

Abstract
Background
Skeletal muscle regeneration is an adaptive response to injury that is crucial to the maintenance of muscle mass and function. A p21‐activated kinase 4 (PAK4) serine/threonine kinase is critical to the regulation of cytoskeletal changes, cell proliferation, and growth. However, PAK4's role in myoblast differentiation and regenerative myogenesis remains to be determined.
Methods
We used a mouse model of myotoxin (notexin)‐induced muscle regeneration. In vitro myogenesis was performed in the C2C12 myoblast cell line, primary myoblasts, and primary satellite cells. In vivo overexpression of PAK4 or kinase‐inactive mutant PAK4S474A was conducted in skeletal muscle to examine PAK4's kinase‐dependent effect on muscle regeneration. The regeneration process was evaluated by determining the number and size of multinucleated myofibres and expression patterns of myogenin and eMyHC. To explore whether PAK4 inhibition improves muscle regeneration, mice were injected intramuscularly with siRNA that targeted PAK4 or orally administered with a chemical inhibitor of PAK4.
Results
p21‐activated kinase 4 was highly expressed during the myoblast stage, but expression gradually and substantially decreased as myoblasts differentiated into myotubes. PAK4 overexpression, but not kinase‐inactive mutant PAK4S474A overexpression, significantly impeded myoblast fusion and MyHC‐positive myotube formation in C2C12 cells, primary myoblasts, and satellite cells (P < 0.01). Conversely, PAK4 silencing led to an 8.7% and a 20.3% increase in the number of multinucleated larger myotubes in C2C12 cells and primary myoblasts. Further, in vivo overexpression of PAK4 by adenovirus injection to mice prior to and after myotoxin‐induced injury led to a 52.6% decrease in the number of eMyHC‐positive myofibres on Day 5 in tibialis anterior muscles as compared with those injected with control adenoviruses (P < 0.01), while Ad‐PAK4S474A showed comparable muscle regeneration parameters. PAK4‐induced repression of muscle regeneration coincided with an increase in phosphatase and tensin homologue (PTEN) expression and a decrease in phosphoinositide 3‐kinase‐Akt signalling. In contrast, PAK4 silencing reduced PTEN expression in mice. Consistent with these findings, prodrug of PAK4 inhibitor CZh‐226 (30 mg/kg) orally administered to mice repressed PTEN expression and accelerated myotube formation. Subsequent mechanistic studies revealed that PAK4 directly phosphorylates PPARγ at S273 to increase its transcription activity, thereby up‐regulating PTEN expression. Importantly, an analysis of the Genotype‐Tissue Expression database showed a positive correlation between PAK4 and PTEN in human skeletal muscle tissues (P < 0.01).
Conclusions
p1‐activated kinase 4 is a new member of PPARγ kinase, and PAK4 inhibition may have a therapeutic role as an accelerant of muscle regeneration.
Keywords: PAK4, PPARγ, PTEN, Muscle regeneration, Myogenesis
Introduction
Skeletal muscle is the most abundant tissue in the human body, accounting for about 40% of total body mass. Adult skeletal muscle displays a remarkable regenerative capability, which is crucial to the maintenance of homeostasis. 1 Muscle regeneration is an adaptive response to traumatic injury and disease, and deregulation of this process may aggravate pathological situations, including muscular dystrophy and sarcopenia. 2 Regenerative myogenesis is a highly coordinated process that encompasses progenitor cell activation, proliferation, differentiation, and fusion. 3 These sequential events are dynamic, and a disturbance in any of these steps leads to regenerative failure. Despite the importance of this process, the underlying molecular mechanisms for skeletal myogenesis are still largely unknown.
Myogenesis must be tightly regulated at different levels by a variety of extracellular and intracellular signalling pathways. The phosphoinositide 3‐kinase (PI3K)–Akt pathway is considered a key mediator in the regulation of diverse cellular processes, including cell proliferation, the cell cycle, cell differentiation, and cell death, 4 which suggests a relationship to skeletal muscle regeneration. Specifically, the PI3K–Akt–mTOR pathway is considered indispensable to the regulation of various stages of regenerative myogenesis. 5 , 6 , 7 Phosphatase and tensin homologue (PTEN) is a lipid phosphatase that converts PtdIns(3,4,5)P3 to PtdIns(4,5)P2, thereby opposing the action of PI3K. Accordingly, deletion of PTEN activates the Akt–mTOR signalling pathway, which accelerates myoblast proliferation and differentiation. 8
p21‐activated kinase 4 (PAK4) is a member of the PAK family, which includes serine/threonine kinases. Based on sequence similarities and functional characteristics, PAK4 classified as a Group II PAK (PAK4, PAK5, and PAK6), which are distinct from Group I PAKs (PAK1, PAK2, and PAK3). 9 PAK family members have isotype‐specific functions through the phosphorylation of their own targets. These functions include the regulation of cellular processes such as cytoskeleton remodelling, cell cycle progression, cell polarity, and cell death 9 ; it must be said, however, that this list is not comprehensive, as PAK4's biological role has mostly been studied in of the context of cancer biology. PAK4 expression is up‐regulated in patients with various types of tumours, and its overexpression may contribute to cancer development and progression. 10 , 11 , 12 Despite the known close relationship between PAK4 and cell fate, the possibility of a role for PAK4 in myogenic differentiation and muscle regeneration has never been explored.
In this paper, we used genetic and pharmacological approaches to show for the first time that PAK4 is a negative regulator of skeletal muscle regeneration. Mechanistically, PAK4 directly phosphorylated peroxisome proliferator‐activated receptor (PPAR)‐γ, which leads to the PTEN induction that is responsible for the inhibition of the PI3K–Akt pathway. Notably, we observed positive correlations between PAK4 and its downstream targets, PPARγ and PTEN in human skeletal muscle tissues. Our findings provide a proof of concept that the inhibition of PAK4 expression and/or activity may offer therapeutic benefits as an accelerant for muscle regeneration.
Materials and methods
Animal studies
Six‐ to eight‐week‐old male C57BL/6J mice were purchased from Orient Bio (Seoul, Korea). The mice had free access to food and water and were maintained in a room with controlled humidity (50%) and temperature (22°C) on a 12 h light/dark cycle. To induce experimental muscle injury, 50 μL of notexin (NTX, 4 μg/mL in saline, Latoxan, Valence, France) was injected into their tibialis anterior (TA) muscles. Wild‐type (Ad‐PAK4) and dominant negative mutant PAK4 (Ad‐PAK4S474A) adenoviruses were prepared as described previously. 11 For siRNA knockdown in mice, PAK4 siRNAs were preincubated with Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA). Mice were injected in their TA muscles with adenoviruses (2 × 109 pfu) or siRNA (600 pmol) targeting PAK4 on Days −2, 2, 6, and 10 after NTX. To pharmacologically inhibit PAK4 in mice, prodrug of PAK4 inhibitor CZh‐226 (30 mg/kg) 13 dissolved in 5% DMSO + 5% Tween 80 + 90% PBS was administered orally once daily for 15 days in NTX model. Mice were sacrificed, and TA muscles were harvested for biochemical and histological analyses.
Histology
Tibialis anterior muscle tissues were immediately placed in 30% sucrose solution and embedded with liquid nitrogen‐cooled isopentane. For immunofluorescence staining of the regenerating myofibres, frozen sections (10 μm) were incubated overnight at 4°C with primary antibodies against laminin (Sigma‐Aldrich, St. Louis, MO, USA), eMyHC (DSHB, Iowa City, IA, USA), myogenin (Santa Cruz Biotechnology, Dallas, TX, USA), and desmin (Abcam, Cambridge, UK). After washing with PBS, secondary antibodies (Alexa Fluor 488‐conjugated goat anti‐rabbit IgG1 and Alexa Fluor 594‐conjugated goat anti‐mouse IgM, Thermo Fisher Scientific, Waltham, MA, USA) were incubated for 1 h at 37°C. Sections were counterstained with DAPI, and the mean cross‐sectional area (CSA) of each of the fibres was determined in five fields from each animal using iSolution DT 36 software (Carl Zeiss, Oberkochen, Germany). To determine the number of myonuclei, the number of DAPI‐positive nuclei within the lamina‐positive sarcolemma was counted. For immunohistochemical staining of the Ki67, frozen sections (10 μm) were incubated overnight at 4°C with primary antibody against Ki67 (Novus Biologicals, Centennial, CO, USA).
