
Figure 3. Hybrid genome and transcriptome status results in distinct splicing patterns and HPV oncogene expression levels.
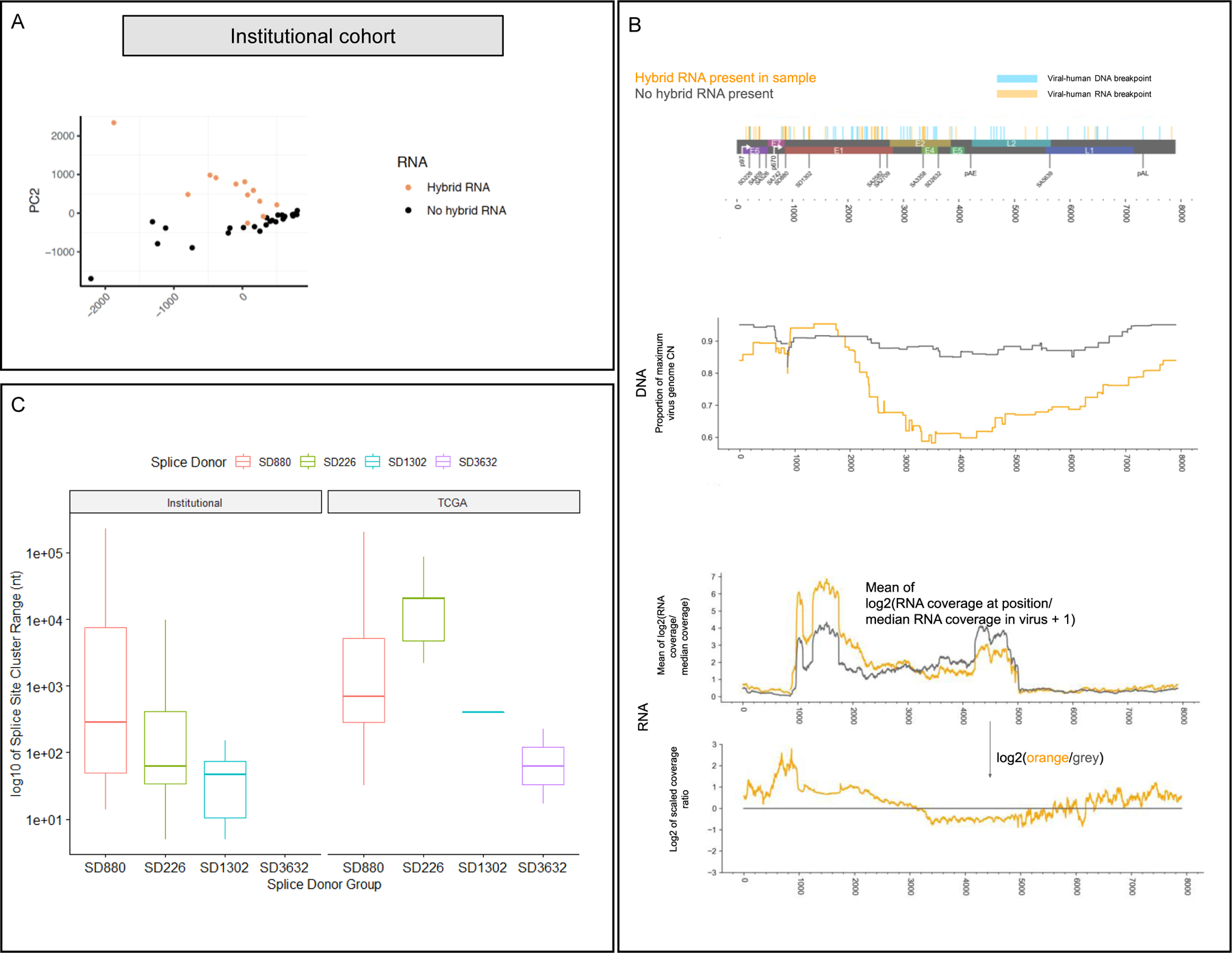
A) Principal component analysis of splice junction expression in individual tumors with annotation of tumors by presence or absence of human-viral hybrid transcripts. PCA of the tumor cohort supports the finding that hybrid transcriptome tumors (orange dots) exhibit distinct splicing patterns compared to non-hybrid transcriptome tumors (black dots).
B) Top: genome map of HPV16, indicating human-viral DNA breakpoints and RNA breakpoints. Middle (DNA): Mean proportion of the CN at each position over the maximum HPV16 CN for each sample with hybrid RNA transcripts (orange) and those without (grey), based on AA copy number estimates. We observed selective enrichment of viral genomic copy number in the E6/E7 region, and 5’ end of E1 in hybrid RNA tumors, while those without hybrid RNA showed much more uniform enrichment of viral copy number throughout the genome. Bottom (RNA): RNA-seq coverage for hybrid RNA tumors (orange) and those without hybrid RNA (grey). Coverage is rescaled by the median coverage across the virus, and the log2 value is shown. The lower plot shows the log2 ratio of scaled coverage between orange and grey from the upper plot, representing a log2 fold-change in mean scaled coverage. The highest peak overlaps SD880, indicating selectively increased transcription of that location in tumors with hybrid RNA. We also observed that the E4/E5 region, including the 5’ end of L2 were far less likely to have selective enrichment for genomic copy number in hybrid RNA tumors, and that there was decreased expression of those regions as compared to tumors without hybrid RNA.
C) Splice acceptor cluster quantification of hybrid transcripts for Institutional and TCGA cohorts stratified based on splice donor location. We found that the median splice acceptor range varied from 29 to 20,399 nucleotides wide across the TCGA and institutional cohort, and were narrowest for SD1302 (median 28.8 in TCGA; 406 in institutional) and widest for SD226 (20,399 in institutional cohort).