

SUMMARY
Stalled DNA replication fork restart after stress as orchestrated by ATR kinase, BLM helicase, and structure-specific nucleases enables replication, cell survival, and genome stability. Here we unveil human exonuclease V (EXO5) as an ATR-regulated DNA structure-specific nuclease and BLM partner for replication fork restart. We find elevated tumor EXO5 correlates with increased mutation loads and poor patient survival, suggesting EXO5 upregulation has oncogenic potential. Structural, mechanistic and mutational analyses of EXO5 and EXO5-DNA complex reveal a single-stranded DNA binding channel with an adjacent ATR phosphorylation site (T88Q89) that regulates EXO5 nuclease activity and the BLM binding identified by mass-spectrometric analysis. EXO5 phospho-mimetic mutant rescues the restart defect from EXO5 depletion that decreases fork progression, DNA damage repair, and cell survival. EXO5 depletion furthermore rescues survival of FANCA-deficient cells and indicates EXO5 functions epistatically with SMARCAL1 and BLM. Thus, an EXO5 axis connects ATR and BLM in directing replication fork restart.
Keywords: Exonuclease, replication stress, fork restart, interstrand crosslink repair, Bloom, genetic instability, Fanconi anemia, ATR phosphorylation, cancer biology, tumor proliferation
eTOC Blurb
Hambarde et al. reveal EXO5 structures and its heretofore missed structure-specific nuclease activity for replication fork restart orchestrated with ATR kinase and BLM helicase. EXO5 depletion rescues FANCA-deficient cell survival and fork protection defects. High EXO5 expression predicts high mutation loads and poor cancer patient survival, implying therapeutic opportunities.
Graphical Abstract
INTRODUCTION
DNA replication requires stalled replication fork protection and restart to avoid fork collapse, which leads to genome instability, a cancer hallmark (Yeeles et al., 2013). Human replication protein A (RPA) binds single-stranded DNA (ssDNA) exposed at stalled forks. Replication stress response kinase ATR (ataxia-telangiectasia and Rad3-related) is activated by RPA-coated ssDNA (Saldivar et al., 2017). RPA and ATR recruit fork remodelers (SMARCAL1, ZRANB3, HLTF) and RAD51, which together with specialized nucleases (DNA2, MRE11, EXO1) and helicases (BLM, WRN) mediate fork degradation and restart (Kolinjivadi et al., 2017; Lemacon et al., 2017; Liao et al., 2018; Quinet et al., 2017; Thangavel et al., 2015; Nimonkar et al., 2011).
BLM (Bloom syndrome RecQ-like helicase) is recruited to stalled forks after phosphorylation by ATR enabling efficient fork restart and suppression of origin firing (Davies et al., 2007). BLM deficiency results in Bloom’s syndrome, a genetic disorder characterized by hyper-recombination, increased sister chromatid exchange (SCE), and radial chromosome formation: all indicating BLM’s anti-recombination role in crossover suppression (Wu and Hickson, 2003). To resolve stalled forks, BLM works with RPA and nucleases regulated by ATR to avoid mutagenic off-target incisions (Davies et al., 2007). Yet, evidence that ATR directly regulates any structure-specific nuclease for fork restart is sparse (Dibitetto et al., 2020).
Human exonuclease V (EXO5) is biochemically characterized as a ssDNA-specific sliding exonuclease with an iron-sulfur cluster (Sparks et al., 2012). EXO5 loads at ssDNA ends with 5’ to 3’ polarity enforced by RPA and slides along ssDNA prior to cutting, analogously to DNA2 (Zhou et al., 2015), but distinct from FEN1 and EXO1, which cut ssDNA flaps by first binding and loading onto adjacent dsDNA (Orans et al., 2011; Tsutakawa et al., 2017), or MRE11, which cuts from 3’ to 5’ polarity with endo- and exonuclease activities (Paull, 2018; Shibata et al., 2014). In budding yeast EXO5 exonuclease is vital for mitochondrial genome maintenance (Burgers et al., 2010). Yet, most eukaryotic EXO5 proteins, including fission yeast and human, function in the nuclear DNA damage response (DDR) (Sparks et al., 2019). EXO5-deficient cells are sensitive to interstrand cross-linking (ICL) agents such as cisplatin and mitomycin C that cause radial chromosome structures at metaphase (Sparks et al., 2012). Furthermore, EXO5 is a susceptibility gene in testicular and prostate cancers, implying a role in genome integrity (Ali et al., 2019; Paumard-Hernandez et al., 2018). Yet, with no reported EXO5 structures and limited knowledge of EXO5 structure-specific DNA interactions, molecular mechanism, regulation, and biological functions, EXO5 is an enigmatic DDR nuclease.
Here we computationally analyzed The Cancer Genome Atlas (TCGA) data and found elevated EXO5 in tumors correlates with increased mutation loads and decreased patient survival. We solved EXO5 structures with and without bound ssDNA to determine its mechanism for structure-specific nuclease activity. We discovered that ATR-dependent phosphorylation of EXO5 adjacent to its DNA binding channel enables BLM interaction to orchestrate restart of SMARCAL1 regressed replication forks stalled by replication stress associated with tumor proliferation.
RESULTS
High EXO5 Gene Expression Predicts Poor Patient Survival and High Mutation Loads
As low EXO5 mRNA levels occur in some tumors ( Knijnenburg et al., 2018), we tested whether EXO5 mRNA expression correlates with patient outcome. Of 26,735 informative subjects, EXO5 gains from copy number alterations (CNAs), predominantly gene amplifications, were associated with shorter survival (Figure 1A). Analysis of TCGA transcriptomic data indicated that in two-thirds of tumor types EXO5 mRNA levels were higher than in matched controls (Figure 1B). Furthermore, predictive analysis by the Kaplan-Meier estimator revealed worse outcome in adrenocortical carcinoma (ACC) patients with high EXO5 mRNA levels (Figure 1C). Although EXO5 resides within a frequently overexpressed domain on chromosome 1p32–1p36 (Figure 1D), we found little evidence to exclude EXO5 for a role in poor prognosis in ACC patients (see STAR Methods).
Figure 1. High EXO5 Gene Expression Predicts Poor Patient Survival and High Mutation Loads.

(A) Kaplan-Meier survival curve for patients with and without EXO5 copy number alterations (CNAs); median months survival: with CNAs 51.2, without CNAs 106.9. (B) Normalized EXO5 mRNA levels between tumor and matched normal tissues from TCGA. Top, p-values from Wilcoxon tests sorted by difference in mRNA level. (C) Kaplan-Meier survival probability curve and hazard ratio (HR) in ACC patients with low (below mean, orange) and high (above mean, blue) EXO5 mRNA levels. (D) Number of TCGA tumors with significant EXO5 coexpression with genes within cytogenetic band 1p32–1p36. In parenthesis, distance from EXO5 transcription start site (TSS) in Mbp. Brown, TSSs of refSeq genes. (E) GSEA for highly expressed genes in ACC. (F) Check plot of TCGA tumor types with significant GSEA enriched terms for all genes associated with poor outcome when expressed above mean levels. Left. GO terms; right, genes within (top) and outside (bottom) GO terms enriched in at least 6 tumor types and consistently overexpressed in all 15 tumor types relative to matched controls (see panel B). (G) Correlation between EXO5 mRNA level and somatic mutations in ACC; x-axis, EXO5 mRNA levels, y-axis, number of exome-wide somatic single-base substitutions and small indels. r = 0.337, P(α)0.05 = 0.829. (H) BRCA signature 3 mutations. Left, box plot of EXO5 expression for 200 subjects with highest (brown) and lowest (green) RNA-seq values. Right, bar plot of number of subjects with (black) and without (hatched) signature 3 mutations for low and high EXO5 gene expression groups, respectively, from left panel. P-value from Fisher’s exact test. See Figure S1.
We reasoned that if EXO5 expression were an informative marker of patient outcome, expression of other genes along its pathway may bear similar prognostic power. Therefore, we computed Kaplan-Meier and Cox proportional hazards regressions (hazard ratios) for all 20,500 RNA-seq entries surveyed in TCGA for ACC. At a threshold of p <0.001, there were 316 genes associated with poor outcome in patients with high mRNA levels, which were strongly enriched in gene ontology (GO) terms associated with DNA replication, cell division, DNA repair and DNA repair-dependent DNA synthesis (Figure 1E). Thus poor outcome in ACC may arise from uncontrolled cell division and subsequent replication stress-induced DNA damage, and EXO5 may be an integral DDR component.
We assessed if upregulation of cell proliferation and subsequent DDR would manifest in poor survival also in other cancer types by extending hazard ratios to the other 32 TCGA tumor types and performing GSEA on all genes with p-value <0.001 (LGG, brain lower grade glioma; KIRC, kidney renal clear cell carcinoma; MESO, mesothelioma) or <0.01 (remaining sets; Figure S1A). Of the 15 tumor types in which patient survival was negatively impacted by high mRNA levels, about half exhibited enrichment in GO terms related to cell cycle, and four to DNA damage/repair (Figure 1F, left). In these GO terms, 59 genes were in common in at least 6 tumor types, and 30 were significantly overexpressed in all tumor types when compared to matched controls (Figure 1F, right). As the total number of genes overexpressed in all tumor types was 191, the fraction of 30/59 compared to 191/20500 was significant (p = 2.2 × 10−37; Fisher exact test). Thus, these 30 genes fulfill the definition of tumor-type oncogenes. Three of them (CENPA, MYBL2 and TPX2) were among the top 10 genes whose expression correlated most strongly with exome-wide mutational loads (Bacolla et al., 2019), and 13 of them have been included in clinical trials as targets for inhibitor drugs in immunotherapy or as biomarkers for chemotherapy (https://clinicaltrials.gov). In sum, our bioinformatics analyses suggest that correlation of high EXO5 expression with poor outcome arises from a broad response to DDR following cell cycle hyperactivation and replication stress.
In the ACC cohort, EXO5 mRNA levels also correlated positively with load of exome-wide single base substitutions and small indels (Figure 1G). Similarly, in the three largest TCGA datasets (BRCA, breast invasive carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma), ~15–50% more cases with high EXO5 mRNA levels incurred increased single-base substitutions conforming to COSMIC signature 3, associated with insufficient homologous recombination (HR)-dependent repair (Alexandrov et al., 2020; Davies et al., 2017; Polak et al., 2017), than those with low EXO5 mRNA levels. This prevalence was strongest in BRCA patients (Figure 1H), in which signature 3 mutations account for up to 60% of all single-base substitutions, and in which ~50% patients display signature 3 mutations (Eckelmann et al., 2020). In BRCA samples, signatures 13, 20, 21 and 24 were also more frequently associated with high EXO5 expression, whereas signature 16 occurred more frequently in low EXO5-expressing patients (Figure S1B). The link of signature 13 to base excision repair (BER) suggests diverting strand breaks from defective HR to the BER pathway. In sum, high EXO5 levels may contribute to mutations by shunting repair of replication errors away from error-free HR and into error-prone pathways.
EXO5 Suppresses Genomic Instability during DNA Replication by Promoting Fork Restart
As higher EXO5 mRNA levels may impact cancer prognosis and mutation frequency, we tested EXO5 roles in DNA replication stress responses. Using DNA fiber assays we measured fork progression in cells with and without EXO5 depletion (Roy et al., 2018; Schlacher et al., 2011; Singh et al., 2018). EXO5 depletion reduces fork progression and replication recovery relative to control cells during and after fork stalling with either hydroxyurea (HU) or cisplatin in HeLa and HEK293 cells (Figures 2A–2C, S2A, and S2B). While EXO5 knockdown (KD) does not significantly change fork termination events or fork speed in unchallenged cells, it significantly increases stalled replication forks while decreasing restarted forks (Figures S2C and S2D). EXO5 knockout (KO) in LNCaP prostate cancer cells (Ali et al., 2020) also resulted in slower replication recovery after HU-induced stalling (Figures 2D and 2E) and higher sensitivity to cisplatin-induced cell death (Figure 2F). The collective data show that EXO5 is required for efficient restart of stalled replication forks.
Figure 2. EXO5 Is Required for Replication Stress Recovery and ICL Repair.

(A) Western blots showing EXO5 KD using a pool of 4 siRNA (pooled), and two different siRNA (1, 2) sequences in HeLa and HEK293 cells. (B) DNA fiber analysis of IdU track lengths of CldU+IdU containing fibers after hydroxyurea (HU) treatment; top, experimental scheme. (C) IdU tract length of CldU+IdU tracts after cisplatin treatment in HeLa cells. (D) Western blot of CRISPR EXO5 KO in LNCaP cells. (E) DNA fiber analysis in LNCaP cells as described in (B) (**** p<0.0001, Mann-Whitney test). (F) Clonogenic cell survival of LNCaP cells with and without EXO5 KO after drug treatment (* p<0.05, ** p<0.01, student t test). (G–I) Immunofluorescence staining with anti-γH2AX antibody and (J–L) with anti-RPA1 antibody after removing noted drugs. Each experiment, 50 cells were analyzed. Data are represented as mean ± SEM (* p<0.05, ** p<0.01). See Figure S2.
Unresolved replication stress leads to genomic instability (Zeman and Cimprich, 2014). Consistently, we observed increased SCE with EXO5 depletion (Figure S2E), and treatment with cisplatin or camptothecin (CPT), a topoisomerase I inhibitor, further enhanced SCE ~ 3-fold and radial formation ~ 3–5 fold (Figures S2F–S2I), similar to what is observed in BLM deficient cells (Hemphill et al., 2009). Moreover, drug treatment delayed γ-H2AX foci disappearance in EXO5-depleted cells (Figures 2G–2I and S2J), supporting an EXO5 role in suppressing DNA damage arising from stalled replication forks.