Chromatin immunoprecipitation
C2C12 cells were lysed and cross‐linked by incubating cells in 1% formaldehyde for 15 min at room temperature. Cross‐linking was stopped by 5 min of incubation with 125 mM glycine. Chromatin immunoprecipitation (ChIP) assay was performed using ChIP Enzymatic Chromatin IP Kits (Cell Signaling Technology, Beverly, MA, USA). Chromatins were immunoprecipitated overnight at 4°C with antibodies to PPARγ and non‐specific IgG (Cell Signaling Technology). Data were normalized to input. All primer sequences are listed in Supporting Information, Table S1.
In vitro kinase assay
Recombinant human PPARγ (0.5 μg, RP‐75683, Invitrogen) were incubated with active PAK4 (ab96405, Abcam) in assay buffer (50 mM Tris–HCl at pH 7.6, 10 mM MgCl2, 2 mM DTT, and 0.1 mM EDTA) containing 5 μCi of [γ‐32P]ATP and 50 μM cold ATP at 30°C for 30 min. Reaction mixtures were then subjected to SDS‐PAGE, and 32P‐labelled proteins were detected by autoradiography. For Coomassie blue staining, gel was stained in Coomassie protein stain buffer (ab119211, Abcam) for 1 h.
Site‐directed mutagenesis
Phosphorylation mutant of PPARγ was generated using a site‐directed mutagenesis kit (GeneAll, Seoul, Korea) by converting serine residue (S273) to alanine (codon change from TCA to GCA). Mutations were confirmed by DNA sequencing performed at Bioneer (Daejeon, Korea).
Statistical analysis
Data are expressed as the mean ± standard deviation. Statistical comparisons were made using one‐way analysis of variance followed by Fisher's post hoc analysis. The significance of differences between groups was determined using Student's unpaired t‐test. A P value less than 0.05 was considered significant.
Additional methods
Additional details concerning our methods are provided in the Methods section in the Supporting Information.
Results
p21‐activated kinase 4 is down‐regulated during myoblast differentiation
To test the hypothesis that PAK4 expression may be implicated in skeletal muscle regeneration, we first examined PAK4 protein levels in C2C12 cells during myoblast differentiation. PAK4 was highly expressed during the myoblast stage, but levels gradually and substantially decreased as myoblasts differentiated into myotubes (Figure S1A). The characteristic expression kinetics of the myogenic markers [an early induction of myogenin (Myog) and a subsequent increase of MyHC] were in sharp contrast with those of PAK4. qPCR analyses of PAK4 and MyHC mRNA showed the consistent patterns (Figure S1B). Similar findings were observed in primary myoblasts obtained from the hindlimb muscles of mice (Figure S1C and S1D). Reciprocal PAK4 and MyHC expression patterns in myoblasts and myotubes were further confirmed by immunofluorescence imaging analysis (Figure S1E and S1F), confirming the inverse relationship between PAK4 and myogenesis.
p21‐activated kinase 4 inhibits myogenic differentiation
We next investigated whether overexpressing PAK4 changed myogenic ability and, if so, whether its kinase activity was necessary to this process. C2C12 myoblasts were infected with Ad‐LacZ, wild‐type (Ad‐PAK4), or kinase‐dead mutant of PAK4 (Ad‐PAK4S474A) prior to differentiation. Forced PAK4 expression significantly impeded myoblast fusion and MyHC‐positive myotube formation compared with the Ad‐LacZ‐infected cells (Figure 1A). Western blotting and qPCR analyses of molecular markers verified impaired myogenesis in PAK4‐overexpressing cells (Figure 1B and 1C). PAK4's negative effects on myogenic differentiation were further confirmed in primary myoblasts (Figure 1D–1F). Infecting cells with Ad‐PAK4S474A, however, resulted in no changes to myoblast differentiation and fusion relative to the Ad‐LacZ group, suggesting that PAK4's kinase function is essential to the action.
Figure 1.
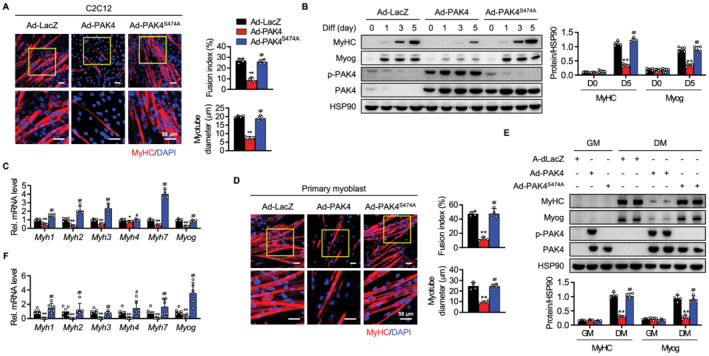
Suppression of myogenesis in PAK4‐overexpressing C2C12 cells and primary myoblasts. C2C12 myoblasts (A–C) and primary myoblasts (D–F) were infected with adenoviruses expressing β‐galactosidase (LacZ), wild‐type PAK4, or a PAK4 kinase‐inactive S474A mutant and differentiated for 5 days. (A, D) Cells were immunostained with anti‐MyHC antibody. Myogenic conversion was scored by quantifying the fusion index and myotube diameter. (B, E) Protein levels of myogenic markers and (C, F) mRNA levels of myosin heavy chains and myogenin were determined by western blotting and qPCR, respectively. Values are mean ± SD. * P < 0.05 and ** P < 0.01 vs. Ad‐LacZ; # P < 0.05 and ## P < 0.01 vs. Ad‐PAK4.
Having confirmed the negative role of PAK4 in myogenic differentiation, we next examined whether PAK4 silencing would have the opposite effect. An increase in the number of multinuclear cells with larger myotubes was observed in PAK4‐silenced C2C12 cells (Figure S2A). The gradual increases in protein and mRNA levels of Myog and MyHC were accelerated by PAK4 silencing during differentiation (Figure S2B and S2C). Associated changes to cell morphology and myogenic marker expression were also found in PAK4‐silenced primary myoblasts (Figure S2D–S2F).
Some recent reports have suggested that Group I PAKs, including PAK1 and PAK2, may induce muscular differentiation that protects against freeze injuries and cancer‐associated muscle atrophy. 14 , 15 We therefore questioned whether PAK4 silencing instigated any compensatory changes in PAK1 and PAK2. Our tests revealed no difference in the levels of these proteins in the PAK4‐silenced C2C12 cells (Figure S2B), indicating PAK4's bona fide inhibitory effect on muscle cell differentiation.
Satellite cell (SC) differentiation from a muscle stem cell into a skeletal myocyte is an initial step that eventually leads to myotube formation and muscle regeneration. 16 Thus, we further determined the inhibitory effect of PAK4 on myogenesis in SCs. Consistent with the results in the myoblasts, PAK4 was highly expressed in proliferating PAX7‐positive SCs, but markedly decreased in differentiating cells, as shown by western blotting and immunostaining (Figure 2A–2C). Moreover, infection of SCs with Ad‐PAK4, but not Ad‐PAK4S474A, distinctly reduced the expression of myogenic markers (Figure 2D and 2E). In sum, our results support the conclusion that PAK4 acts as a negative regulator of myogenesis.
