
FIGURE 5.
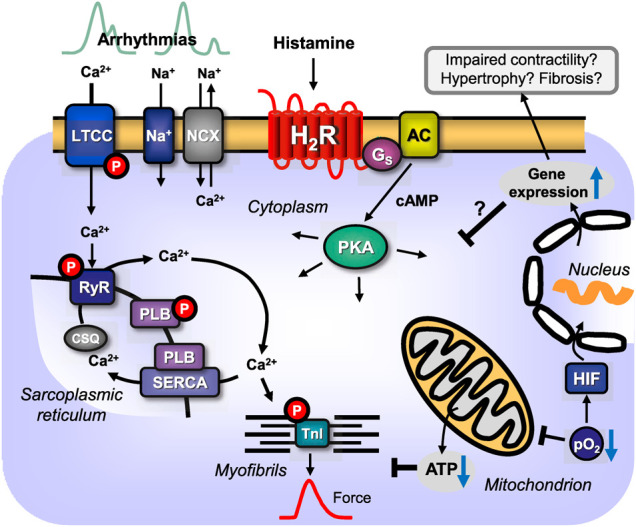
Scheme: putative pathophysiological role(s) of cardiac H2-histamine-receptors (H2R). H2R via stimulatory GTP-binding proteins (Gs) can activate adenylyl cyclases (AC) which would enhance the 3′,5′-cyclic adenosine-phosphate (cAMP)-levels in central compartments of the cardiomyocyte and activate cAMP-dependent protein kinases (PKA), which would increase the phosphorylation state and thereby the activity of various regulatory proteins in the cell (see Figure 1A). PKA-stimulated phosphorylation might also increase the current through the L-type Ca2+ channel (LTCC) and/or release of Ca2+ from the sarcoplasmic reticulum (SR) via the cardiac ryanodine receptor (RYR), which can occur in a non synchronous way that leads to early (top left) or delayed (top right) afterdepolarizations and thus to arrhythmias. In diastole, Ca2+ is pumped via the SR-Ca2+-ATPase (SERCA) from the cytosol into the SR. Activity of SERCA is increased by phosphorylation of phospholamban (PLB). PKA can enhance nuclear gene transcription. In this context, the expression of putatively detrimental proteins may be enhanced and that may impair cardiac function by fostering fibrosis and hypertrophy, reduce cardiac contractility and may lead to heart failure. Hypoxia (reduced oxygen partial pressure: pO2) and ischaemia impair respiration in the mitochondrion and thus formation of ATP in mitochondria or might activate directly hypoxia-inducible transcription factors (HIF). Increased expression or altered function of sarcolemmal ion channels like the sodium cation channel (Na+) or the sodium/calcium exchanger (NCX) but also increased expression of H2-histamine receptors, can lead to supraventricular or ventricular arrhythmias by alteration of Ca2+ homeostasis.