EXO5 Binds Replication Stress Proteins RPA and BLM
We reasoned that if EXO5 acts in replication fork processing, it may affect replication stress responders such as RPA. In EXO5-depleted cells, the frequency of residual RPA foci was higher after drug treatment (Figures 2J–2L and S2K) with no effect on cell cycle progression, suggesting higher RPA foci are not due to cells accumulating in S-phase (Figure S1C). Concomitantly, BrdU foci detected under non-denaturing conditions, which indicate nascent ssDNA formation, were higher in EXO5-depleted cells (Figures S2L–S2N), suggesting EXO5 resection reduces ssDNA accumulation.
To identify EXO5 partners in replication fork processing, we employed mass-spectrometric analysis of immunoprecipitated EXO5 complexes which detected BLM and RPA1 (RPA70 subunit) among the top 10 hits (Table S1). The EXO5 interactions with BLM and RPA1 were confirmed by immunoprecipitation, with BLM or RPA1, and by co-localization of GFP-EXO5 foci with BLM and RPA1 in drug-treated cells (Figures 3A–3D). Unexpectedly, BLM depletion reduced EXO5 foci following cisplatin treatment (Figure 3E). We reasoned that significant EXO5 foci formed after 12 hrs because these drug-treated cells accumulated in S/G2 phase (Figure S1C). These data suggest damage-induced EXO5 foci formation depends on BLM, and EXO5 cooperates with BLM and RPA1 during replication stress response to process stalled forks.
Figure 3. EXO5 Forms a Complex with BLM and RPA.

(A) Western blots of Flag immunoprecipitation (IP) with noted antibodies. Anti-HA detected overexpressed HA-Flag-EXO5. Western blots of (B) BLM and (C) RPA1 IP with anti-HA antibody to detect IP HA-Flag-EXO5. (D) Colocalization of GFP-EXO5 with RPA1 or BLM foci after noted drug treatment. Scale bar: 5 μm. (E) EXO5 foci per cell measured by overexpressing GFP-EXO5 in BLM-depleted cells before drug treatment (representative EXO5 foci in inset, nuclei stained with blue DAPI). Each experiment, 50 cells were analyzed. Data are represented as mean ± SEM (** p<0.01). See Table S1.
EXO5 Interweaves Fold and Active Channel with [4Fe-4S] Cluster Region
To elucidate EXO5 function as a structure-specific nuclease, we solved X-ray crystal structures of N-terminally truncated human EXO5 (Δ30-EXO5) at 2.5 Å (DNA- and metal-free) and three DNA-bound complexes at 2.71–2.88 Å resolution with and without metal ion bound (Table 1). EXO5-DNA Complex 1 is metal-free. Complex 2 is EXO5 with Na+1 ion and Sm3+ (inactive) to capture a metal-bound substrate complex. Complex 3 includes phosphor-mimetic mutant T88E-EXO5 and Mg2+ for DNA interaction insights for ATR- and BLM-dependent experiments (see Figure 6). All EXO5-DNA complexes are substrate structures with nucleotides (nts) on either side of the scissile phosphate.
Table 1.
X-ray data collection and refinement statistics of human EXO5 structures
| Δ30-EXO5 DNA-free Metal-free |
SAD Peak (Fe) |
Δ30-EXO5 ssDNA-1 (metal-free) Complex 1 |
Δ30-EXO5 ssDNA-2 (Sm/Na) Complex 2 |
T88E-EXO5 ssDNA-1 (Mg) Complex 3 |
|
|---|---|---|---|---|---|
| Data collection | ALS BL12.3.1 | SSRL BL12–2 | ALS BL8.3.1 | ALS BL8.3.1 | ALS BL8.3.1 |
| Wavelength (Å) | 1.1158 | 1.7369 | 1.1159 | 1.1161 | 1.1158 |
| Space group | P22121 | P22121 | P22121 | P22121 | P22121 |
| Cell dimensions | |||||
| a, b, c (Å) | 50.82, 83.84, 95.96 |
49.81, 83.82, 95.99 |
51.17, 83.98, 96.35 |
51.07, 83.66, 96.15 |
50.51, 84.72, 95.03 |
| α, β, γ (°) | 90, 90, 90, | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å)a | 47.98 – 2.50 (2.60 – 2.50) |
49.68 – 2.91 (3.09 – 2.91) |
48.18 – 2.88 (2.98 – 2.88) |
45.10 – 2.71 (2.84 – 2.71) |
47.52 – 2.85 (3.00 – 2.85) |
| Observationsa | 94088 (10495) |
165338 (26639) |
127096 (18250) |
151991 (19957) |
129975 (19268) |
| Unique observationa | 14791 (1617) |
9185 (1426) |
9874 (958) |
11691 (1516) |
9998 (1423) |
| R pim a | 0.104 (0.531) | 0.040 (0.223) | 0.126 (0.585) | 0.090 (0.870) | 0.103 (0.655) |
| Mean I/σI a | 8.2 (1.6) | 17.2 (4.0) | 6.4 (1.6) | 8.9 (1.1) | 8.1 (1.4) |
| Completeness (%)a | 100 (100) | 99.3 (98.4) | 100 (100) | 99.7 (99.7) | 99.7 (99.6) |
| Multiplicitya | 6.4 (6.5) | 18 (18.7) | 12.8 (13) | 13 (13.2) | 13 (13.5) |
| CC(1/2)a | 0.988 (0.570) | 0.998 (0.969) | 0.986 (0.620) | 0.995 (0.394) | 0.992 (0.605) |
| Refinement | |||||
| Rwork/Rfree (%) | 18.20/22.7 | 20.47/25.90 | 19.37/25.34 | 20.26/25.00 | |
| No. of atoms | |||||
| Protein | 2230 | 2095 | 2115 | 2076 | |
| DNA | – | 184 | 150 | 60 | |
| Water | 89 | 41 | 32 | 25 | |
| rmsd bond length (Å) | 0.003 | 0.002 | 0.003 | 0.002 | |
| rmsd Bond angles (°) | 0.530 | 0.490 | 0.610 | 0.440 | |
| Average B-factor | 41.62 | 40.46 | 60.41 | 54.25 | |
| Protein | 41.61 | 37.36 | 58.37 | 52.53 | |
| Solvent | 38.26 | 35.98 | 52.09 | 47.23 | |
| DNA | – | 77.09 | 88.98 | 117.68 | |
| Ramachandran (%) | |||||
| Favored | 97.44 | 96.09 | 96.14 | 98.03 | |
| Allowed | 2.20 | 3.91 | 3.47 | 1.97 | |
| Outliers | 0.37 | 0 | 0.39 | 0 | |
| PDB code | 7LW7 | 7LW8 | 7LW9 | 7LWA |
Values in parentheses are the statistics for the highest resolution shell of data.
Figure 6. ATR Phosphorylation of EXO5 T88 Promotes BLM Interaction and Damage Foci Formation.

(A) Multiple EXO5 sequence alignment shows invariant -TQ- ATR recognition motif. (B) Detection of EXO5 phosphorylation and BLM interaction by western blot of Flag IP with anti-SQ/TQ antibody and BLM antibody in cells treated with or without HU or ATR inhibitor VE-821. (C, E) Quantitative analysis of EXO5 (GFP-EXO5 expression) and BLM foci formation in HeLa cells treated with HU or cisplatin with or without VE-821. (D) Western blotting with phospho-CHK1 antibody detects ATR inhibition by VE-821. (F) Interaction of HA-Flag-EXO5 variants with BLM by IP without drug treatment. (G) Phosphorylation of EXO5 variants and BLM interaction after drug treatment. Anti-HA antibody detected HA-Flag-EXO5 expression in (B), (F) and (G). (H) Co-localization of drug-induced DNA damage foci for EXO5 variants with BLM. Scale bar: 10 μm. (I) Quantitative foci analysis of EXO5 variants with HU treatment. (J) Binding affinity between BLMcat and EXO5 (WT and T88E) measured by MST. Atto-647-labeled BLMcat was titrated with EXO5 variants in triplicates. MST fluorescence changes were normalized as fraction bound. (K) EXO5-T88E-DNA Complex 3 (DNA, orange sticks) bound to Mg2+. (L) Electrostatic surface view of Complex 3 showing local negative charge near DNA binding path (disordered crossover-helix, blue dash-lines). (M-O) Clonogenic survival assays for EXO5-depleted cells with WT or EXO5 T88 mutant’s complementation after HU, cisplatin, or CPT treatment. For foci, 50 cells were analyzed for each experiment. Foci and cell survival data are averaged from three independent experiments (** p<0.01). See Figure S6.
The EXO5 structure forms an integrated single domain (69–358 aa, overall dimensions 57 × 45 × 42 Å) including 11 α-helices and 5 β-strands that form an oval-shaped disc (Figures 4A, S3A, and S3B). Two helical bundles sandwich a central β-sheet. One helical region contains an [4Fe-4S] cluster formed by N- and C-terminal helical elements (Figures 4B and 4C), protected primarily by conserved hydrophobic residues and stabilized by a E93 to R330 salt bridge. An E93L substitution formed inclusion bodies, showing the protein is sensitive to the [4Fe-4S] cluster fold, consistent with biochemical results showing that the C343A/C346A mutant loses nuclease activity (Sparks et al., 2012). The central 5-strand anti-parallel β-sheet is separated near the center, such that three strands (β3 –β5) continue in the twisted sheet whereas the two N-terminal strands (β1 and β2) bend ~80° to take the hairpin through the opposite face and pack between the two helical bundles (Figure S3B). The two longest helices (α5 and α6) cross the domain top to bottom and side to side while the bent β-hairpin provides a front to back connection, topologically connecting EXO5 into a single folded domain.
Figure 4. EXO5 Crystal Structures Reveal Selection of ssDNA Ends.

(A) Color-coded diagram of indicated EXO5 domains. (B) DNA-free and metal-free EXO5 crystal structure exhibits a [4Fe-4S] cluster region and a splayed β-sheet nuclease fold with the active site. Disordered connections to crossover-helix (blue dash-lines) and the [4Fe-4S] cluster (ball-and-sticks, Fe: brown; S: yellow). (C) [4Fe-4S] cluster region connects N-terminal C92 with C-terminal cysteines as stabilized by the α10 helix (Hydrogen-bonds shown in magenta-dashed lines). (D) Active site D182 and E196 interact with α4 crossover-helix in the absence of metal ion and ssDNA. (E) Overlay of EXO5 DNA-free and DNA Complex 1 structures. Crossover-helix (yellow) in DNA-free structure moves outward and is disordered when ssDNA (cyan, ribbon) is bound. (F) EXO5-DNA Complex 1 structure (DNA, cyan ball-and-sticks) narrow entrance and active channel allows only ssDNA access with base-stacking (R344, W91, and Y80; yellow sticks). (G) EXO5-DNA Complex 2 structure. DNA (ball-and-sticks) colored by B-factor (blue most rigid to white to red most flexible. (H) EXO5-DNA Complex 2 structure electrostatic surface showing clustered positive charged residues in the [4Fe-4S] cluster region (omitted DNA structure shown as yellow trace). (I) EXO5-DNA Complex 2 structure shows active site Sm(III) ion coordination. Sodium ion binds D182 and E165. (J) EXO5 model binding to DNA fork. 5’-end threads via [4Fe-4S] cluster region into active channel. Three base-stackings plus phosphate backbone interactions stabilize ssDNA. Y221 and T84 anchor ssDNA to position the downstream scissile phosphate in the active site. See Figures S3, S4, and Table 1.
Over the active site center is a α4-helix (120–129 aa) and a flexible loop (130–135 aa), only seen in the DNA-free structure: these elements cross over the channel to link into a disordered connection over the active channel (107–119 aa) collectively defined here as the “crossover-helix” (Figures 4B and 4D). Structure similarity searches identify EXO5 as a member of PD-(D/E)XK nuclease superfamily (with a αβββαβ topology) (Steczkiewicz et al., 2012). The conserved active site D-EXK motif is blocked by the α4-helix in the DNA-free structure. To open space for this crossover-helix, the EXO5 four-helix bundle is in a different position from the Dna2 structure (Figure S3C). Remarkably, the relative locations of the [4Fe-4S] cluster and D-EXK motif are well conserved with Dna2 (Zhou et al., 2015), CRISPR-associated Cas4 (Lemak et al., 2013), and AddB nucleases (Krajewski et al., 2014), despite low overall sequence identity (< 20%, RMSD = 3.3~4.5 Å), supporting their functional importance.
Dynamic Crossover-Helix Opens to Thread a ssDNA End
The crossover-helix is absent in electron density maps of the DNA complexes, suggesting that it moves outward as an open flexibly tethered gateway over the active site channel (Figure 4E). In the DNA-free structure, conserved R124 and H121 of the α4 crossover-helix interacted with D182 and E196 active site residues and formed a H-bonding network with Y221, Q217, and K198 (Figure 4D). To see if the crossover-helix that undergoes an order-to-disorder transition is key for EXO5 function, we tested H121A and R124A mutants, which nearly abolished nuclease activity (Figure S3D) and decreased DNA binding affinity ~2- and 3-fold, respectively (Figure S3E), suggesting a key crossover-helix role for transient EXO5 ssDNA interactions.
The crossover-helix order-to-disorder transition (~30 aa) upon DNA binding recalls the reverse disorder-to-order transition in FEN1 (~45 aa) induced by DNA binding (Figures S4A–S4C) (Tsutakawa et al., 2011) that selects for 5’ ssDNA flaps and a free 5’ terminus while allowing room for threading dsDNA or loops. We therefore tested if gapped and fork substrates can be processed by EXO5. EXO5 cuts any free-end DNA substrates, including replication fork-mimics or D-loops, but not a gapped substrate, which has duplex DNA on both ends (Figure S4D). Thus, the open crossover-helix and/or narrow channel near the [4Fe-4S] cluster region stringently select for a free ssDNA terminus.