Figure 2.
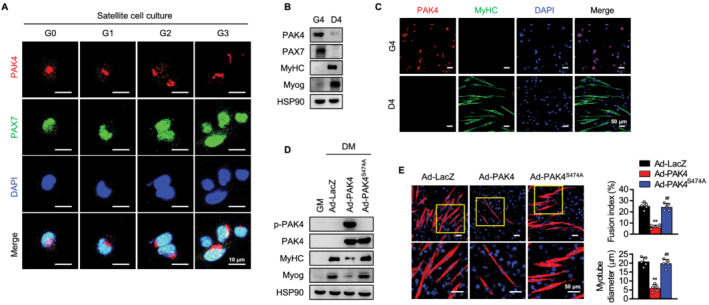
Suppression of myogenesis in PAK4‐overexpressing satellite cells (SCs). (A–C) Immunofluorescence staining and western blotting analysis for PAK4 in SCs cultured in growth medium for different time periods (from G0 to G4) followed by 4 day culture in differentiation medium (D4). (D, E) SCs were infected with PAK4 and mutant PAK4 adenoviruses and cultured in growth medium (GM) for 6 h and in differentiation medium (DM) for another 4 days. Protein levels of myogenic markers (MyHC and Myog) and myotube formation were determined. Values are mean ± SD. ** P < 0.01 vs. Ad‐LacZ; ## P < 0.01 vs. Ad‐PAK4.
p21‐activated kinase 4 overexpression delays while p21‐activated kinase 4 silencing accelerates skeletal muscle regeneration
The surprising finding of increased muscle cell differentiation by PAK4 silencing in vitro prompted us to explore the role of PAK4 in skeletal muscle regeneration in vivo. To induce skeletal muscle injury, mice were injected with NTX as depicted in Figure 3A, which is a well‐established model for studying skeletal muscle regenerative response. When we examined PAK4 expression levels in the damaged muscles, PAK4 had significantly increased between Day 1 (24 h after NTX exposure) and Day 3, but had then disappeared during the regenerative period, consistent with the reciprocal patterns with MyoG and eMyHC (Figure 3B).
Figure 3.
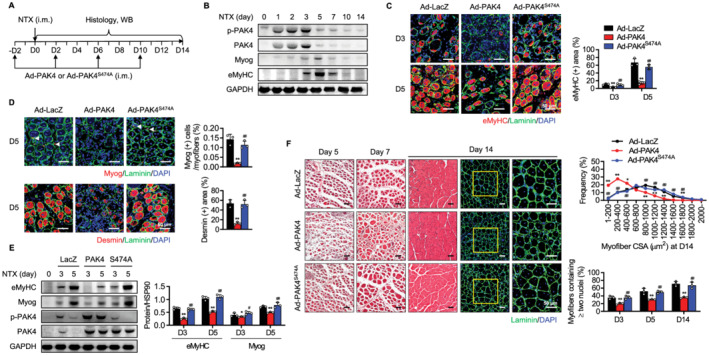
Impairment of skeletal muscle regeneration in PAK4‐overexpressing mice. (A) The tibialis anterior (TA) muscles of C57BL/6 mice were injected with Ad‐LacZ, Ad‐PAK4, or Ad‐PAK4S474A and then injured by intramuscular injection of NTX. (B) Time course analysis of PAK4 and myogenic markers after NTX injection by western blotting. (C) Immunofluorescence analysis of eMyHC‐positive fibres in TA muscles. (D) Immunofluorescence staining of Myog‐positive or desmin‐positive myofibres at Day 5. Arrowheads indicate MyoG‐positive myofibres. (E) Western blot analysis of Myog and eMyHC in injured TA muscles of control and PAK4‐overexpressing mice at 3 or 5 days after injury (n = 3). (F) H&E and immunofluorescence analyses of sections. Average cross‐sectional area (CSA) of regenerating myofibres and the percentage of myofibres containing two or more centrally located nuclei per field were determined from immunofluorescence sections. Values are mean ± SD. * P < 0.05 and ** P < 0.01 vs. Ad‐LacZ; # P < 0.05 and ## P < 0.01 vs. AdPAK4.
Forced expression of PAK4 by Ad‐PAK4 injection led to an appreciable decrease in the number of eMyHC‐positive myofibres in post‐injury TA muscles (Figure 3C). Consistently, the proportion of Myog‐positive or desmin‐positive myofibres and protein levels of eMyHC and Myog were significantly down‐regulated in PAK4‐overexpressed mice (Figure 3D and 3E). Ad‐PAK4S474A‐injected mice, however, showed comparable muscle regeneration parameters to those injected with Ad‐LacZ. In addition, PAK4 overexpression increased Ki67‐positive myofibres during the transition period to differentiation (Figure S3), suggesting that PAK4 acted as a switch to induce regeneration that balanced proliferation and differentiation. To monitor the time course of muscle regeneration, cryosections at 5, 7, and 14 days after injury were stained with H&E and the distribution of myofibre CSA was compared. H&E and immunofluorescence analyses further supported these findings: overexpression of PAK4, but not mutant PAK4, shifted the regenerating myofibres towards smaller diameters and decreased the proportion of multinucleated myofibres (Figure 3F).
To strengthen the effect of PAK4 modulation in vivo on muscle regeneration, we also introduced local silencing of PAK4 in muscle tissues. The siRNA against PAK4 was repeatedly injected into TA muscles before and after the NTX injection according to the protocol shown in Figure 3A, and levels of PAK4 were significantly reduced (Figure 4A). Immunofluorescence observations of TA muscle sections in control mice showed an increase in the number of regenerating myofibres, which were identifiable by the presence of eMyHC‐positive myofibres at 5 days after injury (Figure 4B). Importantly, the regenerative response was started earlier in PAK4 silenced mice, as revealed by the presence of regenerating myofibres at 3 days after injury (Figure 4B). In addition, PAK4 silencing resulted in a significant shift towards regenerating myofibres of larger diameters, and increased the number of myofibres that contained two or more nuclei (Figure 4C), suggesting that muscle fibre differentiation was enhanced by the PAK4 signalling blockade. Consistently, protein levels of myogenic markers and the proportion of Myog‐positive or desmin‐positive myofibres were significantly higher in PAK4‐silenced mice than control mice on Day 5 after injury (Figure 4D and 4E). Collectively, our results clearly establish that PAK4 suppresses skeletal muscle regeneration by inhibiting myogenic differentiation.
Figure 4.
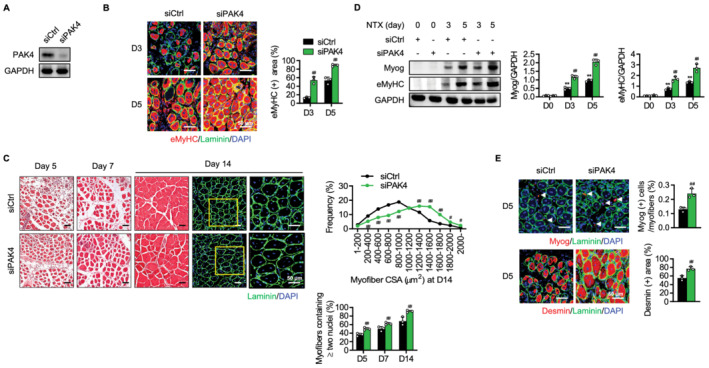
Acceleration of skeletal muscle regeneration in PAK4‐silenced mice. The tibialis anterior (TA) muscles of C57BL/6 mice were injected with scrambled siRNA (siCtrl) or siRNA against PAK4 (siPAK4) and then injured by intramuscular injection of NTX as shown in Figure 3A. (A) Western blotting analysis for PAK4 in PAK4 silenced muscles. (B) Immunofluorescence analysis of eMyHC‐positive fibres in TA muscles. (C) H&E and immunofluorescence analyses of sections. Average cross‐sectional area (CSA) of regenerating myofibres and the percentage of myofibres containing two or more centrally located nuclei per field were determined from immunofluorescence sections. (D) Time course analysis of myogenic markers by western blotting. (E) Immunofluorescence staining of Myog‐positive or desmin‐positive myofibres at Day 5. Arrowheads indicate MyoG‐positive myofibres. Values are mean ± SD. ** P < 0.01 vs. D0; ## P < 0.01 vs. siCtrl.
