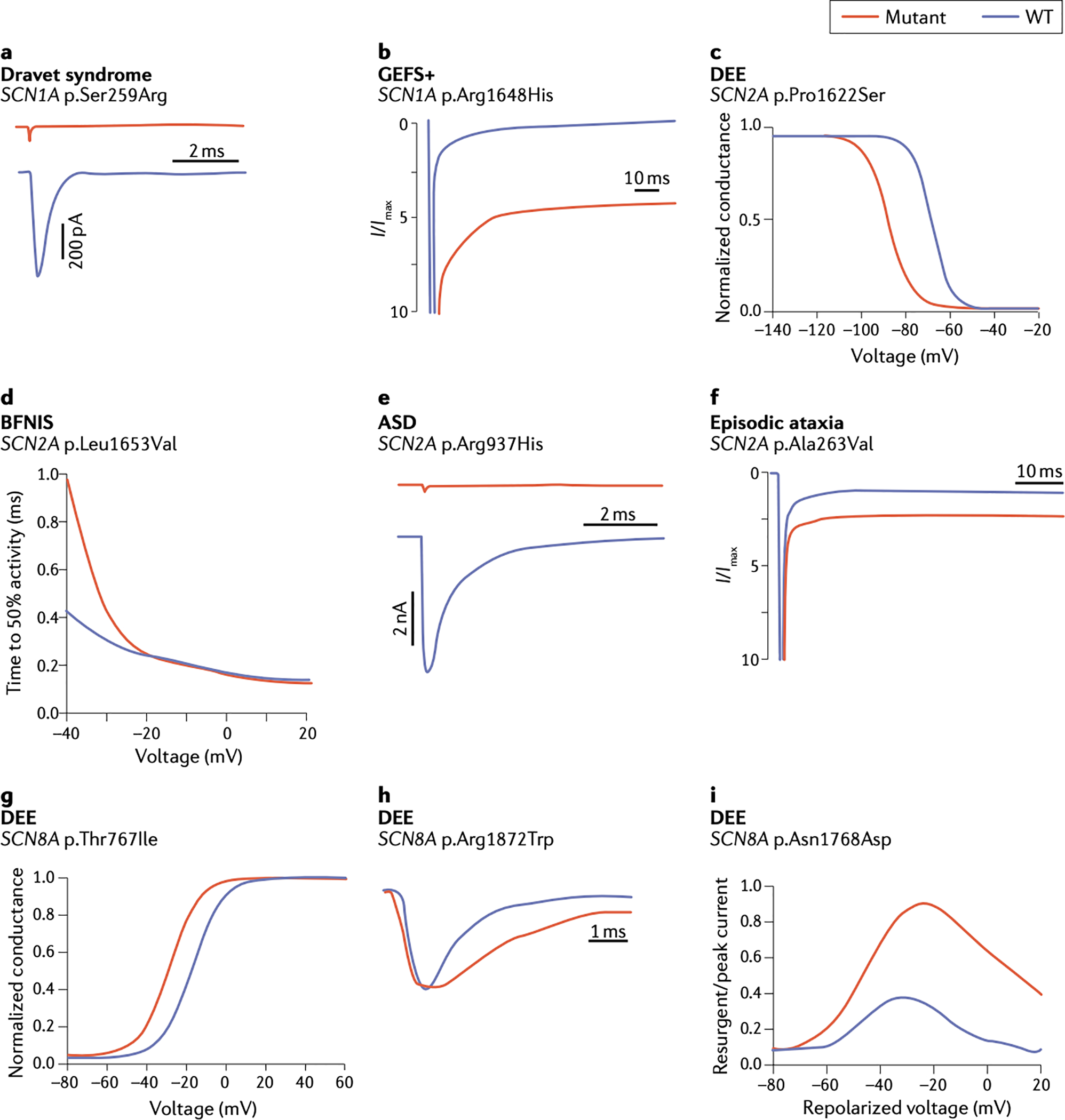
Representative examples adapted from the indicated publications, which contain experimental details. A. The Dravet Syndrome mutation p.S259R in SCN1A causes complete loss of channel function 83. B. The inherited variant p.R1648H in SCN1A in a family with GEFS+ causes increased persistent current 99. C. p.P1622S in SCN2A in a patient with late-onset DEE causes a hyperpolarizing shift in the voltage dependence of inactivation 104. D. p.L1653V in SCN2A in a family with benign familial neonatal-infantile seizures (BFNIS) causes rapid channel activation 114. E. p.R937H in SCN2A in a patient with autism spectrum disorder (ASD) causes loss of channel function 116. F. p.A263V in SCN2A in a patient with episodic ataxia causes increased persistent current 120. G. The mutation p.T767I in SCN8A in a patient with DEE causes premature channel activation 122. H. De novo mutation p.R1872W in SCN8A in a patient with DEE causes delayed channel inactivation 163. I. De novo mutation p.N1768D in SCN8A in a patient with DEE causes elevated resurgent current 164.