DNA Binding Restricts Polarity and Activity to ssDNA Substrates
The EXO5-DNA complex structures reveal ssDNA bound in the narrow curved channel, primarily via sequence-independent, sugar-phosphate backbone interactions, and aromatic stacking to the bases (Figure 4F). At either end of the binding groove, the ssDNA nts −4, +3, and +5 stack against side chains of Y80, W91, and R344, respectively. The 3’-ssDNA was positioned close to the [4Fe-4S] cluster and polarity was set at the active site by electrostatic register and steric molding to the phosphodiesters. These geometric requirements disfavor ssDNA from binding in the opposite polarity. Furthermore, this specificity region (nts +1, −1, −2) is most strongly bound, based on low B-factors (Figure 4G). The C2’-endo sugar puckers observed are energetically unfavorable for RNA, which favors C3’-endo sugar pucker (Figures S4E and S4F), explaining reported specificity against RNA (Burgers et al. 2010).
Both narrowing the entranceway and blocking base pairing allow EXO5 to discriminate against dsDNA. The channel entrance is narrowed to a minimum diameter of ~10 Å by the [4Fe-4S] cluster (Figure 4H) and a triple arginine loop from R200 to R202 (Figure 4F). The positive arginine patch interacts with ssDNA phosphodiesters and positions the bases down into the groove. We tested the importance of conserved Q210, which forms a wedge between nts +2 and +3 in the metal free DNA Complex 1; but shifts to brace R200 in the Sm3+-coordinated Complex 2. We found that Q210A significantly reduced nuclease activity and caused a ~4-fold decrease in DNA binding affinity (Figures S3D and S3E), showing this channel acts in proper substrate loading.
Central Metal-Coordinated D-EXK Active Site in the DNA-Binding Channel
Analysis of the different DNA complexes reveal active site metal ion coordination with uncleaved substrates at the scissile phosphate. Notably, bound metals shifted ssDNA substrate ~2.3 Å towards the metal ion and in position for incision with a ~45° bend at the scissile phosphate cut site (Figures 4I and S4F). In Complex 2, one Sm3+ ion is coordinated to D182, E196, the carbonyl of L197, and the scissile phosphate oxygen; one Na+ is coordinated to D182 and E165. E165A retained nuclease activity, while D182A had no detectable activity (Figure S3D), suggesting the Na binding site is not important for activity. The Sm3+ ion site corresponds to one of two Ca2+ sites in Dna2 (Zhou et al., 2015; Yang, 2011), representing a single catalytic metal binding site. Yet, based on other nuclease structures, a second metal site likely exists. Tests showed nuclease activity for Mn2+, Fe2+ and Mg2+, but not Zn2+, Ca2+ or Sm3+ ions (Figure S4G). The strong nuclease activity with Fe2+ hints that Fe2+ may allosterically activate the nuclease activity, possibly via crossover-helix residue H121.
At the channel bottom, conserved K198 (part of the D-EXK motif) and Q217 stabilize the scissile phosphate, while Y221 and T84 stabilize the upstream phosphate (Figures 4G and S4H). Supporting this point, Y221F showed no nuclease activity, indicating that substrate positioning by Y221 is critical for activity. Interestingly, Y221F, but not the metal-binding D182A, showed ~ 6-fold lower DNA binding affinity (Figure S3E), reinforcing its key role in substrate binding. These results suggest that EXO5 stringently positions the phosphodiester to doubly ensure ssDNA specificity and efficient hydrolysis.
DNA Product is a 5 nt 5’ Overhang
The channel geometry and rigid positioning near the scissile phosphate for catalysis implies a minimal number of ssDNA nts is needed to reach the active site. We therefore varied the 5’ overhang length in a nuclease assay. EXO5 incised substrates longer than 7 nts but not 5 nts or shorter (Figure S4I). Based on Complex 1, it takes 6 nts to go 1 nt past the scissile phosphate, and 6 nts give the length needed to reach Y221, the anchoring residue (Figure 4F), consistent with Complex 2 structure (Figures 4G and S4J). Anchoring DNA binding on phosphate groups points the bases upward to interact with and displace the regulatory crossover-helix (Figures 4G and S4A). The EXO5 product will always be a minimum 5 nt 5’ overhang from the dsDNA or fork, which has implications for resection and the next enzyme in the pathway (Figure 4J). Together the EXO5-DNA structures and mutational analyses unveil a metal-ion-dependent catalysis that requires crossover-helix opening and the [4Fe-4S] cluster region to specifically thread ssDNA.
EXO5 is Epistatic to BLM Helicase for Fork Restart
To test if EXO5 and BLM act in the same pathway for fork restart, we depleted either EXO5 or BLM or both (Figure S5A); the double-KD did not enhance cell death (Figures 5A–5C and S5F–S5G) nor did it further reduce fork recovery (Figures 5D and 5E) after drug treatment more than the single KD. These data suggest that the two proteins are epistatic for fork processing. Measuring fork protection of nascent DNA at stalled forks by DNA fiber assays (Schlacher et al., 2011) further revealed that either KD of EXO5, BLM, or both suppresses intrinsic nascent fork degradation in HeLa cells (Figure S5H), showing that EXO5 and BLM process stalled forks.
Figure 5. EXO5 and BLM Act in the Same DNA Replication Stress Pathway.

(A-C) Clonogenic cell survival assays detect effects of EXO5 and/or BLM depletion on drug sensitivity of HeLa cells against HU, cisplatin, and CPT. Data presented as interpolated from three independent experiments (* p<0.05, ** p<0.01, student t test) (D-E) DNA fiber analysis determining replication fork recovery in EXO5 and/or BLM depleted cells with HU or cisplatin treatment (****p<0.0001, Mann-Whitney test). (F-I) Clonogenic cell survival to detect effect of depletion of MUS81, DNA2, MRE11, and EXO1 depletion and with or without EXO5 KD after HU treatment. (J) Western blot with noted antibodies show efficient KD. (K) DNA fiber analysis to determine DNA replication fork recovery in HeLa cell depleted for proteins as indicated with HU treatment. (L) The rhq1Δ exo5Δ double mutant resembles growth rate in the single mutants by tetrad dissection assay. (M) EXO5 end-resection on replication fork mimic substrate with BLMcat and RPA70NAB. Reaction mixture incubated at 30 °C for 30 min. (Cy5-labeled at 3’-end, star). See Figure S5.
We tested if EXO5 has distinct roles in relation to other known fork processing nucleases by testing their epistasis (Figures 5F–5I and S5B–S5E). Upon HU treatment, EXO5 depletion alone induced more cell death than MUS81 depletion, while the combination did not further exacerbate sensitivity (Figure 5F). By contrast, combined EXO5/DNA2, EXO5/MRE11 or EXO5/EXO1 KD further sensitized cells relative to either KD alone (Figures 5G–5I and S5I–S5K), supporting distinct roles for EXO5 and these nucleases. Yet reduced fork recovery in HU treated EXO5- or MRE11- or combined EXO5/MRE11-depleted cells was similar (Figure 5K). We also tested replication fork remodeler SMARCAL1 (Couch et al., 2013) in relation to EXO5. We found that simultaneous KD of SMARCAL1 and EXO5 had similar effects on replication fork restart (Figures 5J and 5K), suggesting they act in the same pathway at stalled forks.
If EXO5-BLM acts in replication restart fundamental to cell survival, this activity may be evolutionary conserved. We performed exo5 genetic epistasis analyses in fission yeast Schizosaccharomyces pombe, (Matsuyama et al., 2006; Sparks et al., 2019; Sparks et al., 2012). Genetic interaction analyses of exo5Δ with fan1Δ (Fanconi anemia-associated nuclease 1), pso2Δ (the human SNM1 nuclease homolog), and rad13Δ (the human XPG nuclease ortholog) upon cisplatin treatment indicated that exo5 functions primarily in a pathway requiring pso2 and rad13, but independent of fan1 (Figure S5L). Furthermore, the exo5Δ and rhq1Δ (the human RecQ and BLM helicase homolog) strains were sensitive to cisplatin treatment and the double-KO did not have an additive effect (Figure S5M). Similar results were seen in tetrad dissection experiments, showing rhq1Δ exo5Δ double mutant did not significantly impact the growth rate compared to single mutants (Figure 5L). Thus, a genetic epistatic relationship between EXO5 and BLM is conserved from fission yeast to humans.
Given the EXO5, BLM, and RPA relationship in cells, we did biochemical nuclease assays with and without BLM and RPA variants. Purified BLMcat (BLM catalytic domain) unwound duplex DNA for EXO5 5’ end-resection with lower efficiency (Figure S5N, lanes 3–4). Adding RPA70NAB (RPA1 DNA binding domain) alone, but not RPA70N or RPA32C (domains known for protein interactions) (Pretto et al., 2010), increased end resection efficiency; yet, EXO5 could not continue processing the duplex without unwinding (Figure S5N, lanes 3 and 6–8). Only adding RPA70NAB with BLMcat facilitated dsDNA unwinding and end-resection (Figure S5N, lanes 10–12). Addition of ATP partially inhibited EXO5 activity (Figure S5N, lanes 2–3), and EXO5 could not efficiently process substrate with full-length RPA, which occupies ~28 nts leaving insufficient space for EXO5 to load and process (Figure S5N, lanes 5 and 9). Next, we tested if EXO5 can process replication fork mimics. EXO5 alone yielded an end-resected ~35 nts product leaving ~5 nts from DNA duplex (Figure 5M, lanes 2 and 3). With BLMcat, it inefficiently unwound the duplex (Figure 5M, lane 4, and S5O). Together with RPA70NAB, the longer end-resection product accumulated, suggesting RPA70NAB was bound to ssDNA, thereby limiting resection by EXO5 (Figure 5M, lane 5). Notably, when both BLMcat and RPA70NAB were present, the replication fork mimic was most efficiently unwound and resected by EXO5 (Figure 5M, lane 6, and S5O), suggesting that BLMcat, RPA70NAB, and EXO5 work synergistically for fork processing. We found similar results using a D-loop substrate (Figure S5P). These collective findings show that EXO5 is epistatic to BLM at stalled replication forks, and that its nuclease activity is enhanced by BLM and RPA70.
ATR-Dependent EXO5 Phosphorylation Promotes BLM Interaction and Foci Formation
We tested if EXO5 is regulated by ATR, the key replication stress response regulator (Saldivar et al., 2017). We identified evolutionary-conserved ATR consensus phosphorylation motifs (SQ/TQ), but only T88Q89 was conserved in mammals (Figures 6A and S6A). Interestingly, we found BLM interaction and T88 phosphorylation were lost in the presence of ATR inhibitor VE-821 with or without HU treatment (Figure 6B). Treatment with ATR inhibitors reduced EXO5 and BLM foci formation after drug treatment (Figures 6C, 6E, S6D, and S6E) and decreased CHK1 phosphorylation (Figures 6D and S6F), confirming the ATR inhibition (Davies et al., 2004; Liu et al., 2000). To validate ATR-mediated EXO5 phosphorylation on EXO5-BLM interaction and EXO5 foci formation, we generated an EXO5 phospho-dead T88A mutant, which disrupted the EXO5-BLM interaction and diminished EXO5 foci formation (Figures 6F–6I). These loss-of-function defects were rescued by a phospho-mimetic T88E mutant (Figures 6F, 6H, 6I).
We assessed T88E-EXO5 in vitro binding to BLM by MicroScale Thermophoresis (MST) (Jerabek-Willemsen et al., 2011). T88E increased binding affinity to BLMcat ~5-fold (from 53 μM to 12 μM) (Figure 6J). It decreased DNA binding affinity ~10-fold (from 157 nM to 1474 nM) compared to WT (Figure S3E) and reduced nuclease activity ~20% (Figure S6C). Addition of BLM and RPA70NAB did not stimulate T88E nuclease activity compared to WT-EXO5 (Figure S6C). EXO5-DNA binding analysis by surface plasmon resonance reveals T88E decreased the association rate (ka) ~10-fold without affecting the dissociate rate (kd) (Figure S6B). Thus, T88E or T88 phosphorylation increases the local negative charge to enable regulation of DNA binding to the EXO5 active site channel (Figures 6K and 6L), consistent with the T88 position on α2-helix adjacent to the DNA binding path.
Complementation of phospho-mimetic mutant T88E-EXO5 mediated the co-localization of damage-induced BLM foci (Figures 6H and 6I) and rescued EXO5-depleted cell survival after drug treatment (Figures 6M–6O, S6G, and S6H). Similarly, the reduced replication recovery in EXO5 depleted cells was rescued by T88E-EXO5 upon HU treatment (Figure S6M). The elevated residual γ-H2AX foci in EXO5-depleted cells was rescued by T88E but not by the T88A mutant (Figures S6I–S6L). Expression of EXO5 nuclease-dead mutant D182A failed to rescue drug induced γ-H2AX foci formation, the fork recovery defect, or cell survival (Figures S6H, S6I, and S6M), confirming EXO5 nuclease activity is critical for stalled fork processing.
EXO5 Depletion Rescues FANCA Deficiency for Fork Protection and Survival
As BLM loss can rescue some FA defects in cells challenged with replication stress or ICL (Moder et al., 2017), we tested if BLM may act with EXO5 in balancing fork processing with fork protection. EXO5 depletion rescued survival in FANCA- (Figure 7A), but not FANCD2-depleted (Figure 7B) cells post-HU treatment. In FANCA KD HeLa cells, we observed degradation of tracts nascently-labeled before stalling, indicating a fork protection defect that was suppressed by EXO5 depletion (Figure 7C). Furthermore, BLM KD rescued FANCA-depleted cell survival resembling EXO5 KD (Figure 7D), however, they differed from FANCD2-depleted cell survival (Figure 7E). Depletion of EXO5 or BLM had no additive effect on cell killing in FANCD2-deficient cells, consistent with FANCD2 functions independent of FA core complex (Chaudhury et al., 2013; Pichierri et al., 2004). These results suggest that EXO5-BLM complex fork processing is restricted by FANCA fork protection, so co-depletion protects from lethality.