p21‐activated kinase 4 suppresses the phosphoinositide 3‐kinase‐Akt pathway via phosphatase and tensin homologue induction in myocytes and skeletal muscle
We next investigated the molecular mechanisms involved in the PAK4‐mediated repression of muscle regeneration. PI3K‐Akt and their downstream effectors are a key intracellular pathway implicated in the act of skeletal muscle regeneration triggered by acute muscle injury. 4 , 5 , 6 , 7 , 8 Several other signalling pathways (e.g. Wnt/β‐catenin, STAT3, p38 MAPK, and NFATc1) have been also reported as regulating muscle regeneration myogenesis. 17 , 18 , 19 , 20 We therefore examined whether PAK4 regulated these pathways and found that while PAK4 overexpression almost entirely suppressed both PI3K and Akt phosphorylation in C2C12 myotubes (Figure 5A), other signalling pathways remained unchanged (Figure S4). As a result, the phosphorylation of downstream targets of Akt (mTOR, p70S6K, and FoxO1) was repressed by PAK4 overexpression (Figure 5A). Conversely, PAK4 silencing in the cells had the opposite effect (Figure 5B). PAK4's inhibitory effect on PI3K–Akt signalling was also validated in the TA muscle of NTX‐treated mice (Figure 5C and 5D).
Figure 5.
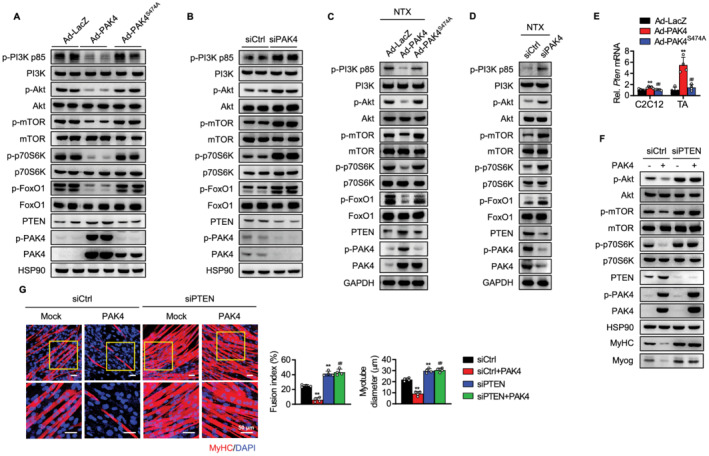
Suppression of PI3K‐Akt pathway by PAK4. (A, B) Pak4 gene was either overexpressed or silenced in C2C12 cells as indicated, and PI3K‐Akt signalling pathway on Day 5 was analysed by western blotting. (C, D) The tibialis anterior (TA) muscle was injected with either PAK4 adenovirus or PAK4 siRNA and PI3K‐Akt signalling pathway on 7 days after NTX injection was analysed by western blotting. (E) mRNA levels of Pten were determined in PAK4‐overexpressing C2C12 cells and TA muscles. (F, G) C2C12 myoblasts were transfected with siCtrl or siPTEN, and PI3K‐Akt signalling and myotube formation were compared. Values are mean ± SD. ** P < 0.01 vs. Ad‐LacZ or siCtrl; ## P < 0.01 vs. AdPAK4 or siCtrl + PAK4.
As PTEN, a PIP3 phosphatase, is a natural PI3K inhibitor, we further examined whether PTEN expression was altered by overexpression or silencing of PAK4, and therefore, if PTEN was implicated in the regulation of PI3K‐Akt signalling. PTEN protein and mRNA levels were substantially increased by PAK4 overexpression in the C2C12 cells and TA muscles of NTX‐treated mice (Figure 5A, 5C, and 5E). Conversely, PAK4 silencing reduced PTEN expression in those conditions (Figure 5B and 5D). Notably, mutant PAK4 had no effect on PTEN induction and the consequent inhibition of PI3K‐Akt signalling, which strongly suggests that PAK4's kinase activity is a necessary prerequisite for these processes. We next sought to determine whether PTEN up‐regulation by PAK4 correlated with the inhibition of Akt signalling and myogenesis and determined that PAK4's ability to inhibit the PI3K‐Akt pathway and the process of myogenic differentiation was completely eliminated by PTEN silencing (Figure 5F and 5G). These results indicate that PAK4 inhibits the PI3K‐Akt pathway through the induction of PTEN and is responsible for the inhibition of myogenesis.
p21‐activated kinase 4 phosphorylates and activates peroxisome proliferator‐activated receptor Υ to induce phosphatase and tensin homologue
Because PAK4's up‐regulation of PTEN occurred at transcriptional level (Figure 5E), we utilized bioinformatic approaches to identify PTEN's potential transcriptional regulators. Among the putative transcription factors that bind to PTEN's proximal promoter region that were predicted by the evolutionary conserved region browser, PPARγ was identified as a candidate possibly associated with PAK4 by analysis of the Genotype‐Tissue Expression (GTEx) database 21 ; RNA‐sequencing data of 564 human skeletal muscle tissues showed positive correlations between PAK4 and PPARG, PAK4 and PTEN, and PPARG and PTEN (Figure 6A). Our next step was therefore to determine whether PAK4 affected PPARγ's transcriptional activity. Forced PPARγ expression stimulated the activity of the PPRE luciferase, an effect that was further increased by PAK4 overexpression (Figure 6B). In addition, PAK4 overexpression alone elevated human PTEN promoter activity and further enhanced PPARγ's effect on PTEN transactivation (Figure 6C). ChIP assay verified PPARγ's binding to the putative PPRE region of the Pten promoter in the C2C12 cells, which was facilitated by PAK4 overexpression (Figure 6D). The kinase‐dead mutation removed PAK4's ability to increase Pten promoter activity and PPARγ enrichment in the Pten promoter (Figure 6B–6D). Further, we evaluated the impact of PAK4 and PPARγ in PTEN regulation. Our results indicated that PPARγ silencing completely repressed both basal and PAK4‐induced PTEN expression (Figure 6E), confirming the role of PPARγ as a transcriptional activator of PTEN expression downstream of PAK4. PAK4's inhibitory effect on PI3K‐Akt signalling and myogenic differentiation was also stymied by PPARγ silencing (Figure 6E and 6F).
Figure 6.