Figure 7. EXO5 Knockdown Rescues Fanconi-anemia FANCA Deficient Cell Survival and Fork Degradation.

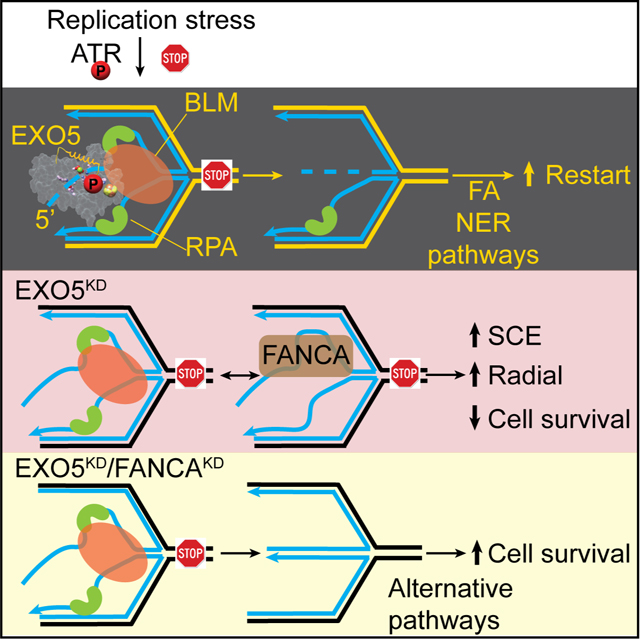
(A–B) Clonogenic survival assays for EXO5 and FANCA or FANCD2-deficient cells after HU treatment. Western blots show KD efficiency in lower panel. Anti-EXO5 and anti-FANCA or anti-FANCD2 antibodies detected noted protein expression (* p<0.05, ** p<0.01, student t test). (C) DNA fiber analysis to determine fork protection showing nascent CldU- tract lengths (**** p<0.0001, Mann-Whitney test). (D–E) Clonogenic survival assays for BLM and FANCA or FANCD2-deficient cells after HU treatment. Western blots show KD efficiency in lower panel. Anti-BLM and anti-FANCA or anti-FANCD2 antibodies detected noted protein expression. (F) Integrated model: EXO5 is phosphorylated by ATR during replication stress and recruited by BLM helicase to RPA-bound stalled forks. 5’-ssDNA from the [4Fe-4S] cluster region threads under EXO5 crossover-helix. RPA (RPA70NAB) guides EXO5 5’–3’ resection while BLM unwinds the reversed fork for restart with FA pathway connections. See Figure S7.
Hazard Ratios Link High EXO5 and FA Gene Expression to Poor Cancer Prognosis
To test relationships between cell proliferation and EXO5-BLM or FA activation, we did a gene coexpression analysis between proliferation-associated MYBL2 and 41 DNA repair genes; the analysis revealed mostly positive correlations for genes related to the FA and BLM interactomes (Figure S7A, top). Analysis of hazard ratios in ACC patients further indicated poor survival for overexpression of genes in these pathways (Figure S7A, bottom). Finally, EXO5 and BLM mRNA levels were positively correlated in 17/33 tumor types (Figure S7B), suggesting that hyperproliferation is linked to impaired survival via unbalanced stalled fork protection, processing, repair, and restart by dedicated DNA repair complexes, which include EXO5.
DISCUSSION
Our findings uncover EXO5 as a structure-specific nuclease that upon ATR-mediated phosphorylation forms a complex with BLM and RPA to resect 5’ ends and restart stalled forks, as balanced by FA fork protection (Figure 7F). Persistent replication and proliferative stress (Macheret and Halazonetis, 2015) increase cell dependence on regulating nucleases for fork restart, and increased SCE and radials in EXO5-depleted cells after drug treatment mirror Bloom’s syndrome cells (German et al., 1974). To our knowledge, EXO5 is the only nuclease reported to affect frequency of both SCE and radial chromosome formation similar to BLM-deficient cells.
RPA70 binds EXO5 to enforce 5’−3’ directionality (Sparks et al., 2012) and stimulates BLM helicase activity via the interaction with BLM N-ter (1–447 aa) (Doherty et al., 2005). BLMcat (640–1290 aa) unwound dsDNA with RPA70NAB and EXO5, suggesting EXO5 binds both BLMcat and RPA70 as a functional complex. Multiple mechanisms enforce EXO5 ssDNA binding, including a crossover-helix regulating access to the ssDNA binding path, like the p62 helix controlling DNA access to XPD helicase in TFIIH (transcription-factor IIH) to regulate a switch between transcription and nucleotide excision repair (Yan et al., 2019; Tsutakawa et al., 2020b). So, EXO5’s crossover-helix order-to-disorder transition upon ssDNA binding may be a regulatory entry point where the ~30 residue disordered region may tolerate bulky damaged bases in contrast to the ordered Dna2 crossover loop (Zhou et al., 2015; Daley et al., 2020). L151P prostate cancer mutation—located in α5-helix adjacent to the active site—would cap the α5-helix, impacting protein folding and active site function and accounting for nuclear localization and nuclease activity defects (Ali et al., 2019).
Interestingly, FAN1 prefers a short 5’ phosphorylated flap substrate with a complementary 3’ flap (Wang et al., 2014), resembling EXO5-BLM product. Strict active site conservation relative to the [4Fe-4S] cluster between EXO5 and other [4Fe-4S]-containing ssDNA nucleases suggests its conserved regulatory role among these nucleases (Fuss et al., 2015; Lemak et al., 2013; Pokharel and Campbell, 2012; Yeeles et al., 2009). Notably EXO5-BLM complex, like Dna2, places the [4Fe-4S] cluster region adjacent the ss-dsDNA junction, where it may act in conformational control and signaling (Fuss et al., 2015). Well-stacked ssDNA bases buttressed against the [4Fe-4S] helical bundle suggest a possible entry and exit point for charge transfer (Sontz et al., 2012).
Our cell survival data suggest EXO5 has distinct roles from MRE11, DNA2, EXO1, and MUS81 nucleases during HU treatment. Biochemically, a reversed fork is a preferred EXO5 substrate while gapped, flap, blunt-end, or nicked Holliday-junction substrates are preferred for MRE11, DNA2, FEN1, EXO1, and MUS81 (Cejka, 2015; Cotta-Ramusino et al., 2005; Hu et al., 2012; Pepe and West, 2014; Schlacher et al., 2011; Taylor and McGowan, 2008; Tsutakawa et al., 2011). EXO5-depletion results in accumulation of nascent ssDNA, γ-H2AX, RPA, and increased SCE: this resembles cell phenotypes of ATR inhibition (Couch et al., 2013) explaining why ATR with its direct role in DNA end-resection specifically regulates EXO5, but not other fork-processing nucleases (Dibitetto et al., 2020). ATR phosphorylates SMARCAL1 to regulate stalled fork regression activity that prevents aberrant fork processing by SLX4- and CtIP-dependent nucleases (Couch et al., 2013). Our findings suggest ATR may reduce fork collapse during replication stress by phosphorylating both SMARCAL1 to limit its fork reversal activity and EXO5 to promote fork restart.
Yet, replication fork restart and fork protection are balanced. Thus depletion of EXO5 or BLM can rescue FANCA-deficient cells supporting and extending the finding that BLM depletion rescues FANCC-deficient cells (Moder et al., 2017). Indeed, hazard ratios correlate high expression of EXO5, FA genes and BLM to poor cancer patient survival. These TCGA data suggest EXO5 with BLM participate in tumor cell proliferative replication stress responses along with FA proteins, and tumor hyperproliferation may cause high mutation rates from rebalancing stalled fork protection, processing, repair, and restart.
Our overall findings advance paradigms for ATR-mediated replication stress responses and BLM helicase partners. EXO5-BLM balances fork protection by FANCA and acts as an anti-recombinase for fork restart to allow replication continuation. Thus, EXO5 inhibitors merit investigation for cancer biology and as a possible FA gene specific strategy to rescue cell survival that may reduce rather than increase cancer risk in FANCA patients.
Limitations of the Study
Our cell assays were done in HeLa, HEK293, and LNCaP cell lines. EXO5 may act differently in other cell types and cancer tissues due to differences in gene expression and protein levels. High mutation loads seen with high EXO5 expression may be due to excessive fork restart or high cancer cell proliferation. Here we define EXO5 replication fork restart functions, but not its activities at other DNA damage sites. EXO5 structures with DNA product and trapped cross-over helix as well as BLM interactions will complement this work. Additional biochemical assays with post-translational modifications and other partner interactions present in cells are required for further investigations. E.g. for cisplatin repair, EXO5 may act with genetically-implicated partners XPG and SNM1 (Tsutakawa et al., 2020a; Baddock et al., 2020).
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John A. Tainer (JTainer@mdanderson.org).
Materials availability
The plasmids and yeast strains generated from this study will be available upon material transfer agreement (MTA) completion. All reagents including antibodies, siRNA, and plasmids can be found in KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-EXO5 Rabbit | LifeSpan BioSciences | Cat#LS-C410042/105176 |
| Anti-BLM Rabbit | Abcam | Cat#Ab2179 |
| Anti-RPA1 Rabbit | Abcam | Cat#Ab79398 |
| Anti γH2AX antibody Mouse | EMD-Millipore | Cat#05–636 |
| Anti-CldU antibody Rat | BD Biosciences | Cat#347580 |
| Anti-IdU antibody/Anti-BrdU antibody Mouse | Novus Bio | Cat#NB-500–169 |
| Anti-HA-HRP conjugated antibody | Roche | Cat#12 013 819 001 |
| Anti-Rabbit-HRP | Thermo Scientific | Cat#32460 |
| Anti-Rabbit-Alexa fluor 488 | Life Technologies | Cat#A11008 |
| Anti-Mouse-Alexa fluor 488 | Life Technologies | Cat#A11001 |
| Anti-Rat-Alexa fluor 488 | Life Technologies | Cat#A11006 |
| Anti-GAPDH antibody | Cell Signaling | Cat# 8884S |
| Anti-beta actin antibody | Santa Cruz Biotechnology | Cat# Sc-8432 |
| Anti-Mre11 antibody | Bethyl Laboratories | Cat# A303–998A |
| Anti-MUS81 antibody | Abcam | Cat# ab14387 |
| Anti-DNA2 antibody | Abcam | Cat# ab197283 |
| Anti-EXO1 antibody | Abcam | Cat# ab69874 |
| Anti-FANCA antibody | Abcam | Cat# ab201457 |
| Anti-FANCD2 antibody | Abcam | Cat# ab108928 |
| Anti-SMARCAL1 antibody | Abcam | Cat# ab154226 |
| Bacterial and Virus Strains | ||
| E. coli Rosetta 2 cells | Novagen/Sigma-Aldrich | Cat# 71402 |
| E. coli BL21(DE3) cells | Novagen/Sigma-Aldrich | Cat# 70235 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Samarium (III) chloride, hexahydrate | Sigma | 248800 |
| Propidium Iodide | Sigma | Cat#P4864 |
| Cisplatin | Fisher Scientific | Cat#AC193762500 |
| Hydroxyurea | Sigma Aldrich | Cat#C9911 |
| Camptothecin | Sigma | Cat#H8627–5G |
| VE-821 | Sigma | Cat#SML1415–5MG |
| VE-822 (Berzosertib) | Selleckchem | Cat#S7102 |
| BrdU | Sigma Aldrich | Cat#B5002 |
| CldU | Sigma Aldrich | Cat#C6891 |
| IdU | Sigma Aldrich | Cat#I7125 |
| PureLink RNase | ThermoFisher | Cat#12091021 |
| DMEM | Corning | Cat#10-013 CV |
| Deposited Data | ||
| Coordinate and x-ray data of EXO5 | This paper | PDB: 7LW7 |
| Coordinate and x-ray data of EXO5 in complex with dT12 (Complex 1) | This paper | PDB: 7LW8 |
| Coordinate and x-ray data of EXO5 in complex with 12mer, Sm, and Na (Complex 2) | This paper | PDB: 7LW9 |
| Coordinate and x-ray data of T88E-EXO5 in complex with dT12 and Mg (Complex 3) | This paper | PDB: 7LWA |
| EXO5 IP mass-spectrometry data | This paper; PRIDE Archive | PXD015460 (https://www.ebi.ac.uk/pride/archive/) |
| Original unprocessed imaging data | This paper; Mendeley Data | http://dx.doi.org/10.17632/y82wnzcwg8.1 |
| Genomic analysis code | This paper ; Bacolla et al., 2019 | https://github.com/abacolla/tcgaAnalyses |
| Experimental Models: Cell Lines | ||
| HeLa cells | ATCC | Cat#CCL-2 |
| HEK293 cells | ATCC | Cat#CRL-1573 |
| EXO5 KO LNCaP cells | Binghui Shen Lab (Ali et al., 2019) | N/A |
| Experimental Models: Organisms/Strains | ||
| OM811 (h+ leu1-32) | Russell Lab stock | N/A |
| OL5616 (h+ leu1-32 exo5::kanMX4) | This paper | N/A |
| OL5617 (h+ leu1-32 fan1::kanMX4) | This paper | N/A |
| OL5618 (h+ leu1-32 pso2::natMX6) | This paper | N/A |
| OL5619 (h+ leu1-32 ura4-D18 rad13::ura4+) | This paper | N/A |
| OL5620 (h+ leu1-32 exo5::kanMX4 fan1::kanMX4) | This paper | N/A |
| OL5621 (h+ leu1-32 exo5::kanMX4 pso2::natMX6) | This paper | N/A |
| OL5622 (h+ leu1-32 ura4-D18 exo5::kanMX4 rad13::ura4+) | This paper | N/A |
| OL5623 (h− leu1-32 exo5::kanMX4) | This paper | N/A |
| OL5624 (h+ leu1-32 ura4-D18 exo5::kanMX4 rqh1::ura4) | This paper | N/A |
| OL5563 (h+ leu1-32 ura4-D18 pso2::natMX6) | Boddy Lab | N/A |
| NB3167 (h+ leu1-32 ura4-D18 rqh1::ura4+) | Boddy et al., 2003 | N/A |
| Oligonucleotides | ||
| See Table S2 | This paper | N/A |
| Recombinant DNA | ||
| 1C | a gift from Scott Gradia (Unpublished) | RRID:Addgene_29654 |
| 1GFP | a gift from Scott Gradia (Unpublished) | RRID:Addgene_29663 |
| pET15b | Novagen/Sigma-Aldrich | 69661 |
| pBG100 | Vanderbilt Center for Structural Biology | N/A |
| GFP-EXO5 WT | This paper | N/A |
| HA-Flag-EXO5 WT | This paper | N/A |
| GFP-EXO5 T88A | This paper | N/A |
| GFP-EXO5 T88E | This paper | N/A |
| HA-Flag-EXO5 T88A | This paper | N/A |
| HA-Flag-EXO5 T88E | This paper | N/A |
| HA-Flag-EXO5 D182A | This paper | N/A |
| Software and Algorithms | ||
| Chimera | https://www.cgl.ucsf.edu/chimera/ | Version 1.13.1 |
| Phenix | https://www.phenix-online.org/ | Version 1.11.1-2575 |
| Coot | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | Version 0.8.9.2 |
| CCP4 suite | https://www.ccp4.ac.uk/ | Version 7.0.074 |
| XDS | http://xds.mpimf-heidelberg.mpg.de/ | Version 0.85 |
Data and Code Availability
The x-ray diffraction data and coordinates of EXO5 structures in complex with ssDNA are deposited in Protein Data Bank (PDB) (https://www.rcsb.org). PDB ID: 7LW7 (DNA-free), 7LW8 (ssDNA-1, Complex 1), 7LW9 (ssDNA-2 with Sm/Na, Complex 2), and 7LWA (T88E mutant with ssDNA and Mg, Complex 3).
The original western blots and gel images are deposited in Mendeley data. http://dx.doi.org/10.17632/y82wnzcwg8.1
The MS proteomics data (dataset identifier PXD011727) have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository.
https://www.ebi.ac.uk/pride/archive/ Reviewer account details:
Username: reviewer99034@ebi.ac.uk
Password: x3aDXMZ9
The codes used for genomic analyses are available at https://github.com/abacolla/tcgaAnalyses
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and growth media
Rosetta 2, BL21(DE3) E. coli cells were used for protein expression. Selection and growth of the cells were performed in Lysogeny broth (LB) or Terrific broth (TB) media with appropriate antibiotics [Kanamycin 50 μg/mL for the 1GFP (Addgene), 1C (Addgene), and pBG100 (Vanderbilt Center for Structural Biology) vectors; Ampicillin 100 μg/mL for the pET15b (Novagen/Sigma-Aldrich) vector] at 37 °C.
Yeast strains and growth media
Schizosaccharomyces pombe strains were generated and manipulated using standard techniques as previously described (Forsburg and Rhind, 2006). Schizosaccharomyces pombe strains were maintained on yeast extract with supplements (YES) media (0.5% yeast extract, 3% glucose, 225 mg each of adenine, L-histidine, L-leucine, and uracil) and grown at 32 °C unless otherwise indicated. Deletion mutants generated for this study were made by electroporation in 1 M sorbitol of a DNA fragment consisting of a nourseothricin (nat) resistance marker flanked by 300 –400 base pairs upstream and downstream of the pso2+ coding sequence. Positive clones were selected by growth on YES plates containing 100 μg/ml nourseothricin. Nat+ colonies were checked for proper integration by PCR. Double mutants were generated by mixing single-mutants of opposite mating types on nitrogen-limiting media (SSA) for 3 days at 25 °C, followed by performing tetrad dissection of the resulting spores. Plates were incubated for 4 days at 32 °C, and genotypes were assessed by replica plating on selective media. Yeast strains used are listed in KEY RESOURCES TABLE.
Mammalian cell culture
HEK293 and HeLa cells were purchased from ATCC (Manassas, VA) and cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a humidified atmosphere with 5% CO2. EXO5 KO LNCaP and WT LNCaP cells (a gift from Dr. Binghui Shen lab, Beckman Research Institute of City of Hope, Duarte, CA) were cultured in RPMI media with 10% FBS and 1% penicillin-streptomycin at 37°C in a humidified atmosphere with 5% CO2. Plasmid DNA or siRNA transfection was performed using an Amaxa Nucleofector electroporation kit and following the manufacturer’s protocol (Lonza, Allendale, NJ). GFP-EXO5 and HA-Flag-EXO5 WT and mutant plasmids are listed in KEY RESOURCES TABLE.
METHOD DETAILS
Genomic analyses
Analyses of tumor samples. The survival curve for patients with EXO5 CNAs versus those without EXO5 CNAs was obtained from cBioPortal (http://www.cbioportal.org/) by choosing the “Curated set of non-redundant studies” tab, followed by the “Copy number alterations” tab and “All (42,027)” subjects from 158 studies. Choosing the “Mutation” tab alone yielded a nonsignificant log rank test p-value for patients with and without EXO5 mutations.
Normalized rsem values from RNA-seq-transformed data and exome-wide mutations were downloaded from The Cancer Genome Atlas (TCGA) using the TCGA-Assembler utility (Wei et al., 2018) and analyzed with in-house scripts. For the comparison of EXO5 gene expression levels between tumor and matched normal tissues, we choose the 15/33 datasets in which at least 10 controls were available. Gene coexpression analyses were performed with in-house scripts. Gene Set Enrichment Analyses (GSEA) were conducted using the database for annotation, visualization and integrated discovery (DAVID), version v6.8, at https://david.ncifcrf.gov. Significant GO terms were those with p-value <0.05 after Benjamini correction for multiple testing. Survival curves for TCGA data with high and low EXO5 expression were assessed for significance using log rank tests.
Signature mutations for TCGA samples were from https://cancer.sanger.ac.uk/cosmic/signatures. The percent mutations attributed to signature 3 in TCGA patients with high (above mean) versus low (below mean) EXO5 expression was: for BRCA 20.8 ± 20.4 versus 12.5 ± 16.5 (n = 197, p-value 1.3 × 10−5), for LUAD 5.3 ± 8.6 versus 2.4 ± 5.7 (n = 218, p-value 4.0 × 10−5) and for LUSC 9.8 ± 10.2 versus 7.6 ± 9.2 (n = 215, p-value 1.8 × 10−2). P-values were from Welch’s t-tests.
TCGA tumor abbreviations: ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors, THCA, thyroid carcinoma; THYM, thymoma, UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma.
Details on analysis of putative cancer-related genes near EXO5. Gross focal amplifications involving band 1p34.2, where EXO5 resides, is common in some malignancies, such as small cell lung cancer (Iwakawa et al., 2013). Therefore, we computed correlation coefficients between EXO5 expression and that of all other genes in the 33 TCGA tumor types. For each dataset, we selected the top 10 positive correlations based on p-values. In BLCA, BRCA, LUAD, LUSC, and OV cancers at least 8/10 top correlations fell within 1p32–1p36. Of all genes in this region, the most frequently co-expressed (18/33 tumor types) was ZFP69, the closest upstream neighbor of EXO5 (Figure 1D). NFYC, the second highest (7/33 tumor types), was the third downstream of EXO5, and it encodes a putative oncogene (Cui et al., 2016; Tong et al., 2015). However, NFYC was only moderately overexpressed in a few (3/15) tumor types (p-values of 0.00045 in LIHC, 0.0037 in LUAD and 0.0032 in BLCA). Two splicing factors, PRPF38A, which is required in triple negative breast cancers (Chan et al., 2017), and PPIE, occurred in 6/33 tumor types and were located further away. Highly co-expressed genes were interspersed with other genes that displayed limited coregulation; e.g. RIMS3, located between EXO5 and NFYC, was consistently downregulated in the TCGA dataset, and coexpression with EXO5 was absent in 13/33 tumor types, whereas weak coexpression was seen in other types.
Molecular cloning and protein purification
Purification of EXO5, RPA, and BLM
Full-length human EXO5 in the pGEX6p-1 vector was expressed in E coli. However, the final purification results showed the aggregation of FL-EXO5. Therefore, N-terminal region (1 –30 a.a.), predicted to be disordered in PONDR program (http://www.pondr.com/) was truncated in the construct of Δ30-EXO5 and cloned into the 1GFP expression vector (Addgene #29663). The Δ30-EXO5 protein was expressed in Rosetta 2 cells until OD600 = 0.6 –0.8, and induced with 0.5 mM IPTG at 22 °C for overnight. The harvested cell pellets were brought into an anaerobic Coy chamber (oxygen level < 0.1 ppm) for purification, and resuspended in the lysis buffer (30 mM Tris (pH 8.0), 0.5 M NaCl, 5% glycerol, 2 mM BME, 5 mM MgCl2, 20 ug/mL DNase, 200 μg/mL lysozyme, and 0.5 mM PMSF). The lysate was homogenized using Douncer (x10 strokes), and sonicated for 45 sec (x8 cycles). After centrifugation, the supernatant was loaded onto an HisTrap FF crude 5 mL column using Akta purifier 10 FPLC system (GE healthcare), and the protein was eluted in the imidazole buffer (30 mM Tris (pH 8.0), 0.25 M NaCl, 5% glycerol, 2 mM BME, and 300 mM imidazole). The green fractions containing 6His-GFP-Δ30-EXO5 were collected and cleaved with TEV protease at 4 °C for overnight. The mixture was concentrated using Amicon centrifugal filter (10 kDa MWCO) and diluted with Buffer C (50 mM Tris (pH 8.0), 5% glycerol, and 1 mM DTT) to a final NaCl concentration of ~ 30 mM, followed by the purification using HiTrap Heparin HP (5 mL) column. The eluted protein was further purified by size-exclusion Superdex 200 increase column (10/300) with the buffer (25 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM KCl, and 5% glycerol).
Human RPA constructs
RPA70N (RPA70 residues 1–120) and RPA32C (RPA32 residues 202–270) preparation was carried out as described elsewhere (Souza-Fagundes et al. 2012; Feldkamp et al. 2014) with minor modifications. Briefly, the pET15b-RPA70N or the pBG100-RPA32C plasmid was transformed into E.coli BL21(DE3) cells. Cells were grown in Terrific Broth media (RPI) at 37 °C. When the OD reached 0.8 –1.0 at 600 nm, the temperature was lowered to 18 °C, and protein expression was induced by addition of 0.5 mM IPTG. After 16 hours, cells were harvested by centrifugation and the pellets were resuspended in buffer A (50 mM TRIS-HCl at pH 7.5, 500 mM NaCl, 10 mM imidazole, 2 mM β-mercaptoethanol). Lysis was carried out by sonication (10 min; 5 seconds “on” followed by 5 seconds “off”; 50% power) and the lysate was centrifuged at 50,000 x g for 30 min at 4 °C. The supernatant was filtered and loaded onto a HisTrap FF column (GE healthcare) pre-equilibrated in buffer A. The column was washed with 10 column volume (CV) of high salt wash buffer (20 mM Tris-HCl at pH 7.5, 1 M NaCl, 10 mM imidazole, 2 mM β-mercaptoethanol) and 10 CV of buffer A. The protein was eluted using a 20 CV linear gradient to buffer B (20 mM Tris-HCl at pH 7.5, 1M NaCl, 300 mM imidazole, 2 mM β-mercaptoethanol). Elution fractions containing the protein of interest were then concentrated using an Amicon® ultra-filtration unit with a 3000 MW cutoff and loaded onto a Superdex 75 gel filtration column (GE healthcare) pre-equilibrated in SEC buffer (25 mM HEPES at pH 7.5, 100 mM NaCl). Gel filtration was carried out in the SEC buffer with elution fractions analyzed by SDS-PAGE to confirm purity. Fractions containing the protein of interest were pooled together, aliquoted, and flash frozen in liquid nitrogen. Samples were stored at −80 °C until needed. Protein purity and integrity was confirmed by SDS-PAGE and mass spectrometry analysis.
The preparation of RPA70NAB (RPA70 residues 1–422) has been described elsewhere (Arunkumar et al. 2003; Brosey et al. 2009). Expression was carried out in a manner similar to RPA70N and RPA32C, from a pBG100-RPA70NAB plasmid. Purification followed the same steps with an additional heparin chromatography purification step prior to gel filtration. Essentially, HisTrap elution fractions containing RPA were dialyzed against dialysis buffer (25 mM HEPES at pH7.5, 100 mM NaCl, 5 mM β-mercaptoethanol, 10% glycerol). The dialyzed protein was loaded onto a Heparin FF column (GE Healthcare) pre-equilibrated in buffer hepA (25 mM HEPES at pH 7.5, 50 mM NaCl, 5 mM β-mercaptoethanol, 10% glycerol). After a wash step with 10 CV of hepA buffer, the protein was eluted with a linear gradient toward hepB (25 mM HEPES at pH 7.5, 1 M NaCl, 5 mM β-mercaptoethanol, 10% glycerol) over 20 CV. Elution fractions containing the protein of interest were identified by SDS-PAGE, pooled together and concentrated using an Amicon® ultra-filtration unit with a 10,000 MW cutoff prior to the gel filtration step. The rest of the purification was as described above for RPA70N and RPA32C. Protein purity and integrity was confirmed by SDS-PAGE and mass spectrometry analysis.
Preparation of RPA full length RPA was carried out as described elsewhere (Brosey, Chagot, and Chazin 2012). Protein purity and integrity was confirmed by SDS-PAGE and mass spectrometry analysis.