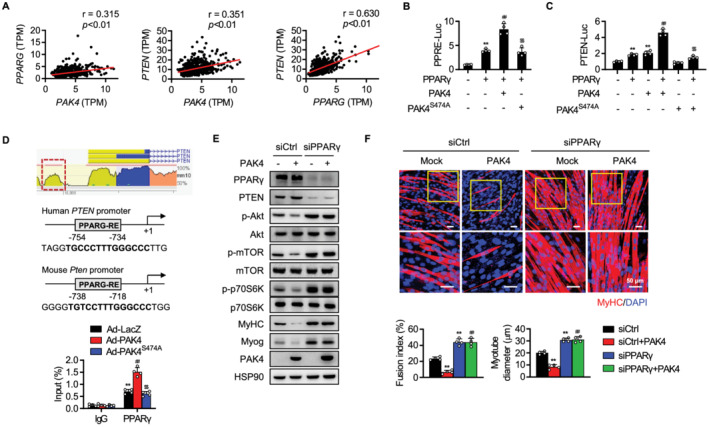
Increase of PPARγ‐mediated PTEN transcription by PAK4. (A) Genotype‐Tissue Expression (GTEx) analysis of human skeletal muscle. Pearson correlation coefficients between PAK4 and PTEN, PAK4 and PPARG, and PPARG and PTEN in human skeletal muscle were calculated. TPM, transcripts per million. (B, C) After transfection of HEK293T cells as indicated, PPRE‐luciferase and PTEN‐luciferase activities were determined. (D) Maps of human and mouse Pten promoters and ChIP‐qPCR assay showing binding of PPARγ to the Pten promoters. (E, F) C2C12 myoblasts were transfected with scrambled siRNA (siCtrl) or siRNA against PPARγ (siPPARγ), and protein levels of PTEN and PI3K‐Akt pathway and myotube formation were compared. Values are mean ± SD. ** P < 0.01 vs. none, IgG, or siCtrl; ## P < 0.01 vs. PPARγ, Ad‐LacZ, or siCtrl + PAK4; $$ P < 0.01 vs. PAK4 + PPARγ or Ad‐PAK4.
Having learned that PAK4's kinase activity is essential for myogenic processes and for PTEN regulation, we wondered whether PAK4 affects PPARγ transcriptional activity through its phosphorylation. Recombinant protein of PAK4 directly phosphorylated PPARγ as shown by the result of cell‐free kinase assay (Figure 7A). Consistently, co‐immunoprecipitation experiments revealed that PAK4 physically binds to PPARγ (Figure 7B). Out of two known PPARγ phosphorylation sites (i.e. S112 and S273), 22 , 23 only S273 residue was phosphorylated by PAK4 (Figure 7B). We further determined the effect of PAK4's phosphorylation status and kinase activity on the binding and phosphorylation of PPARγ. Wild‐type PAK4, but not kinase‐dead mutant PAK4S474A, bound to PPARγ and phosphorylated it at S273 (Figure 7B); this effect was further enhanced by phospho‐mimetic mutant PAK4S474D (Figure 7C). The addition of neutralizing pS474‐PAK4 antibody to the immunoprecipitation mixture consistently prevented the binding and phosphorylation of PPARγ (Figure S5A). Treatment of the cells with PAK4 inhibitor CZh‐226‐P resulted in similar effects on both WT PAK4 and mutant PAK4S474D transfected cells (Figure S5B and S5C), indicating that active‐form phosphorylated PAK4 is responsible for the physical interaction with and phosphorylation of PPARγ. The S273 phosphorylation of PPARγ by PAK4 was also validated in the C2C12 myotubes and TA muscles of NTX‐treated mice (Figures 7D and S6). Because extracellular signal‐regulated kinase (ERK) and cyclin‐dependent kinase (CDK)‐5 were reported to mediate S273 phosphorylation of PPARγ, 22 , 24 , 25 we checked the effect of PAK4 on these kinases. Levels of p‐ERK and p‐CDK5 were not affected by PAK4 overexpression or knockdown in myotubes and TA muscles (Figures 7D and S6). Additionally, CDK5 silencing and ERK inhibition had no effect on PAK4‐mediated PPARγ phosphorylation (Figure 7E and 7F), supporting the direct action of PAK4 on PPARγ phosphorylation in muscle cells. In contrast, substituting serine with alanine at position 273 (S273A) in PPARγ ended PAK4's phosphorylation of PPARγ, induction of PTEN expression, and inhibition of PI3K–Akt signalling and myoblast differentiation (Figure 7G and 7H). Phosphorylation at S273 had either activating or repressing effects on PPARγ depending on the target genes and activates Gdf3 26 while represses Nr1d1 25 in macrophages and adipocytes. Consistent with this, overexpression of PAK4 significantly increased the mRNA level of Gdf3 but decreased Nr1d1 in C2C12 cells (Figure S7). These findings demonstrate that PAK4 directly phosphorylates PPARγ at S273 residue, thereby activating its transcriptional activity and promoting PTEN expression in the myoblasts of both mice and humans.
Figure 7.
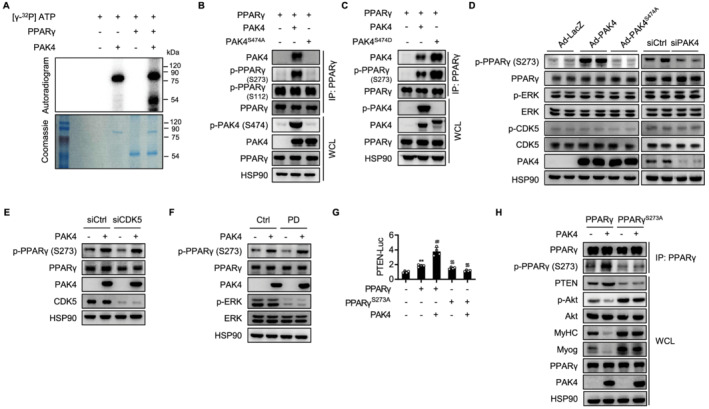
Direct phosphorylation of PPARγ by PAK4. (A) Recombinant PPARγ was incubated with active PAK4 and [32P]ATP for 30 min at 30°C, and proteins in the mixture were resolved by SDS‐PAGE. The band was visualized by autoradiography of 32P‐labelled protein. Loading of proteins was confirmed by Coomassie blue staining. (B, C) After transfection of HEK293T cells as indicated, co‐IP was performed to determine PAK4 interaction with and phosphorylation of PPARγ. (D) C2C12 myoblasts were PAK4 overexpressed or silenced, and then phosphorylation of PPARγ, ERK, and CDK5 on Day 5 was analysed by western blotting. (E) C2C12 myoblasts were transfected with scrambled siRNA (siCtrl) or siRNA against CDK5 (siCDK5), and then PAK4 phosphorylation of PPARγ was analysed by western blotting. (F) C2C12 myoblasts were pretreated with ERK inhibitor PD98059 (10 μM), and PAK4 phosphorylation of PPARγ was analysed. (G) PTEN‐luciferase activities were determined after transfection of HEK293T cells as indicated. (H) C2C12 cells were transfected with wild‐type or mutant PPARγ (S273A) along with PAK4, and then PI3K–Akt signalling and myogenic markers on Day 5 were determined. Values are mean ± SD. ** P < 0.01 vs. none; ## P < 0.01 vs. PPARγ; $$ P < 0.01 vs. PAK4 + PPARγ.
Treatment of p21‐activated kinase 4 inhibitor accelerates skeletal muscle regeneration in mice
Because PAK4 silencing accelerates myogenic differentiation (Figure S2) and muscle regeneration (Figure 4), we finally examined whether chemically inhibiting PAK4 promoted muscle regeneration in response to acute injury. We orally administered prodrug of PAK4 inhibitor CZh‐226‐P to mice 13 2 days before NTX injection and throughout muscle regeneration period (Figure 8A). No adverse effects from treatment with CZh‐226‐P were observed, as shown by the activities of transaminases in sera (Figure 8B). Consistent with our previous experimental results, PAK4 inhibitor treatment repressed S273 phosphorylation of PPARγ (and thus PTEN suppression) and increased p‐Akt (Figure 8C). Accordingly, treatment with a PAK4 inhibitor increased myogenic markers, accelerated myotube formation, and shifted CSA distribution rightward (Figure 8C and 8D). However, CZh‐226‐P's promoting effect on muscle regeneration was not further observed in mice administered an intramuscular injection with siPAK4 (Figure 8E), affirming the importance of muscular PAK4 inhibition to the chemical's action. These results suggest that pharmacological inhibition of PAK4 offers a novel therapeutic approach to the amelioration of muscular diseases associated with defects of regenerative capacity.
Figure 8.