Human BLMcat purification
Human BLMcat (640–1290 aa) was subcloned into 1C vector with 6His-MBP tag (Addgene #29654). The plasmid was transformed into Rosetta 2 cells. The single colony was used to inoculate 50 mL of LB media overnight, followed by 10 mL inoculation in 1L of LB media with 50 μg/mL kanamycin. Cells were grown at 37 °C until OD600 ≅ 0.6–0.8, and the protein expression was induced with 0.5 mM IPTG at 28 °C overnight. The cells were harvested and frozen at −80 °C until use. Cells were resuspended in buffer A (50 mM Tris (pH 8), 500 mM NaCl, and 5% glycerol) with a protease inhibitor tablet (Roche), DNase (0.02 mg/mL) and lysozyme (0.2 mg/mL). The protein was eluted in a gradient of 9–240 mM imidazole. The fractions containing BLMcat protein were pooled and mixed with TEV protease overnight to remove His-MBP tag. The mixture was passed through HiTrap Heparin 5 mL column (GE lifesciences) to separate His-MBP from BLMcat, which was eluted in a gradient of 30 –700 mM NaCl. The cleaved BLMcat protein was further purified on Superdex 200 (10/300) size-exclusion column in a buffer of 50 mM Tris (pH 8), 0.5 M NaCl, and 5% glycerol.
Nuclease assay
wt-Δ30-EXO5 (150 nM) was mixed with BLM (160 uM) and RPA (200 nM) variants in the buffer of 50 mM Tris (pH8), 50 mM NaCl, 2.5% glycerol, 2.5 mM DTT, and 0.1mg/mL of BSA for 20 min at 20 °C before adding the Cy5-labeled DNA substrates (10 nM). The nuclease reaction was initiated by adding final concentration of 5 mM Mg(OAc)2 or 2 mM ATP / 5 mM Mg(OAc)2. The mixture was incubated at 30 °C for the times indicated. The reaction was then stopped by adding final concentration of 20 mM EDTA, 0.5% SDS, 6.7% glycerol, and 24% formamide. The mixture was boiled at 95 °C for 5 min and either immediately transferred on ice or slowly cooled down (by turning off PCR machine) for footprint experiment with addition of 100-fold excess of non-labeled template strand to avoid re-annealing. Each mixture (7 μl) was loaded on the 15% TBEurea gels, which was pre-run at 200 V for 1 hour in 1x TBE buffer and separated at 180V for 45 min. The gels were visualized at 648 nm on ChemiDoc imaging system (Bio-Rad). For Δ30-EXO5 mutants or metal-dependent nuclease activity assay, 75 and 150 nM EXO5 variants were used with 3’-Cy5-labeled 45-mer ssDNA substrate using the same protocol as described above at 30 °C for 20 min. At least three independent experiments were run to confirm the activity unless otherwise specified. Although the molecular weight markers were run together with the samples, to enhance the contrast of the samples’ bands the molecular weight markers with 6-FAM-labeled fluorophore visualized at 495 nm, different absorption channel to Cy5-DNA substrates and products, were cropped and merged separately to gel figures shown in 6K, S2D, S3D, S3H, S3I, S4F, S4G and S5A (right panel).
Binding assay using MicroScale Thermophoresis (MST)
Δ30-EXO5 variants ~ 100 μM or 200 μM for Q210A, T88E, and WT (in the buffer 25 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM KCl, and 5% glycerol) were titrated in 1:1 dilution series with DNA binding buffer (25 mM Tris (pH 8), 50 mM NaCl, 5 mM DTT, 0.02 mg/mL BSA, and 0.05% tween-20). Protein samples were then mixed equally with 100 nM DNA (with 3’-Cy5 labeled from Integrated DNA Technologies, Inc.) in a total volume of 20 μL and loaded in 16 thin glass capillaries. The experiments were measured at 25 °C using Monolith NT.115 (NanoTemper, Germany). The binding affinity KD were calculated based on the changes of initial fluorescence intensity quenching upon the titration of the protein concentration using equation (1).
| (1) |
b = fluorescence in completely bound state
u = fluorescence in completely unbound state
x = fraction bound
y = output normalized fluorescence
A = concentration of labeled molecule in nM
T = titrant concentration in nM
K = dissociation constant (KD)
To confirm that the initial fluorescence quenching was caused by EXO5 binding, SD test was performed. 10 μL of sample from tube 1 (most concentrated protein) and 16 (least concentrated protein) was transferred to tube each containing 10 μl of a 2× SD mix (4 % SDS, 40 mM DTT). The mixtures were incubated for 5 min at 95 °C to denature the protein and loaded into capillaries for fluorescence intensity measurement. Both samples had the same fluorescence intensity, therefore confirming the proteininduced quenching.
For the binding of BLMcat and WT variants, BLMcat was labeled with Atto-647N NHS ester dye (Sigma-Aldrich) via amine-coupling based on manufacture’s protocol. The resulting Atto-647-labeled BLMcat (200 nM) was mixed with equal volume of EXO5 variants (400 μM of WT or T88E), which was titrated in 1:1 dilution series with buffer (20 mM Tris (pH 7.5), 250 mM NaCl, 5% glycerol, 0.05% tween-20, and 10 mM KCl). The mixture was incubated at 37 °C for 30 min before loading into capillaries. The capillaries were incubated at 25 °C before starting MST measurements. MST was measured at 25 °C using 40% LED power and 40% MST power. No fluorescence quenching was observed from varied titrant concentrations. The resulting MST trace was fitted as KD binding using above equation (1).
Kinetic binding assay using Surface Plasmon Resonance (SPR)
The surface plasmon resonance experiments were performed using a BIACORE T200 (GE Healthcare) with SA sensor chip. The immobilization protocol used a wizard tool to target the response unit (RU) level to 100 on flow cell 2. Basically, the Flow cell 2 was activated with 1 M NaCl and 50 mM NaOH, followed by the injection of ligand consisting of 20 nM of 3’-labeled biotinylated 45mer ssDNA (14 kDa, Integrated DNA Technologies, Inc.). The final immobilized RU level reached to 118. The flow cell was washed with 50% isopropanol, 1 M NaCl, and 50 mM NaOH. The system was primed with the experimental buffer (25 mM Tris (pH 8), 50 mM NaCl, 0.05% P-20, 5 mM DTT, and 0.5 mM EDTA) three times. Flow cells 1–2 were equilibrated with experimental buffer overnight at 10 μL/min flow rate. To collect kinetic binding data, the analyte (WT and T88E-EXO5, 38 kDa) was desalted with the experimental buffer and injected (from 2.44 nM to 2.5 μM for WT and 9.77 nM to 10 μM for T88E-EXO5) at a flow rate of 30 μL/min at 25 °C. The contact time and dissociation time of the complex was 120 sec and 600 sec, respectively. The surfaces were regenerated with a 1 M NaCl for 60 sec at a flow rate of 30 μL/min. Duplicate injections (in a 11, 22, 33 order) of each sample and a buffer blank were flowed over two flow cells. The data were collected at a rate of 10 Hz and fit to a 1:1 binding model using global data analysis in BiaEvaluation 3.0 software. The ka, kd, and KD were reported from the two independent runs. And each run was fitted for standard deviation calculation.
Crystallization, data collection, and structure determination
wt-Δ30-EXO5 (6 mg/mL) was crystallized in 1:1 ratio with the reservoir of 16% PolyEthylene Glycol (PEG) 3350, 0.1 M Bis-Tris (pH 5.5), and 3% glucose in an anaerobic chamber at 20 °C. Single crystals were transferred to the cryo-protecting solution in the presence of 16% ethylene glycol. Single wavelength anomalous (SAD) diffraction data were collected at Fe absorption edge of 1.7369 Å at beamline 12–2 at the Stanford Synchrotron Radiation Lightsource (SSRL) in Menlo Park, CA.
Native wt-Δ30-EXO5 (5 mg/mL) was crystallized in 1:1 ratio with the reservoir of 20% PEG 3350 and 0.2 M lithium acetate in the anaerobic chamber at 20 °C. The single crystal was transferred to the cryo-protecting solution in the presence of 16% ethylene glycol, followed by flash frozen in liquid nitrogen. The data were collected at SIBYLS beamline 12.3.1 at the Advanced Light Source (ALS) in Berkeley, CA.
The collected datasets were integrated using XDS program, and scaled and merged using aimless software in CCP4 suite. The de novo structure model was determined by Fe-SAD dataset using AutoSol software in Phenix program. Four Fe sites were found, and the initial model with 207 residues were built with Rwork = 0.34, followed by running the AutoBuild wizard in Phenix. The final model was manually fit using the Coot software for several runs and further refined in native dataset using phenix.refine software.
Co-crystallization trials with ssDNA or overhang substrates were not successful. DNA soaking experiments were therefore performed to obtain the DNA-bound structures. Complex 1 crystals. DNA-free WT-Δ30-EXO5 crystals were grown in 16% PEG 3350 and 0.2 M lithium acetate as described above. Single-strand DNA oligo (3’ thiol modifier C3 S-S dT12, 160 μM, from IDT Inc.) was added to the drop containing the crystallization solution, and crystals were soaked for overnight at 20 °C in the glovebox against crystallization condition. Crystals were then transferred to the new drop with the same concentration of ssDNA plus 16% ethylene glycol, followed by snap frozen in the liquid nitrogen. Complex 2 crystals. DNA-free WT-Δ30-EXO5 crystals were grown in 20 % PEG 3350 and 0.2 M ammonium acetate at 20 °C. Single-strand DNA oligo (12mer, ATT GCT GAA GGG, 157 μM, from IDT Inc.) was added to the drop containing crystallization solution and 5 mM SmCl3. The rest of the procedure was the same as for complex 1 described above. Complex 3 crystals. T88E-Δ30-EXO5 crystals were grown in 20% PEG3350 and 0.1 M magnesium formate at 20 °C. Single-strand DNA oligo (3’ thiol modifier C3 S-S dT12, 320 μM, from IDT Inc.) was added to the drop containing crystallization solution and 16% ethylene glycol for 4 min before flash freezing in liquid nitrogen. The structure determination of the EXO5-DNA complexes used DNA-free EXO5 as searching model for molecular replacement. The ssDNA was manually built in the electron density map using Coot program. The refinement was performed using phenix.refine. The final complex structure has complex 1 (9 of 12 nts), complex 2 (7 of 12 nts), and complex 3 (3 of 12 nts) ) seen by electron density. The data collection and refinement statistics are listed in the Table 1. Structural similarity searches were done using PDBeFold (Krissinel and Henrick, 2004) and Dali (Holm and Laakso, 2016) servers. And the graphic presentation was made using Chimera (Pettersen et al., 2004).
Sister chromatid exchanges and chromosomal aberrations
Sister chromatid exchanges were examined by the previously described procedure (Pandita, 1983). Briefly, after drug treatment (50 μM cisplatin for 1hr in serum free DMEM, 1 μM CPT for 12 hours), HeLa cells were labeled with bromodeoxyuridine (100 μM BrdU) for 48h and metaphase cells were collected after colcemid treatment (0.2 μg/mL for 4 hours). Methanol-acetic acid fixed metaphase cells were then dropped on the slides, and after 48 hours exposed to black light followed by Giemsa staining. Metaphases were photographed at 40x magnification and exchanges were scored manually. Radial chromosomes were assessed using methods described previously (Pandita, 2006; Sparks et al., 2012). In these experiments, 48 hours after siRNA transfection, HeLa cells were treated with mentioned drugs (50 μM cisplatin for 1 hour in serum free DMEM, 1 μM CPT for 12 hours). 12 hours after drug treatment, cells were pulse labelled with 100 μM BrdU, followed by colcemid (0.05 μg/ml) for 3 hours. Cells were then harvested, treated with hypotonic solution (0.075 M KCl, 5% fetal calf serum) for 10 minutes, and fixed with 3:1 methanol acetic acid. Cells were dropped onto slides for metaphase spreads and stained with Giemsa staining. Fifty metaphases were counted for radial chromosomes, and each experiment was repeated three times.
DNA fiber assays
DNA fiber assays were performed to assess replication fork stalling, restart and fork protection as previously described (Schlacher et al., 2011; Singh et al., 2018). Briefly, HeLa, HEK293 or LNCaP (WT and EXO5 KO) cells were pulsed with CldU (100μM, 30min) and replaced by media with or without the drug and followed by IdU (150μM, 60min), or by media containing the drug together with IdU as indiciated in Figures. DNA fibers were spread by gravity and stained using standard immuno-fluorescent procedures with CldU and IdU specific antibodies. Replication recovery was assessed by measuring the tract length of the second label (IdU with or after replication stalling) of tracts that contain both CldU+IdU labels. The percentage of restarted forks after HU was calculated by dividing the number of CldU tracts by the number of (CldU tracts plus CldU+IdU tracts). For DNA fiber analysis without HU, the percentage of terminated forks was calculated by dividing the number of CldU tracts by the number of (CldU tracts plus CldU+IdU tracts), and the fork speed is given as the CldU tract length of CldU+IdU tracts. For determining replication fork protection, HeLa cells were labeled with CldU and their tract length measured following incubation with HU (4mM) as indicated in Figures. DNA fibers were imaged by Carl Zeiss Axio Imager D2 microscope using 63X Plan Apo 1.4 NA oil immersion objective, and statistical analysis was performed using the Mann-Whitney test.