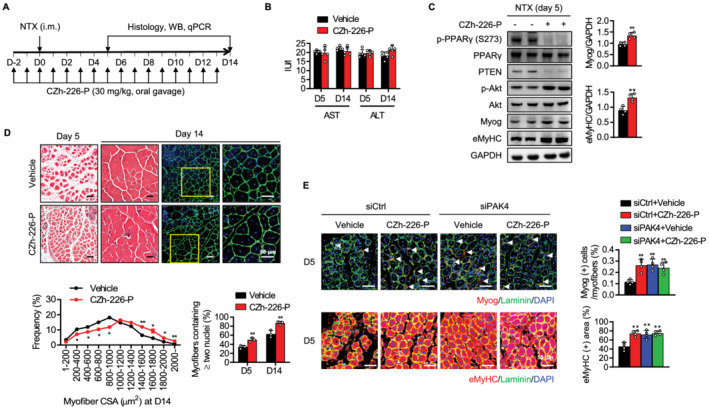
Acceleration of skeletal muscle regeneration by a small molecule inhibitor of PAK4. (A) C57BL/6 mice were treated with prodrug of PAK4 inhibitor CZh‐226‐P (30 mg/kg) via oral gavage 2 days before NTX injection and every day before and after NTX injection until Day 13. (B) Serum levels of AST and ALT were measured. (C) Western blot analysis of PPARγ–PTEN–Akt signalling and myogenic markers in injured TA muscles at 5 days after injury. (D) H&E and immunofluorescence analyses at 5 or 14 days after injury. Average cross‐sectional area (CSA) of regenerating myofibres and the percentage of myofibres containing two or more centrally located nuclei per field were determined from immunofluorescence sections. (E) The tibialis anterior (TA) muscles of C57BL/6 mice were injected with siCtrl or siPAK4 and then injured by intramuscular injection of NTX. CZh‐226‐P (30 mg/kg) was administered once a day via oral gavage for 7 days starting 2 days before NTX injection. Myog (+) and eMyHC (+) fibres in TA muscles were counted at Day 5. Values are mean ± SD. ** P < 0.01 vs. vehicle. CZh‐226‐P, prodrug of CZh‐226.
Discussion
In this study, we uncovered a previously unknown function of PAK4: that of critical myogenesis regulator and contributor to the process of skeletal muscle regeneration. As a first step, we determined that PAK4 expression was suppressed during muscle differentiation in vitro. Forced expression of PAK4 in C2C12 cells and primary myoblasts inhibited myotube formation, while PAK4 silencing promoted this process. Concomitantly, our animal experiments confirmed that PAK4 silencing dramatically enhanced the regenerative capacity of skeletal muscle upon acute myotoxin injury. Crucially, we also provided substantial evidence demonstrating that PAK4 phosphorylates PPARγ at S273 residue to induce PTEN transcription and subsequently suppresses the Akt pathway responsible for myogenesis (Figure S8). We concluded by demonstrating that treating mice with a PAK4 inhibitor down‐regulated PPARγ‐mediated PTEN expression in injured skeletal muscle and promoted muscle regeneration.
Relatively little is known about the effects of each of the six members of the mammalian PAK family on muscle regeneration. Despite the high sequence similarity between each PAK family member, the fact that each has distinct preferred phosphorylation consensus sites suggests that each has a distinct substrate specificity 27 and therefore mediates a different function. Recently, it was suggested that Group I PAKs, including PAK1 and PAK2, may act as positive regulators of myoblast differentiation and promote muscle regeneration, thereby protecting mice against freeze injuries 15 or cancer cachexia‐associated muscle wasting. 14 In contrast, PAK4 (a Group II PAK) exhibits distinct myogenesis suppression behaviour. PAK4's rapidly changing expression patterns during myogenesis (i.e. decrease) and upon muscle injury (i.e. transient increase followed by decrease) imply the rheostatic role of PAK4 in fine‐tuning myogenesis between cell proliferation and differentiation. Unlike the critical function of PAK4 in promoting cell proliferation in cancer, 10 , 11 , 12 it appears in differentiating myoblasts/myotubes that PAK4 down‐regulation may be required to trigger the exit from the cell cycle towards the differentiation/fusion process. Indeed, we showed that PAK4 silencing in myoblasts and skeletal muscles substantially increased myotube formation, which was associated with increases in PI3K‐Akt signalling. In sum, PAK4 inhibition may contribute to skeletal myogenesis and regeneration to maintain homeostasis.
We observed that the protein levels of PAK4 and p‐Akt were reciprocally changed during the myogenic process. Moreover, ectopic overexpression of PAK4 in myoblasts/SCs decreased while depletion of PAK4 in myoblasts increased p‐Akt levels. Accordingly, we observed that PAK4 overexpression decreased protein levels of p‐p70S6K, p‐mTOR, and p‐FoxO1, which are downstream targets of Akt during myoblast differentiation. These results suggest that PAK4 may indirectly suppress Akt phosphorylation. While growth factors are required to activate the PI3K‐Akt pathway in regenerating muscles, negative modulatory factors are also required for fine control of Akt phosphorylation. 8 PTEN is a well‐known example of the latter. To address whether PTEN is involved in the negative regulation of Akt by PAK4, we analysed PTEN expression in PAK4 overexpression or repression conditions. Our results showed that PTEN was highly expressed in PAK4 overexpressing C2C12 cells and TA muscles. Additionally, consistent with the observations reported in other recently published studies, we showed that PTEN was critical for the regulation of myogenic differentiation, as PTEN silencing markedly increased the expression of myogenic markers in C2C12 cells. 8 , 28 Notably, PTEN silencing increased p‐Akt protein to levels similar to those seen during PAK4 silencing, indicating that PAK4 regulates myogenic differentiation in a PTEN‐dependent manner. This suggests a question about the regulatory mechanism responsible for the PTEN expression: What mechanism allows the expression of PTEN by PAK4 during myogenic differentiation?
Phosphatase and tensin homologue is reportedly induced in response to external and endogenous stimuli through various signalling cascades, including PPARγ. 29 While one previous study using glioblastoma cells indicated that PAK4 interacts with PPARγ to induce its binding to PPARγ target gene Nox1 promoter, 30 in this paper, we suggest a novel role for PAK4, by which it increases PPARγ activity in a phosphorylation‐dependent manner towards PTEN expression. In addition, we located a PPARγ binding site, conserved among species, in proximal promoter region of the Pten gene. Co‐immunoprecipitation and ChIP assays showed that PAK4 directly interacted with and phosphorylated PPARγ, thereby increasing PPARγ binding to the Pten promoter and enhancing Pten transcription. GTEx analysis determined that PTEN expression was positively associated with either PAK4 or PPARγ, confirming a PAK4–PPARγ–PTEN axis in human skeletal muscle. We note with interest that PPARγ phosphorylation by PAK4 was observed at S273 in the ligand binding domain, but not at S112 in the activation function 1 domain. S273 phosphorylation of PPARγ has been reported to be mediated by CDK5 and ERK in adipose tissue, 22 , 24 , 25 in which case the impact of S273 phosphorylation on PPARγ target gene transcription varied depending on the different genes involved; S273 phosphorylation suppressed insulin‐sensitizing adipokines (i.e. Adipoq) but resulted in no change to adipogenesis genes (i.e. Pparg2, Fabp4, Cebpa, and Lpl). Transcriptional enhancement by PPARγ S273 phosphorylation also occurs for growth differentiation factor 3, mainly in macrophages, which consistently contributes to impairing insulin sensitivity. 26 In this study, the increases in PPARγ S273 phosphorylation and PTEN expression, and the inhibition of myogenesis, were caused specifically by kinase‐intact PAK4, and these responses were completely reversed by PPARγ depletion. This is strongly suggestive of a causal link between the PAK4–PPARγ–PTEN pathway and the repressed myogenesis, as well as a redundancy in upstream kinases for PPARγ S273 phosphorylation, as PAK4 phosphorylation of PPARγ was not impaired either by CDK5 silencing or ERK inhibition.