Immunoprecipitation and mass-spectrometry
We performed large-scale immunoprecipitation for mass-spectrometry analysis in HEK293 cells. Exponentially growing HEK293 cells were transfected using XtremeGene (Sigma, Cat# 8724105001) with HA-Flag-EXO5 plasmid, lysed in non-denaturing condition (25 mM Tris-HCl, 100 mM NaCl, 10% glycerol, 1% NP-40, 1 mM EDTA) for 48 hours after transfection, and the clarified lysate was incubated with flag M2 magnetic beads (Sigma, M8823) for 4 hours at 4 °C. Immunoprecipitated proteins were eluted in 0.1 M glycine (pH 3.0) and mixed in Laemmli buffer for SDS-PAGE analysis. Immunoprecipitates from 6 independent experiments were pooled and loaded in a 10% SDS-PAGE. Mass-spectrometry of gel sections was performed at Taplin mass spectrometry facility, Harvard Medical School, Boston, MA. Excised gel bands were cut into approximately 1 mm3 pieces. Gel pieces were then subjected to a modified in-gel trypsin digestion procedure (Shevchenko et al., 1996). Gel pieces were washed and dehydrated with acetonitrile for 10 min. followed by removal of acetonitrile. Pieces were then completely dried in a speed-vac. Rehydration of the gel pieces was with 50 mM ammonium bicarbonate solution containing 12.5 ng/μl modified sequencing-grade trypsin (Promega, Madison, WI) at 4 °C. After 45 min., the excess trypsin solution was removed and replaced with 50 mM ammonium bicarbonate solution to just cover the gel pieces. Samples were then placed in a 37 °C room overnight. Peptides were later extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50% acetonitrile and 1% formic acid. The extracts were then dried in a speed-vac (~1 hour). Samples were then stored at 4 °C until analysis.
On the day of analysis the samples were reconstituted in 5 – 10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter x ~30 cm length) with a flame-drawn tip (Peng and Gygi, 2001). After equilibrating the column each sample was loaded via a Famos auto sampler (LC Packings, San Francisco CA) onto the column. A gradient was formed and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid).
As peptides eluted they were subjected to electrospray ionization and then entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher Scientific, Waltham, MA) (Eng et al., 1994). All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD015460 and 10.6019/PXD015460.
For western blotting of immunoprecipitates from HA-FLAG-EXO5 overexpressing HeLa cells, the lysates prepared in the same lysis buffer as mentioned above was used for immunoprecipitation with magnetic Anti-Flag M2 beads.
ATR inhibition experiments
For ATR inhibition experiments, VE-821 or VE-822 was added into HeLa cell growth media at the final concentration 1 μM for 30 min. Inhibitor media was washed three times with warm PBS and drug media was added. Cells were treated with 2mM HU for 24h or with 20 μM cisplatin (in serum free DMEM) for 1 hour. After drug removal and washing, cells were maintained in 0.01μM VE-821 or VE-822 containing media for 12 hours, and fixed with 4% PFA for immunostaining or lysed in IP lysis buffer for immunoprecipitation experiments.
Western blotting and immuno-fluorescence staining
Western blotting and immune-florescence staining (IF staining) was performed as described (Chakraborty et al., 2018). For immune-fluorescence staining HeLa cells grown in chamber slides were fixed with paraformaldehyde (PFA) following drug treatment (HU 2 mM for 24h, cisplatin 50 μM for 1 hour in serum free DMEM, camptothecin 1 μM for 1 hour) at time points mentioned in figures. PFA fixed cells on chamber slides were blocked with 2% BSA or 20% goat serum and stained with mentioned primary antibodies followed by washing and staining with fluorescent conjugated secondary antibodies. A total of 200 –300 cells per samples was analyzed for foci determination. For co-localization experiments, GFP-EXO5 was transfected into HeLa cells using LONZA Nucleofector electroporation system. For determination of ssDNA by BrdU, HeLa cells were incubated with 20 μM BrdU for 24 hours in growth media in chamber slides. Following drug treatment, as mentioned in figures, BrdU cells were pre-extracted with 0.5% tritonX-100 for 5min before fixing with 4% paraformaldehyde. Fixed cells were then permeabilized by a second treatment with 0.5% tritonX-100, blocked with 20% goat serum and stained with BrdU antibody (Novus Bio Cat#NB500–169) and rhodamine red conjugated secondary antibody. Mounting media containing DAPI was used. Images were captured using Z-stacking method in Carl Zeiss Axio Imager2, and standard deconvolution method was applied to remove the background.
Clonogenic survival assays
To determine the effect of cisplatin, CPT or HU on cell survival colony formation assay in HeLa, HEK293, or LNCaP (WT and EXO5 KO) cells was performed as described (Singh et al., 2018). Briefly, 48 hours after siRNA transfection, (no tranfection for LNCaP cells), the cells were seeded in Petri dishes and after overnight incubation media was replace with fresh medium containing cisplatin for 1h, CPT for 12 hours or HU for 24 hours. Cells were washed, replaced with fresh media and colonies were allowed to grow for 12 days before staining with crystal violet. Expression of WT and mutant EXO5 was done by co-transfecting the plasmids and EXO5 UTR siRNA in HeLa or HEK293 cells. Clonogenic assay was performed as mentioned above 48h after co-transfection.
Cell Cycle analysis
HeLa cells at 80% confluence were treated with 50 μM cisplatin in serum free media for 1 hour. After washing the drug the media was replaced with fresh media. Cells were trypsinized and fixed with 70% ethanol at mentioned time points. Fixed cells were washed and stained with staining solution (10% triton X-100, 20mg/ml RNase, 1 mg/ml propidium iodide in PBS) for 1 hour. Stained cells were washed three times with PBS and resuspended in 1 mL PBS. A BD FACS Caliber flowcytometer was used for acquisition and G1/S/G2 cell population was determined using FCS Express7 program.
Fission yeast genetic interaction analyses
For DNA damage sensitivity assays, five-fold serial dilutions of log-phase of the indicated genotypes were spotted onto rich media (yeast extract with supplements,YES) agar plates with the indicated dose of cisplatin and incubated at 32°C for 3 days. Cisplatin stocks were made using a saline solution (0.9% NaCl). For tetrad dissections, rqh1Δ and exo5Δ single mutants were crossed on nitrogen-limiting media (SSA) for 3 days. The resulting spores were individually isolated on YES plates using a micromanipulator and incubated for 4 days at 32°C. Phenotypes were assessed by replica plating on selective media.
Evolutionary analysis of EXO5
The evolutionary history was inferred by using the Maximum Likelihood method and Dayhoff matrix based model. The tree with the highest log likelihood (−28103.56) was selected. The percentage of trees in which the associated taxa clustered together in the Bootstrap method is shown next to the branches. Initial tree for the heuristic search were obtained automatically by applying the Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Dayhoff model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (3 categories (+G, parameter = 3.4014). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 28 amino acid sequences and a total of 75 Bootstrap tree reconstructions. There were a total of 1311 positions in the final dataset. Evolutionary analyses were conducted in MEGA X (https://www.megasoftware.net).
QUANTIFICATION AND STATISTICAL ANALYSIS
The Kaplan-Meier plot for the overall survival of patients with EXO5 CNAs was taken from http://www.cbioportal.org; the p-value was also taken from the same website as obtained from a log rank test. No data were excluded from the 42027 patient’s data available, which originated from 158 nonredundant studies. Other survival curves and Cox regressions were generated in R through the “survminer”,“survival” and “dplyr” packages. Hazard ratios and the associated 95% confidence interval (CI) and p-values were obtained from the Cox PH regression model using the “summary(fitCox)” function in R. Euclidean hierarchical clustering was performed using the “gplots” R package. Regression coefficients for the correlations between MYBL2 log2(normalized rsem + 1) gene expression values and that of other genes were computed from in-house scripts using gene expression data from TCGA. P-values for the differences in mRNA gene expression levels between tumor and matched controls were obtained from Wilcoxon tests in R. P-values for gene expression correlations were obtained through the C++ Boost library (https://www.boost.org), which computes P from the cumulative distribution function (CDF) of the Fisher-Snedecor distribution F, where r is the regression coefficient and n the number of observations For cellular data analysis, Graph pad prism 6 was used for generating graphs and dot plots. T-test was used for determining statistical significance. P values < 0.05 were considered significant.
A minimum 50 metaphase for chromosomes for each experiment were analyzed using Carl Zeiss Axio Imager D2 microscope for chromosomal aberrations, and each experiment was repeated three times. To determine if the differences between conditions are significant, a two-tailed t-test was used.
The cell survival was plotted after combining three to four independent experiments for each experimental condition. To determine if the differences between different conditions are significant, a student t-test was used.
DNA Fibers data analyses were carried out with standard ImageJ software64 in blind. The Mann-Whitney test were applied for statistical analysis.
Supplementary Material
Highlights.
High EXO5 expression links to high mutation loads and low cancer patient survival.
EXO5-DNA structures reveal its structure-specific mechanism for DNA end processing.
Direct ATR phosphorylation of EXO5 regulates the EXO5-BLM complex for fork restart.
Evolutionarily conserved EXO5-BLM fork restart balances FANCA fork protection.
ACKNOWLEDGEMENTS
We thank Arnab R. Chaudhuri, Susan Lees-Miller, Chris Brosey, Aleem Syed, Zu Ye, and Kalpana Mujoo for input plus Xiaoyan Wang and Naga Babu Chinnam for testing full-length EXO5 stability. T.K.P. received WT GFP-EXO5 plasmid from Peter M. Burgers and EXO5 KO LNCaP cells from Binghui Shen. J.A.T. is supported by NIH grants (P01 CA092584, R35CA220430), a Robert A. Welch Chemistry Chair, and Cancer Prevention and Research Institute of Texas (CPRIT) RP180813. T.K.P is supported by NIH RO1 CA129537, RO1 GM109768 and The Houston Methodist Research Institute. K.S. is supported by NIEHS 1R01ES029680 plus CPRIT RP180463 and RP180813. K.S. is a Rita Allen Foundation Fellow and a CPRIT scholar. TCGA research used Texas Advanced Computing Center supported by NSF ACI-1134872. X-ray diffraction data was collected at SSRL beamline 12-2 and ALS beamlines 12.3.1 [supported by NIH project ALS-ENABLE (P30 GM124169) and the Integrated Diffraction Analysis Technologies (IDAT) program] and 8.3.1 (supported by NIH R01 GM124149 and P30 GM124169).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S, Zhang Y, Zhou M, Li H, Jin W, Zheng L, Yu X, Stark JM, Weitzel JN, and Shen B. (2019). Functional deficiency of DNA repair gene EXO5 results in androgen-induced genomic instability and prostate tumorigenesis. Oncogene 39, 12461259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Ye Z, Ahmed Z, and Tainer JA (2019). Cancer mutational burden is shaped by G4 DNA, replication stress and mitochondrial dysfunction. Prog Biophys Mol Biol 147, 47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddock HT, Yosaatmadja Y, Newman JA, Schofield CJ, Gileadi O, McHugh PJ, (2020). The SNMA1 DNA repair nuclease. DNA Repair 95, 102941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley R, Blakemore D, Cancian L, Dumon S, Volpe G, Ward C, Almaghrabi R, Gujar J, Reeve N, Raghavan M, et al. (2018). MYBL2 Supports DNA Double Strand Break Repair in Hematopoietic Stem Cells. Cancer Res 78, 5767–5779. [DOI] [PubMed] [Google Scholar]
- Benitez A, Liu W, Palovcak A, Wang G, Moon J, An K, Kim A, Zheng K, Zhang Y, Bai F, et al. (2018). FANCA Promotes DNA Double-Strand Break Repair by Catalyzing Single-Strand Annealing and Strand Exchange. Mol Cell 71, 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Shanahan P, McDonald WH, Lopez-Girona A, Noguchi E, Yates IJ, Russell P. (2003). Replication checkpoint kinase Cds1 regulates recombinational repair protein Rad60. Mol Cell Biol 23, 5939–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, and Foiani M. (2010). Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11, 208–219. [DOI] [PubMed] [Google Scholar]
- Burgers PM, Stith CM, Yoder BL, and Sparks JL (2010). Yeast exonuclease 5 is essential for mitochondrial genome maintenance. Mol Cell Biol 30, 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P. (2015). DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination. J Biol Chem 290, 22931–22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaganti RS, Schonberg S, and German J. (1974). A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci U S A 71, 4508–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Pandita RK, Hambarde S, Mattoo AR, Charaka V, Ahmed KM, Iyer SP, Hunt CR, and Pandita TK (2018). SMARCAD1 Phosphorylation and Ubiquitination Are Required for Resection during DNA Double-Strand Break Repair. iScience 2, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Sridhar P, Kirchner R, Lock YJ, Herbert Z, Buonamici S, Smith P, Lieberman J, and Petrocca F. (2017). Basal-A Triple-Negative Breast Cancer Cells Selectively Rely on RNA Splicing for Survival. Mol Cancer Ther 16, 2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury I, Sareen A, Raghunandan M, and Sobeck A. (2013). FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res 41, 6444–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, and Foiani M. (2005). Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell 17, 153–159. [DOI] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristini A, Ricci G, Britton S, Salimbeni S, Huang SN, Marinello J, Calsou P, Pommier Y, Favre G, Capranico G, et al. (2019). Dual Processing of R-Loops and Topoisomerase I Induces Transcription-Dependent DNA Double-Strand Breaks. Cell Rep 28, 3167–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Zhang M, Wang Y, and Wang Y. (2016). NF-YC in glioma cell proliferation and tumor growth and its role as an independent predictor of patient survival. Neurosci Lett 631, 40–49. [DOI] [PubMed] [Google Scholar]
- Daley JM, Tomimatsu N, Hooks G, Wang W, Miller AS, Xue X, Nguyen KA, Kaur H, Williamson E, Mukherjee B, et al. (2020). Specificity of end resection pathways for double-strand break regions containing ribonucleotides and base lesions. Nat Commun 11, 3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, et al. (2017). HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SJ, Sheppard KE, Pearson RB, Campbell IG, Gorringe KL, and Simpson KJ (2013). Functional analysis of genes in regions commonly amplified in high-grade serous and endometrioid ovarian cancer. Clin Cancer Res 19, 1411–1421. [DOI] [PubMed] [Google Scholar]