Using this mechanism, we demonstrated that a PAK4 inhibitor could suppress PPARγ phosphorylation and PTEN induction and thereby promote muscle regeneration. In the future, it may be possible to manipulate the muscle regeneration programme by altering the activity of PPARγ through its interacting partner PAK4. In sum, our study provides evidence that PAK4 is an upstream kinase for PPARγ during skeletal muscle regeneration. Keeping in mind that PPARγ is a master regulator of various metabolic homeostasis and cellular proliferation processes, 31 , 32 it would be interesting to investigate whether PAK4‐mediated regulation of PPARγ transcriptional activity is involved in those pathological conditions.
Funding
This work was supported by a grant from the Medical Research Center Program (2017R1A5A2015061) and by grants from the Basic Science Research Program (2020R1A2C2004761, 2020R1C1C1003652, and 2021R1A2B5B02001462) through the National Research Foundation (NRF), which is funded by the Korean government (MSIP). This research was also supported by Korea Drug Development Fund (HN21C0447) funded by Ministry of Science and ICT, Ministry of Trade, Industry and Energy, and Ministry of Health and Welfare.
Conflict interest
None declared.
Author contributions
B‐H.P. and E.J.B. conceived the idea, designed the experiments, and wrote the manuscript. Y.M., C.Y.H., L.H., and I.H.B. conducted the experiments and analysed the data. B‐H.P. and E.J.B. had primary responsibility for the final content. All authors read and approved the final manuscript.
Supporting information
Table S1. Sequences and accession numbers for primers (forward, FOR; reverse, REV) used in qPCR
Table S2. Antibodies used for Western blotting, immunofluorescence and immunohistochemical analyses
Figure S1. PAK4 regulation of myogenesis in C2C12 cells and primary myoblasts. (A, C) Protein levels of PAK4, myosin heave chain (MyHC), and myogenin (Myog) during myogenic differentiation were analyzed in C2C12 cells (A, n=5) and primary myoblasts (B) by Western blotting. (B, D) mRNA levels of PAK4 and myogenesis related genes were quantified by qPCR in C2C12 cells (B) and primary myoblasts (D). (E, F) C2C12 cells and primary myoblasts were induced to differentiate in differentiation medium (DM) for 5 days, and the expressions of PAK4 and MyHC were analyzed by immunostaining. Values are mean±SD. **, p<0.01 versus day 0 or growth medium (GM).
Figure S2. Acceleration of myogenesis in PAK4‐silenced C2C12 cells and primary myoblasts. C2C12 myoblasts (A‐C) and primary myoblasts (D‐F) were transfected with scrambled siRNA (siCtrl) or siRNA against PAK4 (siPAK4). Twenty‐four hours later, the cells were transferred to differentiation medium (DM) for 5 days. (A, D) Cells were fixed and immunostained for MyHC. The fusion index and myotube diameter were determined. (B, E) Expression of myogenic differentiation proteins were assayed by Western blotting. (C, F) mRNA levels of myosine heave chains and myogenin in PAK4 silenced C2C12 cells and primary myoblasts were determined by qPCR. Values are mean±SD. **, p<0.01 versus day 0 or growth medium (GM); ##, p<0.01 versus siCtrl.
Figure S3. Increase of cell proliferation in PAK4‐overexpressing mice. The number of myogenic cell nuclei immunopositive for cell proliferation marker Ki67 was analyzed at days 3 and 5 after notexin treatment. HPF, high‐power fields. Values are mean±SD. **, p<0.01 versus Ad‐LacZ; ##, p<0.01 versus Ad‐PAK4.
Figure S4. Effects of PAK4 overexpression on myogenesis‐related signaling pathways in C2C12 cells. C2C12 myoblasts were infected with Ad‐LacZ, Ad‐PAK4, or Ad‐PAKS474A and differentiated for 5 days. Total and phosphorylation levels of myogenesis‐related proteins were analyzed by Western blotting.
Figure S5. PAK4 kinase‐dependent phosphorylation of PPARγ. HEK293T cells were transfected as indicated and subsequently incubated with either anti‐p‐PAK4 antibody (1 μg/ml) prior to co‐IP (A) or pretreated cells with 5 μM CZh‐26‐P (B, C). Co‐IP was performed to determine PAK4 interaction with and phosphorylation of PPARγ. WCL, whole cell lysate; CZh‐226‐P, prodrug of CZh‐226.
Figure S6. PAK4 phosphorylation of PPARγ in injured TA muscles. Mice were PAK4 overexpressed or silenced and then injured by intramuscular injection of NTX. Phosphorylation of PPARγ, ERK, and CDK5 on day 5 after NTX injection were analyzed by Western blotting.
Figure S7. Genes responsive to PPARγ S273 phosphorylation in C2C12 cells. C2C12 myoblasts were transfected with PAK4 or mutant PAK4 and several genes known to be affected by PPARγ S273 phosphorylation were analyzed by qPCR. Values are mean±SD. **, p<0.01 versus Ad‐LacZ; ##, p<0.01 versus Ad‐PAK4
Figure S8. Proposed summary
Mao Y., Han C. Y., Hao L., Bang I. H., Bae E. J., and Park B.‐H. (2021) p21‐activated kinase 4 phosphorylates peroxisome proliferator‐activated receptor Υ and suppresses skeletal muscle regeneration, Journal of Cachexia, Sarcopenia and Muscle, 12, 1776–1788, 10.1002/jcsm.12774
Contributor Information
Eun Ju Bae, Email: ejbae7@jbnu.ac.kr.
Byung‐Hyun Park, Email: bhpark@jbnu.ac.kr.