- Davies SL, North PS, Dart A, Lakin ND, and Hickson ID (2004). Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol 24, 1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SL, North PS, and Hickson ID (2007). Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol 14, 677–679. [DOI] [PubMed] [Google Scholar]
- Dibitetto D, Sims JR, Ascencao CFR, Feng K, Kim D, Oberly S, Freire R, and Smolka MB (2020). Intrinsic ATR signaling shapes DNA end resection and suppresses toxic DNA-PKcs signaling. NAR Cancer 2, zcaa006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty KM, Sommers JA, Gray MD, Lee JW, von Kobbe C, Thoma NH, Kureekattil RP, Kenny MK, and Brosh RM Jr. (2005). Physical and functional mapping of the replication protein a interaction domain of the werner and bloom syndrome helicases. J Biol Chem 280, 29494–29505. [DOI] [PubMed] [Google Scholar]
- Eckelmann BJ, Bacolla A, Wang H, Ye Z, Guerrero EN, Jiang W, El-Zein R, Hegde ML, Tomkinson AE, Tainer JA, et al. (2020). XRCC1 promotes replication restart, nascent fork degradation and mutagenic DNA repair in BRCA2-deficient cells. NAR Cancer 2, zcaa013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis NA, and German J. (1996). Molecular genetics of Bloom’s syndrome. Hum Mol Genet 5, 1457–1463. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fuss JO, Tsai CL, Ishida JP, and Tainer JA (2015). Emerging critical roles of FeS clusters in DNA replication and repair. Biochim Biophys Acta 1853, 1253–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon J, Ursich S, Lopes M, Hiraga SI, and Donaldson AD (2019). Human RIF1-Protein Phosphatase 1 Prevents Degradation and Breakage of Nascent DNA on Replication Stalling. Cell Rep 27, 2558–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German J, Crippa LP, and Bloom D. (1974). Bloom’s syndrome. III. Analysis of the chromosome aberration characteristic of this disorder. Chromosoma 48, 361–366. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, and Bartek J. (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Hemphill AW, Akkari Y, Newell AH, Schultz RA, Grompe M, North PS, Hickson ID, Jakobs PM, Rennie S, Pauw D, et al. (2009). Topo IIIalpha and BLM act within the Fanconi anemia pathway in response to DNA-crosslinking agents. Cytogenet Genome Res 125, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, and Laakso LM (2016). Dali server update. Nucleic Acids Res 44, W351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, Zhang M, Hu Y, Wang Q, Xu W, et al. (2012). The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 149, 1221–1232. [DOI] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui X, Matsuoka S, Cortez D, Tamai K, Luo G, CarattiniRivera S, DeMayo F, Bradley A. et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for G2/M DNA damage checkpoint. Genes Dev 14, 14481459. [PMC free article] [PubMed] [Google Scholar]
- Iwakawa R, Takenaka M, Kohno T, Shimada Y, Totoki Y, Shibata T, Tsuta K, Nishikawa R, Noguchi M, Sato-Otsubo A, et al. (2013). Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer 52, 802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, and Duhr S. (2011). Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol 9, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep 23, 239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski WW, Fu X, Wilkinson M, Cronin NB, Dillingham MS, and Wigley DB (2014). Structural basis for translocation by AddAB helicase-nuclease and its arrest at chi sites. Nature 508, 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K. (2004). Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemak S, Beloglazova N, Nocek B, Skarina T, Flick R, Brown G, Popovic A, Joachimiak A, Savchenko A, and Yakunin AF (2013). Toroidal structure and DNA cleavage by the CRISPR-associated [4Fe-4S] cluster containing Cas4 nuclease SSO0001 from Sulfolobus solfataricus. J Am Chem Soc 135, 17476–17487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Ji F, Helleday T, and Ying S. (2018). Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep 19:e46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, Kobayashi Y, Hashimoto A, Hamamoto M, Hiraoka Y, et al. (2006). ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol 24, 841–847. [DOI] [PubMed] [Google Scholar]
- Mazouzi A, Velimezi G, and Loizou JI (2014). DNA replication stress: causes, resolution and disease. Exp Cell Res 329, 85–93. [DOI] [PubMed] [Google Scholar]
- McLaughlin LJ, Stojanovic L, Kogan AA, Rutherford JL, Choi EY, Yen RC, Xia L, Zou Y, Lapidus RG, Baylin SB, et al. (2020). Pharmacologic induction of innate immune signaling directly drives homologous recombination deficiency. Proc Natl Acad Sci U S A 117, 17785–17795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moder M, Velimezi G, Owusu M, Mazouzi A, Wiedner M, Ferreira da Silva J, Robinson-Garcia L, Schischlik F, Slavkovsky R, Kralovics R, et al. (2017). Parallel genome-wide screens identify synthetic viable interactions between the BLM helicase complex and Fanconi anemia. Nat Commun 8, 1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, and Halazonetis TD (2010). Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11, 220–228. [DOI] [PubMed] [Google Scholar]
- Nieminuszczy J, Broderick R, Bellani MA, Smethurst E, Schwab RA, Cherdyntseva V, Evmorfopoulou T, Lin YL, Minczuk M, Pasero P, et al. (2019). EXD2 Protects Stressed Replication Forks and Is Required for Cell Viability in the Absence of BRCA1/2. Mol Cell 75, 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, and Kowalczykowski SC (2011). BLM-DNA2-RPA-MRN and EXO1-BLMRPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25, 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orans J, McSweeney EA, Iyer RR, Hast MA, Hellinga HW, Modrich P, and Beese LS (2011). Structures of human exonuclease 1 DNA complexes suggest a unified mechanism for nuclease family. Cell 145, 212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandita TK (1983). Effect of temperature variation on sister chromatid exchange frequency in cultured human lymphocytes. Hum Genet 63, 189–190. [DOI] [PubMed] [Google Scholar]
- Pandita TK (2006). Role of mammalian Rad9 in genomic stability and ionizing radiation response. Cell Cycle 5, 1289–1291. [DOI] [PubMed] [Google Scholar]
- Pattschull G, Walz S, Grundl M, Schwab M, Ruhl E, Baluapuri A, CindricVranesic A, Kneitz S, Wolf E, Ade CP, et al. (2019). The Myb-MuvB Complex Is Required for YAP-Dependent Transcription of Mitotic Genes. Cell Rep 27, 3533–3546. [DOI] [PubMed] [Google Scholar]
- Paull TT (2018). 20 Years of Mre11 Biology: No End in Sight. Mol Cell 71, 419–427. [DOI] [PubMed] [Google Scholar]
- Paull TT, and Gellert M. (1998). The 3’ to 5’ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell 1, 969–979. [DOI] [PubMed] [Google Scholar]
- Paumard-Hernandez B, Calvete O, Inglada Perez L, Tejero H, Al-Shahrour F, Pita G, Barroso A, Carlos Trivino J, Urioste M, Valverde C, et al. (2018). Whole xome sequencing identifies PLEC, EXO5 and DNAH7 as novel susceptibility genes in testicular cancer. Int J Cancer 143, 1954–1962. [DOI] [PubMed] [Google Scholar]
- Peng J, and Gygi SP (2001). Proteomics: the move to mixtures. J Mass Spectrom 36, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Pepe A, and West SC (2014). Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res 42, 3833–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, and Helleday T. (2010). Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol cell 37, 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pichierri P, Franchitto A, and Rosselli F. (2004). BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J 23, 3154–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokharel S, and Campbell JL (2012). Cross talk between the nuclease and helicase activities of Dna2: role of an essential iron-sulfur cluster domain. Nucleic Acids Res 40, 7821–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kubler K, Mouw KW, et al. (2017). A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretto DI, Tsutakawa S, Brosey CA, Castillo A, Chagot M-E, Smith JA, Tainer JA, and Chazin WJ (2010). Structural dynamics and single-stranded DNA binding activity of the three N-terminal domains of the large subunit of replication protein A from small angle X-ray scattering. Biochemistry 49, 2880–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Lemacon D, and Vindigni A. (2017). Replication Fork Reversal: Players and Guardians. Mol Cell 68, 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid F, Harris PD, Zaher MS, Sobhy MA, Joudeh LI, Yan C, Piwonski H, Tsutakawa SE, Ivanov I, Tainer JA, et al. (2017). Single-molecule FRET unveils induced-fit mechanism for substrate selectivity in flap endonuclease 1. Elife 6:e21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Tomaszowski K-H, Luzwick JW, Park S, Li J, Murphy M, and Schlacher K. (2018). P53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLθ pathways. Elife 7:e31723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M. (2011). Doublestrand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, and Mann M. (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, et al. (2014). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Pandita RK, Singh M, Chakraborty S, Hambarde S, Ramnarain D, Charaka V, Ahmed KM, Hunt CR, and Pandita TK (2018). MOF Suppresses Replication Stress and Contributes to Resolution of Stalled Replication Forks. Mol Cell Biol 38:e00484–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Hunt CR, Pandita RK, Kumar R, Yang CR, Horikoshi N, Bachoo R, Serag S, Story MD, Shay JW, et al. (2013). Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol Cell Biol 33, 1210–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontz PA, Mui TP, Fuss JO, Tainer JA, and Barton JK (2012). DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc Natl Acad Sci U S A 109, 1856–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Gerik KJ, Stith CM, Yoder BL, and Burgers PM (2019). The roles of fission yeast exonuclease 5 in nuclear and mitochondrial genome stability. DNA Repair (Amst) 83, 102720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Kumar R, Singh M, Wold MS, Pandita TK, and Burgers PM (2012). Human exonuclease 5 is a novel sliding exonuclease required for genome stability. J Biol Chem 287, 42773–42783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steczkiewicz K, Muszewska A, Knizewski L, Rychlewski L, and Ginalski K. (2012). Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res 40, 7016–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor ER, and McGowan CH (2008). Cleavage mechanism of human Mus81-Eme1 acting on Holliday-junction structures. Proc Natl Acad Sci U S A 105, 3757–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208, 545562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Merino D, Nimmervoll B, Gupta K, Wang YD, Finkelstein D, Dalton J, Ellison DW, Ma X, Zhang J, et al. (2015). Cross-Species Genomics Identifies TAF12, NFYC, and RAD54L as Choroid Plexus Carcinoma Oncogenes. Cancer Cell 27, 712–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo KM, Yuan SS, Lee EY, and Sung P. (1998). Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J Biol Chem 273, 21447–21450. [DOI] [PubMed] [Google Scholar]
- Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, et al. (2011). Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 145, 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Sarker AH, Ng C, Arvai AS, Shin DS, Shih B, Jiang S, Thwin AC, Tsai MS, Willcox A, et al. (2020. a). Human XPG nuclease structure, assembly, and activities with insights for neurodegeneration and cancer from pathogenic mutations. Proc Natl Acad Sci U S A. 117, 14127–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Thompson MJ, Arvai AS, Neil AJ, Shaw SJ, Algasaier SI, Kim JC, Finger LD, Jardine E, Gotham VJB, et al. (2017). Phosphate steering by Flap Endonuclease 1 promotes 5’-flap specificity and incision to prevent genome instability. Nat Commun 8, 15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Tsai C-L, Yan C, Bralic A, Chazin JC, Hamdan SM, Scharer OD Ivanov I, Tainer JA (2020b). Envisioning how the prototypic molecular machine TFIIH functions in transcription initiation and DNA repair. DNA Repair 96, 102972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Brabant AJ, Ye T, Sanz M, German IJ, Ellis NA, and Holloman WK (2000). Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry 39, 14617–14625. [DOI] [PubMed] [Google Scholar]
- Wang AT, and Smogorzewska A. (2015). SnapShot: Fanconi anemia and associated proteins. Cell 160, 354–354. [DOI] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, et al. (2008). Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter P, Hanselmann S, Pattschull G, Schruf E, and Gaubatz S. (2017). Central spindle proteins and mitotic kinesins are direct transcriptional targets of MuvB, B-MYB and FOXM1 in breast cancer cell lines and are potential targets for therapy. Oncotarget 8, 11160–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, and Hickson ID (2003). The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874. [DOI] [PubMed] [Google Scholar]
- Xiong YC, Wang J, Cheng Y, Zhang XY, and Ye XQ (2020). Overexpression of MYBL2 promotes proliferation and migration of non-small-cell lung cancer via upregulating NCAPH. Mol Cell Biochem 468, 185–193. [DOI] [PubMed] [Google Scholar]
- Yan C, Dodd T, He Y, Tainer JA, Tsutakawa SEI (2019). Transcription preinitiation complex structure and dynamics provide insight into genetic diseases. Nat Struct Mol Biol 26, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JT, Cammack R, and Dillingham MS (2009). An iron-sulfur cluster is essential for the binding of broken DNA by AddAB-type helicase-nucleases. J Biol Chem 284, 7746–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JT, Poli J, Marians KJ, and Pasero P. (2013). Rescuing stalled or damaged replication forks. Cold Spring Harb Perspect Biol 5, a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Sengupta S, Yang Q, Linke SP, Yanaihara N, Bradsher J, Blais V, McGowan CH, and Harris CC (2005). BLM helicase facilitates Mus81 endonuclease activity in human cells. Cancer Res 65, 2526–2531. [DOI] [PubMed] [Google Scholar]
- Zhou C, Pourmal S, and Pavletich NP (2015). Dna2 nuclease-helicase structure, mechanism and regulation by Rpa. Elife 4:e09832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The x-ray diffraction data and coordinates of EXO5 structures in complex with ssDNA are deposited in Protein Data Bank (PDB) (https://www.rcsb.org). PDB ID: 7LW7 (DNA-free), 7LW8 (ssDNA-1, Complex 1), 7LW9 (ssDNA-2 with Sm/Na, Complex 2), and 7LWA (T88E mutant with ssDNA and Mg, Complex 3).
The original western blots and gel images are deposited in Mendeley data. http://dx.doi.org/10.17632/y82wnzcwg8.1
The MS proteomics data (dataset identifier PXD011727) have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository.
https://www.ebi.ac.uk/pride/archive/ Reviewer account details:
Username: reviewer99034@ebi.ac.uk
Password: x3aDXMZ9
The codes used for genomic analyses are available at https://github.com/abacolla/tcgaAnalyses