References
- 1. Qazi TH, Duda GN, Ort MJ, Perka C, Geissler S, Winkler T. Cell therapy to improve regeneration of skeletal muscle injuries. J Cachexia Sarcopenia Muscle 2019;10:501–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maleiner B, Tomasch J, Heher P, Spadiut O, Runzler D, Fuchs C. The importance of biophysical and biochemical stimuli in dynamic skeletal muscle models. Front Physiol 2018;9:1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tedesco FS, Dellavalle A, Diaz‐Manera J, Messina G, Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest 2010;120:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glass DJ. PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr Top Microbiol Immunol 2010;346:267–278. [DOI] [PubMed] [Google Scholar]
- 5. Moriya N, Miyazaki M. Akt1 deficiency diminishes skeletal muscle hypertrophy by reducing satellite cell proliferation. Am J Physiol Regul Integr Comp Physiol 2018;314:R741–R751. [DOI] [PubMed] [Google Scholar]
- 6. Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert . Nature 2014;510:393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilson EM, Rotwein P. Selective control of skeletal muscle differentiation by Akt1. J Biol Chem 2007;282:5106–5110. [DOI] [PubMed] [Google Scholar]
- 8. Yue F, Bi P, Wang C, Li J, Liu X, Kuang S. Conditional loss of Pten in myogenic progenitors leads to postnatal skeletal muscle hypertrophy but age‐dependent exhaustion of satellite cells. Cell Rep 2016;17:2340–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumar R, Sanawar R, Li X, Li F. Structure, biochemistry, and biology of PAK kinases. Gene 2017;605:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qasim SL, Sierra L, Shuck R, Kurenbekova L, Patel TD, Rajapakshe K, et al. p21‐activated kinases as viable therapeutic targets for the treatment of high‐risk Ewing sarcoma. Oncogene 2021;40:1176–1190. [DOI] [PubMed] [Google Scholar]
- 11. Park MH, Lee HS, Lee CS, You ST, Kim DJ, Park BH, et al. p21‐activated kinase 4 promotes prostate cancer progression through CREB. Oncogene 2013;32:2475–2482. [DOI] [PubMed] [Google Scholar]
- 12. Dart AE, Wells CM. P21‐activated kinase 4—not just one of the PAK. Eur J Cell Biol 2013;92:129–138. [DOI] [PubMed] [Google Scholar]
- 13. Guo J, Wang T, Wu T, Zhang K, Yin W, Zhu M, et al. Synthesis, bioconversion, pharmacokinetic and pharmacodynamic evaluation of N‐isopropyl‐oxy‐carbonyloxymethyl prodrugs of CZh‐226, a potent and selective PAK4 inhibitor. Eur J Med Chem 2020;186:111878. [DOI] [PubMed] [Google Scholar]
- 14. Cerquone Perpetuini A, Re Cecconi AD, Chiappa M, Martinelli GB, Fuoco C, Desiderio G, et al. Group I Paks support muscle regeneration and counteract cancer‐associated muscle atrophy. J Cachexia Sarcopenia Muscle 2018;9:727–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joseph GA, Lu M, Radu M, Lee JK, Burden SJ, Chernoff J, et al. Group I Paks promote skeletal myoblast differentiation in vivo and in vitro. Mol Cell Biol 2017;37:e00222–e00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmidt M, Schuler SC, Huttner SS, von Eyss B, von Maltzahn J. Adult stem cells at work: regenerating skeletal muscle. Cell Mol Life Sci 2019;76:2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rudolf A, Schirwis E, Giordani L, Parisi A, Lepper C, Taketo MM, et al. β‐Catenin activation in muscle progenitor cells regulates tissue repair. Cell Rep 2016;15:1277–1290. [DOI] [PubMed] [Google Scholar]
- 18. Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, et al. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med 2014;20:1182–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segales J, Perdiguero E, Munoz‐Canoves P. Regulation of muscle stem cell functions: a focus on the p38 MAPK signaling pathway. Front Cell Dev Biol 2016;4:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakuma K, Nishikawa J, Nakao R, Watanabe K, Totsuka T, Nakano H, et al. Calcineurin is a potent regulator for skeletal muscle regeneration by association with NFATc1 and GATA‐2. Acta Neuropathol 2003;105:271–280. [DOI] [PubMed] [Google Scholar]
- 21. GTEx Consortium . The Genotype‐Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, et al. Anti‐diabetic drugs inhibit obesity‐linked phosphorylation of PPARγ by Cdk5. Nature 2010;466:451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase‐mediated phosphorylation of PPARγ. Science 1996;274:2100–2103. [DOI] [PubMed] [Google Scholar]
- 24. Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, et al. Antidiabetic actions of a non‐agonist PPARγ ligand blocking Cdk5‐mediated phosphorylation. Nature 2011;477:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik‐Bogoslavski D, et al. An ERK/Cdk5 axis controls the diabetogenic actions of PPARγ. Nature 2015;517:391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall JA, Ramachandran D, Roh HC, DiSpirito JR, Belchior T, Zushin PH, et al. Obesity‐linked PPARγ S273 phosphorylation promotes insulin resistance through growth differentiation factor 3. Cell Metab 2020;32:665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rennefahrt UE, Deacon SW, Parker SA, Devarajan K, Beeser A, Chernoff J, et al. Specificity profiling of Pak kinases allows identification of novel phosphorylation sites. J Biol Chem 2007;282:15667–15678. [DOI] [PubMed] [Google Scholar]
- 28. Yue F, Bi P, Wang C, Shan T, Nie Y, Ratliff TL, et al. Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat Commun 2017;8:14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamguney T, Stokoe D. New insights into PTEN. J Cell Sci 2007;120:4071–4079. [DOI] [PubMed] [Google Scholar]
- 30. Kesanakurti D, Maddirela D, Banasavadi‐Siddegowda YK, Lai TH, Qamri Z, Jacob NK, et al. A novel interaction of PAK4 with PPARγ to regulate Nox1 and radiation‐induced epithelial‐to‐mesenchymal transition in glioma. Oncogene 2017;36:5309–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, et al. Adipocyte NCoR knockout decreases PPARγ phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell 2011;147:815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mal S, Dwivedi AR, Kumar V, Kumar N, Kumar B, Kumar V. Role of peroxisome proliferated‐activated receptor γ (PPARγ) in different disease states: recent updates. Curr Med Chem 2021;28:3193–3215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences and accession numbers for primers (forward, FOR; reverse, REV) used in qPCR
Table S2. Antibodies used for Western blotting, immunofluorescence and immunohistochemical analyses
Figure S1. PAK4 regulation of myogenesis in C2C12 cells and primary myoblasts. (A, C) Protein levels of PAK4, myosin heave chain (MyHC), and myogenin (Myog) during myogenic differentiation were analyzed in C2C12 cells (A, n=5) and primary myoblasts (B) by Western blotting. (B, D) mRNA levels of PAK4 and myogenesis related genes were quantified by qPCR in C2C12 cells (B) and primary myoblasts (D). (E, F) C2C12 cells and primary myoblasts were induced to differentiate in differentiation medium (DM) for 5 days, and the expressions of PAK4 and MyHC were analyzed by immunostaining. Values are mean±SD. **, p<0.01 versus day 0 or growth medium (GM).
Figure S2. Acceleration of myogenesis in PAK4‐silenced C2C12 cells and primary myoblasts. C2C12 myoblasts (A‐C) and primary myoblasts (D‐F) were transfected with scrambled siRNA (siCtrl) or siRNA against PAK4 (siPAK4). Twenty‐four hours later, the cells were transferred to differentiation medium (DM) for 5 days. (A, D) Cells were fixed and immunostained for MyHC. The fusion index and myotube diameter were determined. (B, E) Expression of myogenic differentiation proteins were assayed by Western blotting. (C, F) mRNA levels of myosine heave chains and myogenin in PAK4 silenced C2C12 cells and primary myoblasts were determined by qPCR. Values are mean±SD. **, p<0.01 versus day 0 or growth medium (GM); ##, p<0.01 versus siCtrl.
Figure S3. Increase of cell proliferation in PAK4‐overexpressing mice. The number of myogenic cell nuclei immunopositive for cell proliferation marker Ki67 was analyzed at days 3 and 5 after notexin treatment. HPF, high‐power fields. Values are mean±SD. **, p<0.01 versus Ad‐LacZ; ##, p<0.01 versus Ad‐PAK4.
Figure S4. Effects of PAK4 overexpression on myogenesis‐related signaling pathways in C2C12 cells. C2C12 myoblasts were infected with Ad‐LacZ, Ad‐PAK4, or Ad‐PAKS474A and differentiated for 5 days. Total and phosphorylation levels of myogenesis‐related proteins were analyzed by Western blotting.
Figure S5. PAK4 kinase‐dependent phosphorylation of PPARγ. HEK293T cells were transfected as indicated and subsequently incubated with either anti‐p‐PAK4 antibody (1 μg/ml) prior to co‐IP (A) or pretreated cells with 5 μM CZh‐26‐P (B, C). Co‐IP was performed to determine PAK4 interaction with and phosphorylation of PPARγ. WCL, whole cell lysate; CZh‐226‐P, prodrug of CZh‐226.
Figure S6. PAK4 phosphorylation of PPARγ in injured TA muscles. Mice were PAK4 overexpressed or silenced and then injured by intramuscular injection of NTX. Phosphorylation of PPARγ, ERK, and CDK5 on day 5 after NTX injection were analyzed by Western blotting.
Figure S7. Genes responsive to PPARγ S273 phosphorylation in C2C12 cells. C2C12 myoblasts were transfected with PAK4 or mutant PAK4 and several genes known to be affected by PPARγ S273 phosphorylation were analyzed by qPCR. Values are mean±SD. **, p<0.01 versus Ad‐LacZ; ##, p<0.01 versus Ad‐PAK4
Figure S8. Proposed summary